
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hardwood versus softwood Kraft lignin – precursor-product relationships in the manufacture of porous carbon nanofibers for supercapacitors†
Philipp
Schlee
 ae,
Omid
Hosseinaei
b,
Christopher A.
O' Keefe
c,
María José
Mostazo-López
d,
Diego
Cazorla-Amorós
d,
Servann
Herou
a,
Per
Tomani
b,
Clare P.
Grey
c and
Maria-Magdalena
Titirici
*a
ae,
Omid
Hosseinaei
b,
Christopher A.
O' Keefe
c,
María José
Mostazo-López
d,
Diego
Cazorla-Amorós
d,
Servann
Herou
a,
Per
Tomani
b,
Clare P.
Grey
c and
Maria-Magdalena
Titirici
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
bRISE Research Institutes of Sweden, Drottning Kristinas väg 61 Stockholm, Sweden
cUniversity of Cambridge, Department of Chemistry, Lensfield Road, Cambridge CB2 1EW, UK
dDepartamento de Química Inorgánica e Instituto de Materiales, Universidad de Alicante, Apartado 99, E-03080 Alicante, Spain
eDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Vuorimiehentie 1, 02150, Espoo, Finland
First published on 22nd October 2020
Abstract
The process of stabilization is essential in the production of carbon fibers from lignins. During stabilization, the initially thermoplastic lignin polymer is converted to a thermoset polymer allowing for high-temperature treatment without a change in shape. In this work, hardwood (HKL) and softwood (SKL) Kraft lignins were stabilized in air at temperatures between 190 and 340 °C before carbonization at 800 °C in a nitrogen atmosphere. Due to the differences in side-chain linkages, functional groups and molar mass, the lignins exhibit different structural changes upon stabilization and hence develop different porosities upon carbonization. Both lignins undergo major crosslinking reactions in the side chains at low temperatures and degradation reactions at high temperatures during stabilization. Crosslinking gives rise to narrow pore size distributions with mainly (sub-) nanometer pores, whereas degradation reactions lead to a more open pore structure with additional mesoporosity (>2 nm). When both types of reactions take place simultaneously, highly accessible (sub-) nanoporosity can be effectively created, which boosts the performance of supercapacitors operating in 6 M KOH(aq). This effect terminates when the crosslinking reactions cease and mainly degradation reactions take place, which occurs in HKL at 340 °C. SKL shows both a lower degree of crosslinking and degradation and hence develops less specific surface area. The optimum performance in an aqueous alkaline supercapacitor is achieved with HKL stabilized at 310 °C. It shows a specific gravimetric capacitance of 164 F g−1 at 0.1 A g−1 and 119 F g−1 at 250 A g−1 with a capacitance retention of more than 90% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
1. Introduction
Kraft lignin, long regarded as a waste stream of low value, presents a promising feedstock for the production of bio-materials, -chemicals and -fuels. Kraft lignin emerges as a by-product from the Kraft process that disintegrates the nature-made composite wood, separating cellulose from lignin and hemicellulose to produce paper.1 Generally, the current use of lignins can be divided into two main branches: (1) as a source for low molecular weight chemicals or fuels and (2) as a macromolecular precursor for materials synthesis.2,3 Since lignin is an oxygenated polymer formed by combinatorial polymerization which is highly altered during the Kraft process, it is chemically and structurally inherently different from hydrocarbons, such as polyolefins. It can be described as a polymer with a crosslinked, complex and three-dimensional structure of phenylpropanoid units bonded through various ether bonds (C–O–C) or carbon–carbon (C–C) linkages.4 Together with its high degree of aromaticity and its high carbon content (>60%) lignin is an ideal candidate for the preparation of porous carbons with good yield, which is a distinct difference to other biopolymers, such as cellulose, hemicellulose or chitosan.5The reactions during the Kraft process yielding new structural arrangements (building units and linkages) and chemical compositions of Kraft lignins have been the focus of several research works in the past.6–9 In particular, advanced spectroscopic and rigorous analytical methods give more and more insights into the diverse structural arrangements of various Kraft lignins.6,8,10 These insights have been proven useful en route to low molecular weight compounds via catalytic depolymerization.3,11 It was found that the higher the number of C–C linkages in the lignin side chains, the less successful the depolymerization; whereas the presence of a large number of more labile arylglycerol-β-O-4′ aryl ether linkages (β-O-4′ ether bonds) has been shown to facilitate the deconstruction of lignin enormously.7,11,12 The different linkages and structural arrangements were also investigated in the use of lignin as a macromolecular precursor for the production of carbon fibers.13–15 Braun et al. investigated the structural and chemical changes of meltspun fibers during the oxidative thermostabilization of hardwood lignin.16 Reactions occurring during thermostabilization in air increase the molar mass and hence the glass transition temperature (Tg).16 It was found that the homolytic cleavage of the α-O-4′ and β-O-4′ ether bonds takes place first during oxidative thermostabilization.16,17 This can be explained by the lower bond dissociation energies (BDE) of the ether bonds (α-O-4′: ∼55 kcal mol−1, and β-O-4′: ∼65 kcal mol−1) compared to the various C–C bonds (∼72–110 kcal mol−1).17–20 The BDEs were shown to vary depending on the number and position of methoxy group substituents on the phenyl rings in the structure of lignin model compounds.21 The bond homolysis yields radical intermediates such as phenethyl and phenoxy radicals which undergo a series of rearrangement, elimination and oxidation reactions.16,20 The scission of methoxy groups was shown to occur only at higher temperatures (≥280 °C) during oxidative thermostabilization.16 The influence of different thermostabilization conditions on the properties of the final carbon fibers has also been investigated by several authors.14,22,23 The authors focused on optimizing the process of oxidative thermostabilization of lignin fibers which is regarded to be one of the bottlenecks towards commercialization of lignin-based carbon fibers. It was found that for meltspun lignin fibers, slow heating rates in the range of 0.01 to 0.5 °C min−1 are needed in order to attain a thermoset lignin and hence retain the fiber shape and properties during carbonization.13,24 Recently, Cho et al. showed that the stabilization temperature and heating rate have only a minor effect on the structure of carbonized electrospun nanofiber mats from softwood lignin.25 These differences did not correlate with changes in mechanical performance.25 Various softwood and hardwood lignin-derived carbon nanofibers (LCNFs) were tested in supercapacitors operating in different electrolytes and cell configurations.26–30 To date, a clear inference concerning the reasons for superior performance of one lignin compared with another is out of reach. Jeon et al. started to address this issue by relating the molar mass of various lignin precursors with the creation of nanoporosity and the performance of the lignin-based carbon powder in aqueous acidic supercapacitors.31 The molar mass, however, has been shown to be an elusive lignin property due lignin's heterogeneity and inadequate measurement techniques.32
Nonetheless, finding precursor-product relationships is decisive to make further progress in the context of Kraft lignin valorization in energy storage applications. Herein, we compare a eucalyptus hardwood Kraft lignin (HKL) with a pine/spruce softwood Kraft lignin (SKL), which are two of the most important types of lignins arising from large scale papermaking, for the manufacture of electrospun CNFs used as free-standing electrodes in aqueous alkaline supercapacitors.33 We show for the first time that the type of Kraft lignin used as precursor determines the properties (e.g. nanoporosity) of the resulting LCNFs and hence their performance in aqueous alkaline supercapacitors. Specifically, we relate the type and relative quantities of the side-chain linkages (β-O-4 ether/C–C linkages) and functional groups (e.g. methoxy groups) of the Kraft lignins to the development of the nanoporosity in the LCNFs, which is the decisive property in electric double layer supercapacitors.
2. Experimental
2.1 Preparation of the lignin precursors
The hardwood Kraft lignin was isolated using the LignoBoost process.34 The black liquor, from eucalyptus wood, was obtained from the ENCE pulp mill (Huelva, Spain). The lignin was fractionated as described in our previous publications.26,27A softwood (pine/spruce mixture) Kraft lignin was obtained from a LignoBoost process (Bäckhammer, Sweden). The lignin was dried at 80 °C under vacuum for about 12 h. A sequential solvent extraction was used to produce a lignin fraction suitable for electrospinning. The solvent extraction method is a simplified version of the method described in Baker and Hosseinaei, 2014.35 The dry lignin (1000 g) was first extracted with methanol by rapidly stirring in 10 L methanol at room temperature for 2 h. The sample was filtrated and the solid recovered on the filter was then extracted using a 70/30 (v/v) mixture of methanol and dichloromethane (1:10 solid to liquid ratio, room temperature, 4 h). The extract solution was then filtered, and lignin from the filtrate was isolated using a rotary evaporator. The isolated lignin was dried in a vacuum oven at 80 °C for 12 h and was used for electrospinning. The final yield for this fraction was 42.5%.
2.2 Preparation of electrospun lignin-based carbon nanofiber mats
The precursor solutions for electrospinning contained either 50 wt% eucalyptus hardwood Kraft lignin (HKL) dissolved in dimethylformamide (DMF) (VWR) and methanol (v/v = 80/20) or 47 wt% pine/spruce softwood Kraft lignin (SKL) dissolved in DMF. These solutions were typically sonicated for 90 min and repeatedly shaken for several minutes and finally stirred for several days in a sealed glass vial until a homogeneous solution was obtained. The electrospinning procedure was similar to the one described in our previous publication.26 After electrospinning, the as-spun lignin nanofiber mats were converted to carbon nanofiber mats by a two-step thermal treatment: (1) stabilization under an air flow (500 ml min−1) and (2) carbonization under N2 flow (500 ml min−1) in a quartz tube (OD: 80 mm) furnace (OTF-1200X, MTI). The lignin nanofiber mats were stabilized at 190 °C, 250 °C, 310 °C and 340 °C for 15 min, where the ramp between RT and 50 °C was 5 °C min−1 and the one between 50 °C and the corresponding stabilization temperature was 0.5 °C min−1. Subsequently, the stabilized nanofiber mats were carbonized at a fixed temperature of 800 °C for 120 min, where the ramp between RT and 110 °C was 5 °C min−1 and the one between 110 °C and the target temperature was set to 3 °C min−1.2.3 Characterization
:1 v/v) and mixed with 100 μL of a solution of N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (20 mg ml−1) and chromium(III) acetylacetonate (5 mg ml−1) as internal standard and relaxation agent, respectively. 31P NMR spectra were acquired using an inverse-gated decoupling pulse sequence with a 90° pulse angle, 10 s relaxation delay, and 512 scans.
000 cycles for the best performing samples (S310-HKL and S340-SKL). In order to gain more insights into the electrochemical properties of the CNFs, a third reference electrode was inserted into the cells for the best performing samples (S310-HKL and S340-SKL). This allows for the measurement of the voltage–time profiles (from GCD measurements) and CV data of the individual electrodes independent of each other as both electrodes are referenced to the unpolarizable reference electrode (saturated calomel electrode, SCE). This is not possible without a reference electrode as then only the total cell voltage can be measured. The results may be seen in the ESI† of this article.
3. Results and discussion
3.1 Lignin precursor characterization
The key properties of the two Kraft lignins used as starting materials for the manufacture of electrospun carbon nanofibers (CNFs) are shown in the lignin property polygon (LPP) in Fig. 1. The characterization methods used to obtain the values presented in the LPP are described in our previous publication.26 The purity of both samples is very high, as indicated by the low ash and carbohydrate content. The low sulfur content (S in Fig. 1) peculiar to Kraft lignins is 2.3% and 1.3% for HKL and SKL, respectively. The carbon content (C in Fig. 1) of SKL is 65.8%, whereas that of HKL is 62.2%. The weight average molar masses (Mw in Fig. 1) of the two lignins (HKL and SKL) are different, where HKL has a molar mass of 1900 g mol−1 and SKL a molar mass of 8600 g mol−1. The polydispersity index (PDI = Mw/Mn) of SKL (2.8) is elevated compared to HKL (1.5). Overall, the PDIs of both lignins are low due to the fractionation process applied which effectively eliminates lower molar mass fractions.35,40,41 Another important difference between the two lignins is the distribution of the hydroxyl groups between aliphatic and phenolic units. The ratio of aliphatic to phenolic OH groups (Al/Ph OH in Fig. 1) is higher for SKL (0.5) compared to HKL (0.3).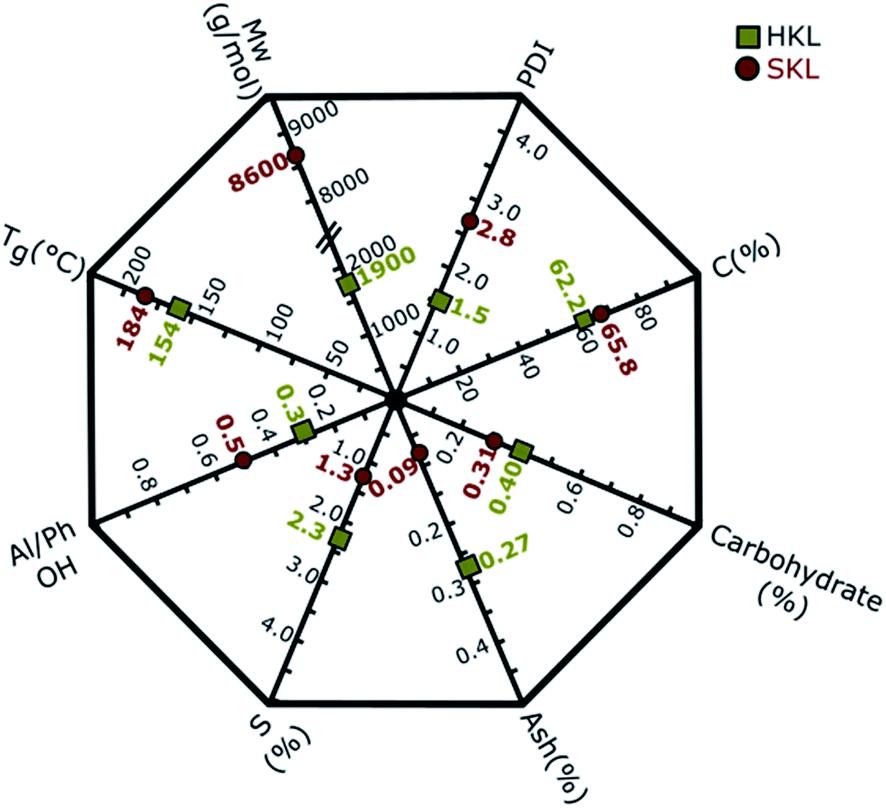 | ||
| Fig. 1 Lignin property polygon (LPP) summarizing key properties of the hardwood (HKL) and softwood (SKL) Kraft lignin precursors: carbon (C), carbohydrate, ash and sulfur (S) content, ratio of aliphatic to phenolic OH groups (Al/Ph OH), weight average molar mass (Mw), polydispersity index (PDI) and glass transition temperature (Tg). | ||
The glass transition temperature (Tg) of HKL is 154 °C, which is lower than that of SKL (184 °C). The disparity in Tg results from the differences in molar mass, number/type of linkages between different building units and functional groups.
2D 1H–13C heteronuclear single quantum correlation (HSQC) NMR measurements were carried out to reveal the structural differences of the SKL and HKL precursors in more detail. The spectra are shown in the ESI (Fig. S.1†). In the aromatic region of the spectrum of HKL, signals corresponding to syringyl (S) and guaiacyl (G) units were found, as expected for hardwood lignins.42,43 The spectrum of SKL exhibits very pronounced signals of G units typical of softwood lignins.42,43 Signals related to stilbenes were identified (Cα–H, Cβ–H) in the spectrum of SKL. These are generated from phenylcoumaran units (β-5′),6 which (Cα–H) were also detected in the side-chain region of SKL. For the development of porosity during heat treatment, the side chains of lignins connecting the stable aromatic units are of great importance. The main linkages determined by 2D 1H–13C HSQC NMR are presented as percentages based on the total number of linkages in Scheme 1. The most pronounced difference between the two Kraft lignin precursors is the relative amount of β-O-4′ ether bonds. In HKL, 63.3% of all linkages are β-O-4′ ether bonds, whereas in SKL, these bonds account for only 35.0% of all linkages. Conversely, the number of more stable C–C bonds in the resinol, epiresinol and phenylcoumaran structures are higher in SKL than in HKL (Scheme 1). Therefore, the ratio of β-O-4 ether to C–C bonds between the aromatic units can be interpreted as an indicator for rigidity/stability of the lignin side chains, which is substantially lower in HKL than in SKL.
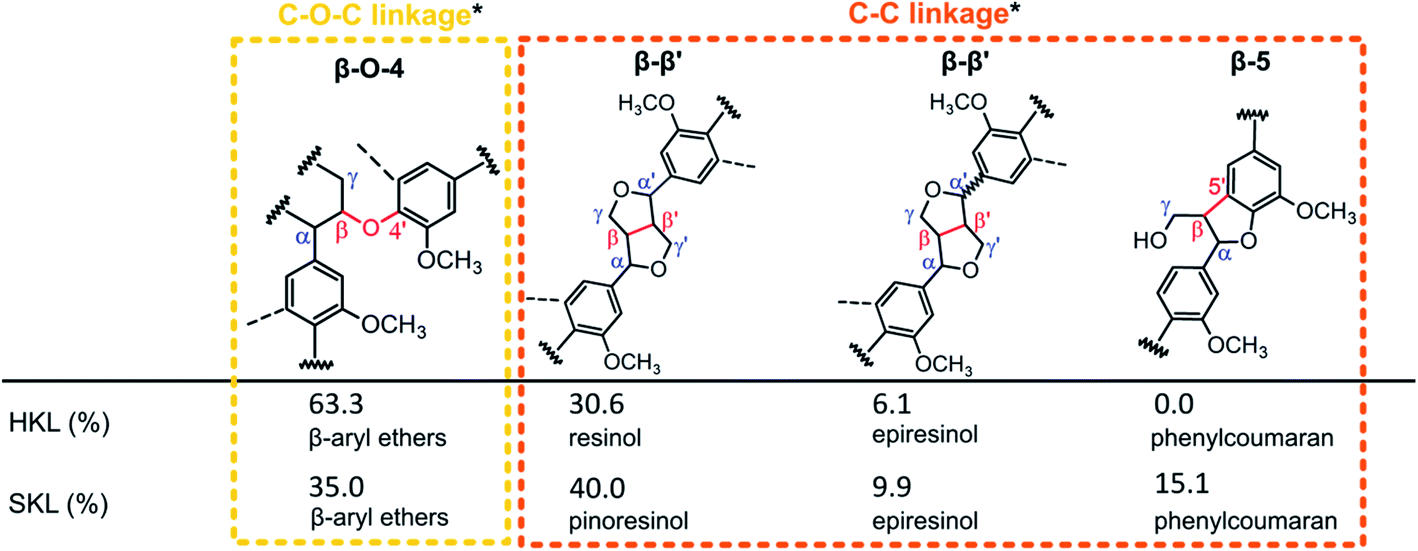 | ||
| Scheme 1 Comparative quantification of linkages based on 2D HSQC NMR (*linkages are categorized based on the strongest bonds connecting two aromatic units). | ||
Next to the quantity of different linkages in the lignin side chains, a second important factor is the type and number of hydroxyl groups present in the lignin polymer. The hydroxyl groups are important in the formation of inter- and intramolecular hydrogen bonds in lignin.44–46 The distribution of OH-groups was investigated by quantitative 31P NMR.38,39 The 31P NMR spectra and the corresponding hydroxyl group distributions of the lignin precursors are presented in Fig. S.2† and Scheme 2, respectively. The total number of hydroxyl functional groups in SKL is 5.35 mmol g−1, which is only slightly lower compared to HKL (5.41 mmol g−1); however, the distribution of hydroxyl groups is different. From the OH-group distributions shown in Scheme 2, it is obvious that HKL is composed of a mixture of phenolic guaiacyl (G) and syringyl (S) moieties and a minor amount of p-hydroxyphenyl (H) units. The SKL precursor contains exclusively G and a minor amount of H moieties, which is consistent with the results from 2D HSQC NMR.
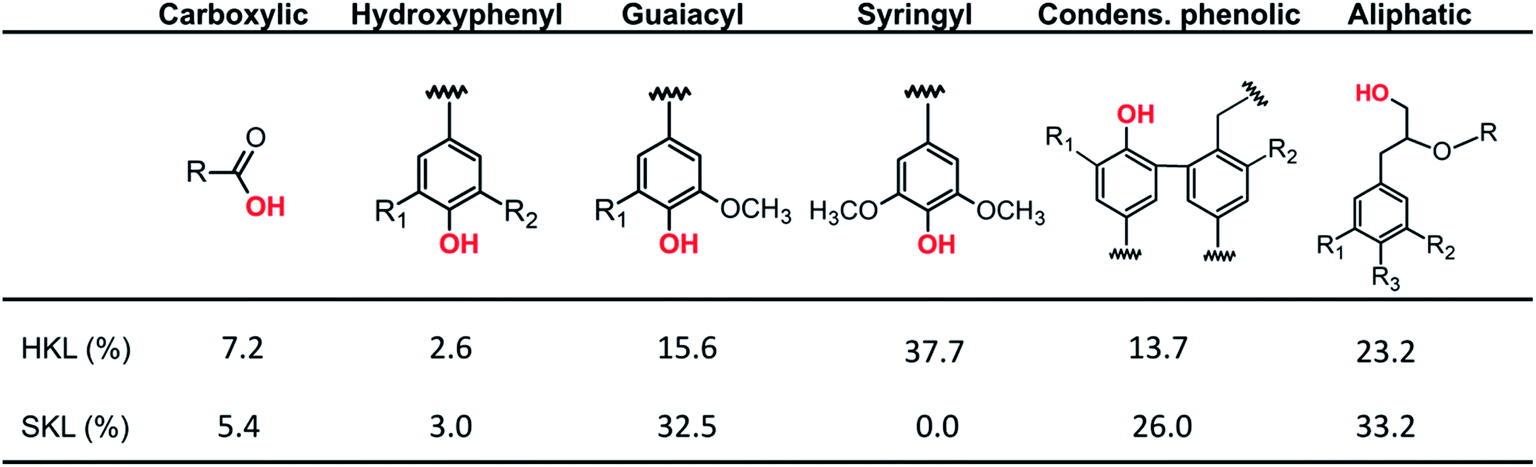 | ||
| Scheme 2 Distribution of hydroxyl functional groups in HKL and SKL precursors determined by 31P NMR. | ||
Additionally, SKL contains considerably more of the condensed phenolic units which is in line with the higher number of C–C bonds in SKL shown by 2D HSQC NMR. Furthermore, there are more aliphatic OH groups in SKL compared to HKL, meaning that SKL can potentially be stabilized at higher heating rates and at lower temperatures than HKL which is an important economic consideration for the carbon fiber production.47 The elevated content of carboxylic acid groups in HKL contributes to a lower thermal stability due to the decarboxylation reaction occurring at low temperatures.48
Stabilizing HKL and SKL at the same heating rate (0.5 °C min−1), but different final stabilization temperatures (190, 250, 310, 340 °C) gives the opportunity to investigate the influence of the starting precursor and stabilization temperature on the development of porosity and oxygen groups in the final LCNFs.
3.2 Stabilization process
To follow the structural and chemical changes upon oxidative thermostabilization FT-IR (SI), 1H–13C CP/MAS NMR and thermogravimetry (TG) coupled with a mass spectrometry system (MS) (TG-MS) were used. It was found that a stabilization temperature of 190 °C is not sufficient to stabilize HKL under the chosen conditions (heating rate/dwell time). It melted and formed a brittle film upon carbonization at 800 °C in N2, whereas SKL was stable during carbonization and retained the fiber shape (Fig. 3).From 1H–13C CP/MAS NMR, it is evident that hardly any structural changes occur in HKL and SKL at such a low temperature. However, in SKL, the aliphatic C–H region (0–40 ppm) changes slightly together with the formation of carboxylic acid groups (∼180 ppm) indicating that even at 190 °C, the oxidation of the SKL side chains takes place. TG-MS clearly shows that the extent of this reaction is small as the amount of gaseous products detected is low (Fig. 2a and b).
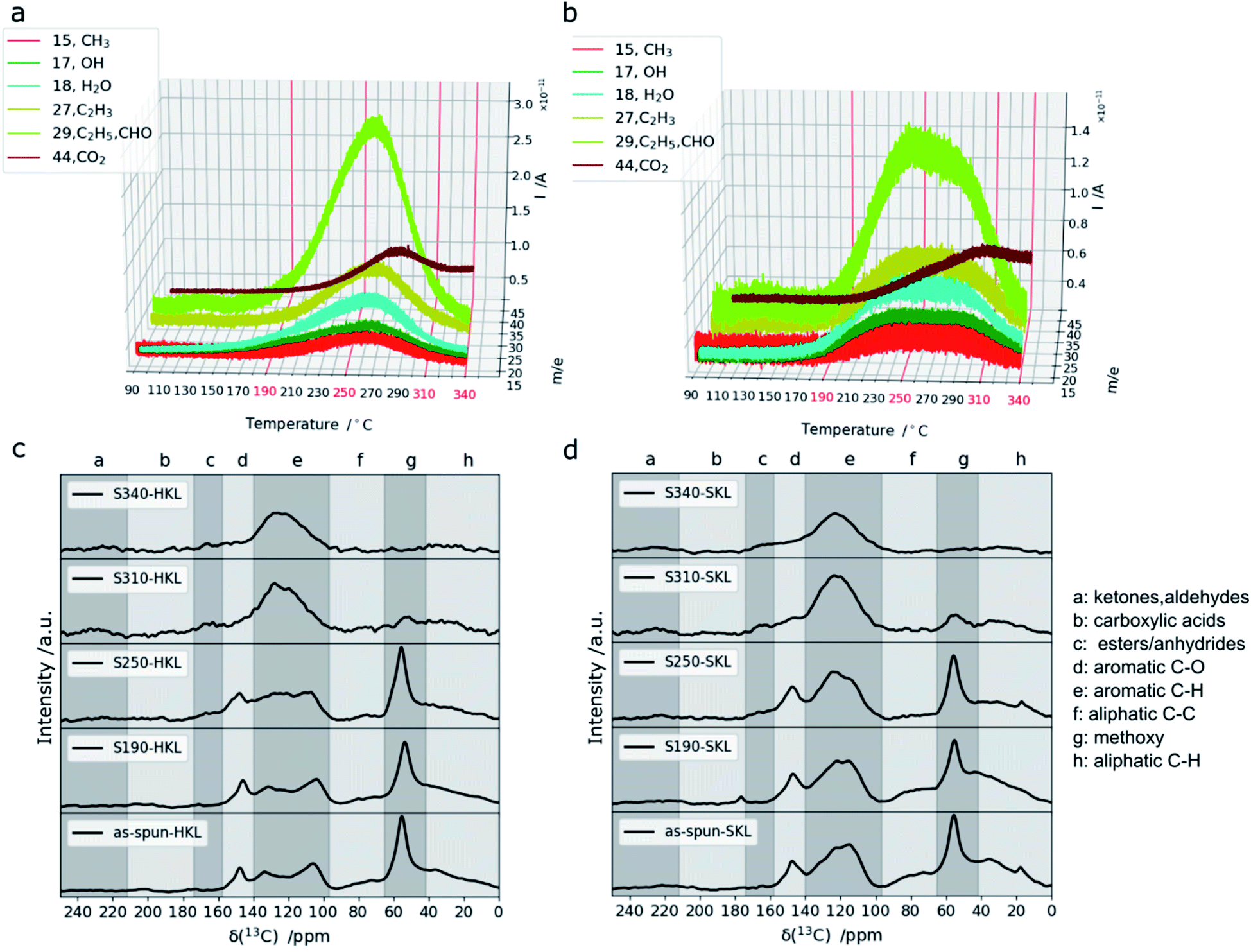 | ||
| Fig. 2 TG-MS signals recorded during stabilization of HKL (a) and SKL (b) and 1H–13C CP/MAS NMR spectra of as-spun fibers and after stabilization at different temperatures of HKL (c) and SKL (d). | ||
At a temperature of 250 °C, both lignins can be stabilized without fusing. For both HKL and SKL, C2H5/CHO (m/z = 29), C2H3 (m/z = 27), H2O (m/z = 18), OH (m/z = 17) and CH3 (m/z = 15) were detected. Additionally, an increasing amount of CO2 (m/z = 44) was measured for both lignins with an onset at 220 °C. The signal-to-noise ratios of all signals recorded by TG-MS are lower for SKL compared to HKL indicating that crosslinking reactions take place to a lesser extent in SKL. This is in line with the more rigid (C–C bonded), hence less reactive side chains of SKL and a lower amount of methoxy groups.17,49 Additionally, the TG-MS signals of HKL are sharp with distinct maxima, whereas the ones of SKL are very broad covering a wider temperature range which may be explained by the higher molar mass (Fig. 1) imposing more diffusional constraints on the volatile products. Moreover, the stable side chains of SKL are composed of bonds with bond dissociation energies that cover a broad range.48 In the 1H–13C CP/MAS NMR spectra of both lignins (Fig. 2c and d), a decrease in intensity in the aliphatic C–H (0–40 ppm) and C–C region (65–97 ppm) can be seen after stabilization at 250 °C. Additionally, a shoulder emerging between 158 and 174 ppm indicates formation of ester/anhydride linkages. These are clear indicators that crosslinking took place. At 310 °C, the drastic decrease in intensity of the signal between 42 and 65 ppm indicates that the scission of methoxy groups took place.
Additionally, the signal between 140 and 158 ppm, indicative of aromatic C–O bonds, disappeared in both lignins and the signals for anhydride and ester linkages are more pronounced than at 250 °C. Hence, the degree of crosslinking in both lignins is enhanced. The TG-MS data shows that at 310 °C, the amount of volatile products decreases more sharply for HKL than for SKL. At 340 °C, there are hardly any structural changes discernible in the 1H–13C CP/MAS NMR spectra in line with the low amount of volatile products detected. Only the signal for CO2 detected in TG-MS rises at high stabilization temperatures. It shows a maximum at around 280 °C for HKL and around 300 °C for SKL and stays at a constant level between 310 and 340 °C for both lignins. In this high temperature region, it can be expected that C–C bonds in the resinol (epiresinol and pinoresinol) structures are affected. For instance, the β-5′ bond in the phenylcoumaran units only present in SKL was shown to be one of the strongest bonds in lignins with a bond dissociation energy of approximately 110 kcal mol−1.17 Breaking these more stable bonds under air flow results in CO2 as a volatile product explaining the reverse trend for CO2 in TG-MS. The weight loss upon stabilization at 310 and 340 °C increases enormously for both lignins as shown in Fig. S.4.† This suggests that stabilization in this temperature region involves increasing degradation and decreasing crosslinking.
3.3 Carbon nanofiber characterization
After stabilization at different temperatures in air, all samples were carbonized at 800 °C for 120 min in nitrogen. SEM images of the resulting LCNFs can be seen in Fig. 3. The overall micro-morphology is similar for HKL and SKL derived LCNFs. Both consist of a randomly entangled nanofiber network. The average LCNF diameters for the samples stabilized at 190 °C (SKL) and 250 °C (HKL) are similar (460 ± 115 nm and 470 ± 109 nm, respectively). As the temperature is increased, the LCNFs derived from HKL show substantially smaller diameters compared to their low temperature counterparts. The LCNFs based on HKL stabilized at 340 °C have an average diameter of 280 ± 69 nm. | ||
| Fig. 3 SEM micrographs of the carbonized HKL (top) and SKL (bottom) fibers stabilized at different temperatures. | ||
This effect was not observed for the LCNFs based on SKL which showed no significant change over the entire temperature range (Table S.2†). This can be related to the higher thermal stability (Tg) of the SKL-derived CNFs compared to the HKL-derived ones. The micro-morphological differences are not expected to affect the electrochemical performance but may be of interest for the mechanical properties. The main factors influencing the performance of LCNFs in electric double layer supercapacitors (EDLC) are the accessible surface area, pore size distribution and the surface functional groups.50,51 The development of nano- and mesoporosity with increasing stabilization temperature is shown in Fig. 4. The overall specific surface area values (m2 g−1) and the corresponding pore size distributions upon increasing the stabilization temperature are considerably different for both lignins. For HKL the specific surface area derived from nitrogen sorption increases linearly with rising stabilization temperature from 250 to 340 °C (Fig. 4b). From the pore size distributions shown in Fig. 4c, it becomes clear that the increase is accompanied by a pore widening. When stabilized at 250 °C (major crosslinking event), the majority of pores is within a very narrow range of around 1.5 nm in diameter (Fig. 4a). Upon increasing the stabilization temperature to 310 °C, which was shown to result in the scission of methoxy groups and further crosslinking reactions, a more open pore structure with bigger nano- and mesopores (1.5–5 nm) is created. At the same time, the porosity in the (sub-) nanometer range probed by CO2 sorption measurements increases too. The latter is assigned to the crosslinking reactions that give rise to a denser polymeric framework with smaller pores (<2 nm).52,53 Interestingly, increasing the stabilization temperature further to 340 °C changes the pore size distribution so that mainly mesopores between 2 and 5 nm are present. This correlates well with 1H–13C CP/MAS NMR and TG-MS data which show that no crosslinking reactions take place at 340 °C, but the cleavage of rigid C–C bonds releasing CO2 dominates. This can be considered as a pre-activation step that leads to increasing pore diameters of pre-existing (sub-) nanopores. This is also confirmed by CO2 sorption measurements. The specific surface areas derived from these measurements show that stabilization at lower temperatures involving crosslinking reactions contributes to an increase in number of pores with diameters below 1 nm. However, the fibers stabilized at 340 °C (no crosslinking) show no increase in (sub-) nanopore surface area (Fig. 4b). This will have implications on the capacitive performance in symmetric supercapacitors operating with 6 M KOH(aq) as electrolyte. Turning to SKL subjected to the same stabilization conditions, a completely different trend can be observed. The differences in the number of C–C linkages in the side chains, fewer methoxy functional groups and higher molar mass of SKL affects the development of porosity tremendously.
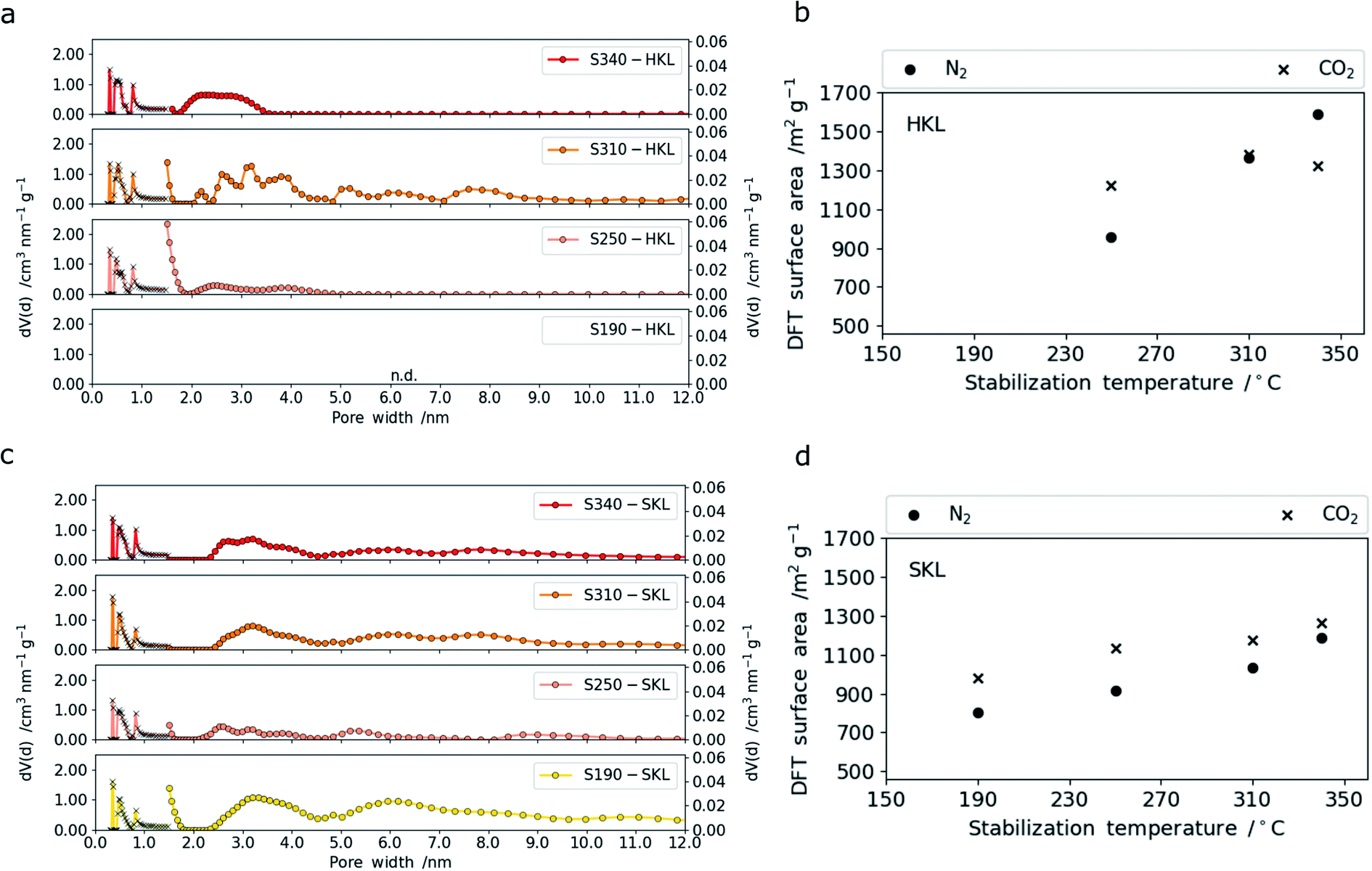 | ||
| Fig. 4 Pore size distributions of HKL (a) and SKL (c) with increasing stabilization temperatures from bottom (190 °C) to top (340 °C) (traces with cross-shaped (x) symbols are derived from CO2 sorption and circle-shaped symbols from N2 sorption measurements). Development of the DFT surface area with increasing stabilization temperature for HKL (b) and SKL (d). | ||
Overall, the specific surface areas determined for SKL are lower than those for HKL. This is a direct consequence of the difference in linkages in the side chains and the number of functional groups (especially methoxy groups). For SKL, both specific surface areas determined from N2 (nano-mesopores) and CO2 ((sub-) nanopores) sorption measurements increase nearly in a linear fashion with rising stabilization temperature. This indicates that the pore widening effect is by far less present in SKL. The observation is in line with the TG-MS data which shows that between 310 and 340 °C minor crosslinking and degradation reactions take place giving rise to a slight increase in both (sub-) nanometer and mesopores.
At very low stabilization temperatures of 190 °C, which did not lead to major structural changes in the polymer, both mesopores and nanopores are present. Stabilization at 250 °C results in a densification of the polymeric framework through major crosslinking reactions and to a narrower pore size distribution with a reduced amount of mesopores in the resulting LCNFs. This is analogous to HKL which indicates that the denser polymeric network created by the crosslinking reactions (most intense at 250 °C) for both lignins yield narrow pore size distributions in the final LCNFs. As SKL is stabilized at higher temperatures (310 and 340 °C), which involves scission of methoxy groups and cleavage of more stable C–C bonds together with further crosslinking reactions, the meso-porosity increases (pore size: 2–5 nm) together with an increase in (sub-) nanoporosity. Overall, the less reactive SKL with more rigid C–C-rich side chains shows a gradual increase in porosity with rising stabilization temperature, whereas the more reactive HKL composed of more ether bonds in the side chains and more methoxy groups shows tremendous changes upon stabilization in a narrow temperature range between 250 and 340 °C. As the two lignins are stabilized, two types of reactions can be distinguished, namely crosslinking and degradation. The former leads to an increase in (sub-) nanoporosity, the latter to an increase in mesoporosity and larger nanopores (>1 nm). Depending on the type of lignin, these reactions take place to different extents and over different temperature ranges.
The oxygen content on the LCNF surface dependent on stabilization temperature was determined by temperature programmed desorption (TPD) shown in Fig. 5 and XPS (Fig. S.7 and S.8†). The total oxygen content of both types of lignins varies only slightly between 4 and 6 wt% over the entire temperature range. Hence, the stabilization temperature does not have a major impact on the oxygen content of the carbonized fibers. The only notable variation is the type of oxygen functional groups in HKL. Stabilized at 310 °C, HKL contains more CO2 desorbing, whereas at 340 °C, more CO desorbing O-functional groups are present. This will have an impact on the capacitance retention as it was shown that CO2 desorbing functional groups are less stable in aqueous alkaline electrolytes than CO desorbing oxygen groups.54 It was shown that increasing oxygenation of the carbon electrodes results in a higher affinity towards alkali and alkaline earth metal ions, such as K+ or Ca2+, contributing to an increase in capacitance.55
 | ||
| Fig. 5 Total amount of desorbed CO and CO2 from TPD measurements dependent on stabilization temperature for HKL (a) and SKL (b). Total oxygen content in wt% derived from the sum of CO + 2CO2 desorbed from the samples. | ||
3.4 Electrochemical characterization
The LCNFs derived from HKL and SKL stabilized at different temperatures and carbonized at 800 °C for 120 min under nitrogen atmosphere were used as free-standing electrodes in supercapacitors. The LCNFs were used without the need of a binder, conductive additive and metallic substrate. The measurements were carried out in symmetric two-electrode supercapacitor cells operating with a 6 M KOH(aq) electrolyte. The resulting trends of capacitance with increasing current densities may be seen in Fig. 6a and b. Comparing the two lignins, it is obvious that HKL shows higher capacitance and rate capability than SKL. This can be related to the higher specific surface area and enhanced accessibility of the nanoporosity. This in turn arises from the more labile side chains which creates accessible nanoporosity at low stabilization temperatures by crosslinking reactions. Additionally, at higher stabilization temperatures, the scission of methoxy groups, which are more abundant in HKL, together with further crosslinking creates both (sub-) nano- and mesoporosity. However, at a stabilization temperature of 340 °C pore widening by cleavage of C–C bonds is detrimental to the capacitive performance in 6 M KOH(aq). Conversely, SKL shows increasing specific surface area in both (sub) nanometer and mesopore range as the stabilization temperature is increased.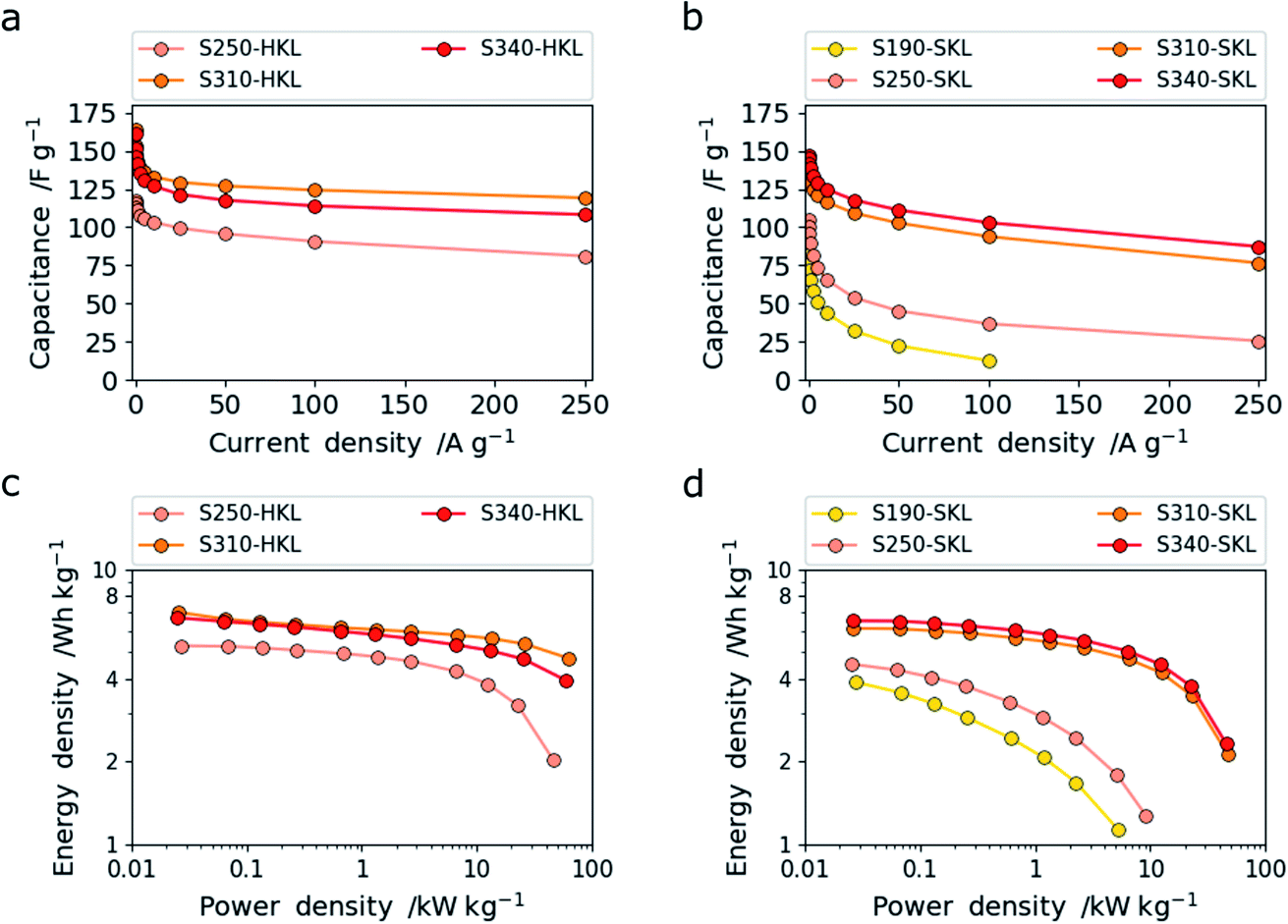 | ||
| Fig. 6 Specific gravimetric capacitance for HKL (a) and SKL (b) with increasing current density and the corresponding Ragone plots for HKL (c) and SKL (d). | ||
This is assigned to the more rigid side chains in SKL that give rise to crosslinking reactions even up to 340 °C leading to enhanced (sub-) nanoporosity rather than the pore-widening effect. At low temperatures, S190- and S250-SKL (SKL stabilized at 190 and 250 °C and carbonized at 800 °C) show very low capacitance and rate capability. A leap in performance can be observed by the scission of methoxy groups in conjunction with crosslinking reactions creating enhanced (sub-) nanoporosity at 310 °C. This is analogous to HKL. S340-SKL shows a small increase in capacitance consistent with the additional increase of (sub-) nanoporosity rather than pore widening. In Fig. 7a and b, galvanostatic charge–discharge (GCD) traces measured at 1 A g−1 are shown. All of them have a triangular shape characteristic of electric double layer supercapacitors (EDLCs).
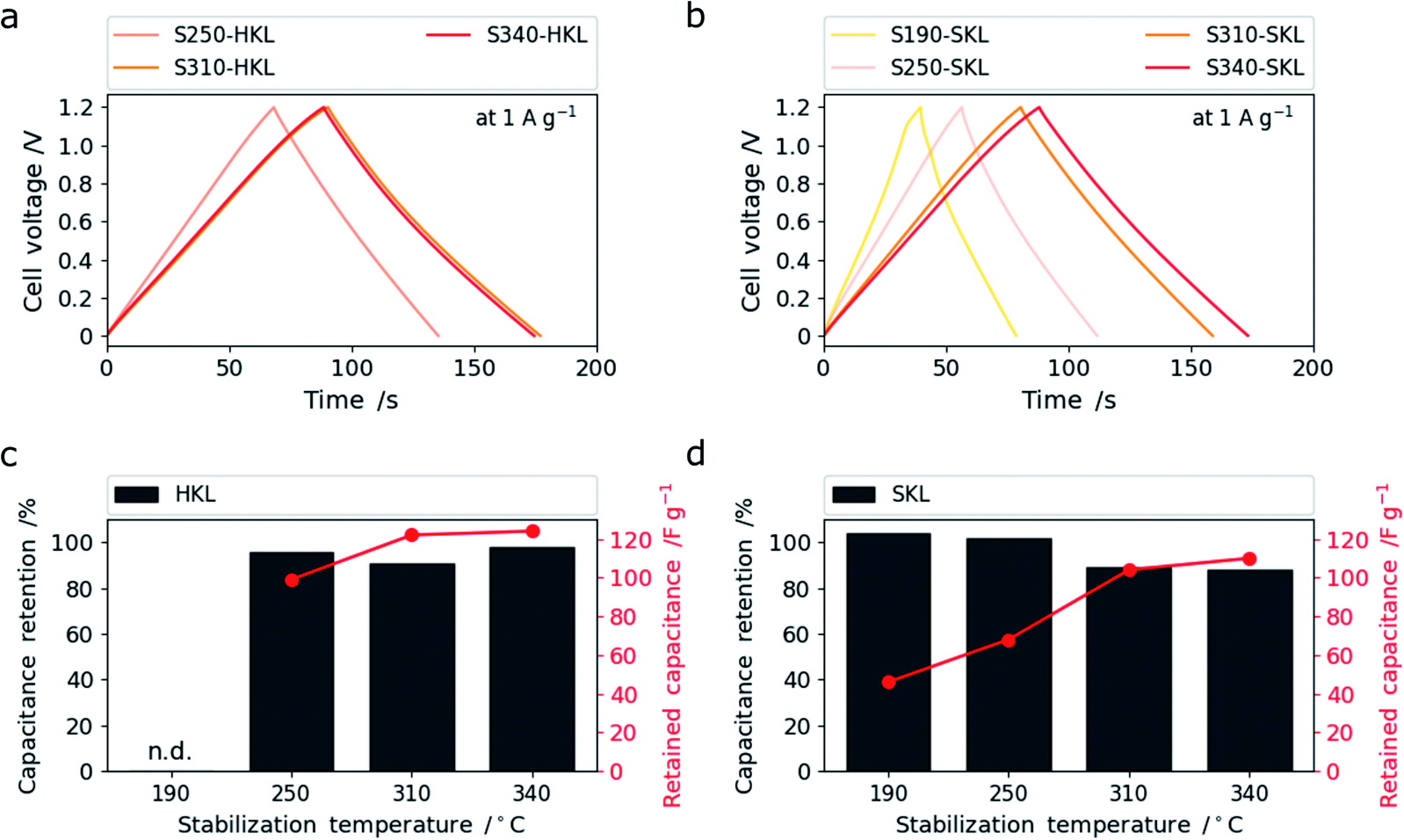 | ||
| Fig. 7 Galvanostatic charge–discharge traces of HKL (a) and SKL (b) at 1 A g−1 and the capacitance retention after 6000 cycles at 10 A g−1 as percentage in form of a bar chart and as absolute values (red curves) for HKL (c) and SKL (d). | ||
The capacitance retention after 6000 cycles at 10 A g−1 is shown in Fig. 7c and d as a relative value (in percent) and as an absolute value (in F g−1). All samples show good capacitance retention with over 90%. S250-HKL and S340-HKL show retention of even more than 95% which makes the difference between the S310-HKL and S340-HKL in terms of capacitive performance only marginal after 6000 cycles. This might be related to the differences in the type of oxygen functional groups determined by TPD. S190- and S250-SKL show capacitance retentions of more than 100% after 6000 cycles which can be assigned to a wetting effect which arises from charging and discharging the material repeatedly.26 This proves the low accessibility of existing (sub-) nanoporosity. S310- and S340-SKL show similar capacitance retention and hence the improved capacitive performance caused by the difference in stabilization temperature is retained even after 6000 cycles. The most promising samples S310-HKL and S340-SKL were further cycled up to 10000 cycles retaining still more than 90% of the initial capacitance (Fig. S.16†).
4. Conclusion
Two Kraft lignins were shown to develop different porosities when subjected to increasing stabilization temperatures before carbonization. The differences in side-chain linkages have a profound effect on the development of porosity during heat treatment and this in turn affects the performance of the resulting lignin-based CNFs used as free-standing supercapacitor electrodes. Upon stabilization two types of reactions occur: (1) crosslinking and (2) degradation. Depending on the nature of the side chains in the lignins, these reactions occur to a different extent and in different temperature windows yielding different porosities. HKL with more β-O-4′ ether bonds, more methoxy groups and lower molar mass undergoes crosslinking reactions at 250 °C yielding a very narrow pore size distribution in the (sub-) nanometer range. At 310 °C, both crosslinking and degradation reactions occur which yields both nano- and mesopores. At 340 °C, mainly degradation reactions (C–C cleavage) take place that lead to a widening of pre-existing nanopores which is detrimental to the capacitive performance. SKL, with fewer β-O-4 ether bonds, but more C–C bonds, fewer methoxy groups and higher molar mass undergoes both crosslinking and degradation reactions over the temperature range of 250–340 °C. Both types of reactions occur to a lesser extent in SKL. Therefore, SKL shows far less pore widening than HKL. The sample with the most suitable porosity (both nano- and mesopores) and hence best performance as free-standing electrode in supercapacitors was prepared by stabilizing HKL at 310 °C. It shows a specific gravimetric capacitance of 164 F g−1 at 0.1 A g−1 and 119 F g−1 at 250 A g−1 with a capacitance retention of more than 90% after 10000 cycles. In the future, these results obtained based on low loadings, which is applicable for comparing various materials with different properties without diffusion effects, will be consolidated by testing the same materials with industrially applicable loadings of up to 10 mg cm−2. Furthermore, the effect of different carbonization conditions (temperature, heating rate, dwell time) need to be investigated in order to find the optimum materials properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. M. L. and D. C. A. thank Spanish Ministry of Science, Innovation and Universities and FEDER (project RTI2018-095291-B-I00) for financial support. C. P. G. and C. O'K. thank Shell. MMT and PS thank RISE AB for co-funding Philipp Schlee's PhD position.References
- C. Houtman, Lessons Learned from 150 Years of Pulping Wood, in Lignin Valorization: Emerging Approaches, The Royal Society of Chemistry, 2018, pp. 62–73 Search PubMed.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- A. Bengtsson, J. Bengtsson, C. Olsson, M. Sedin, K. Jedvert, H. Theliander and E. Sjöholm, Holzforschung, 2018, 72, 1007–1016 CAS.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- C. S. Lancefield, G. M. M. Rashid, F. Bouxin, A. Wasak, W.-C. Tu, J. Hallett, S. Zein, J. Rodríguez, S. D. Jackson, N. J. Westwood and T. D. H. Bugg, ACS Sustain. Chem. Eng., 2016, 4, 6921–6930 CrossRef CAS.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- D. Ibarra, M. I. Chávez, J. Rencoret, J. C. Del Río, A. Gutiérrez, J. Romero, S. Camarero, M. J. Martínez, J. Jiménez-Barbero and A. T. Martínez, J. Agric. Food Chem., 2007, 55, 3477–3490 CrossRef CAS.
- Y. Lu, Y.-C. Lu, H.-Q. Hu, F.-J. Xie, X.-Y. Wei and X. Fan, J. Spectrosc., 2017, 2017, 1–15 CrossRef.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 3552–3599 CrossRef CAS.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS.
- I. Brodin, M. Ernstsson, G. Gellerstedt and E. Sjöholm, Holzforschung, 2012, 66, 141–147 CAS.
- Z. Yue, A. Vakili, O. Hosseinaei and D. P. Harper, J. Appl. Polym. Sci., 2017, 134, 45507 CrossRef.
- M. Foston, G. A. Nunnery, X. Meng, Q. Sun, F. S. Baker and A. Ragauskas, Carbon, 2013, 52, 65–73 CrossRef CAS.
- J. L. Braun, K. M. Holtman and J. F. Kadla, Carbon, 2005, 43, 385–394 CrossRef CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- P. F. Britt, A. C. Buchanan, M. J. Cooney and D. R. Martineau, J. Org. Chem., 2000, 65, 1376–1389 CrossRef CAS.
- P. F. Britt, A. C. Buchanan and E. A. Malcolm, Energy Fuels, 2000, 14, 1314–1322 CrossRef CAS.
- P. F. Britt, A. C. Buchanan and E. A. Malcolm, J. Org. Chem., 1995, 60, 6523–6536 CrossRef CAS.
- A. Beste and A. C. Buchanan III, J. Org. Chem., 2009, 74, 2837–2841 CrossRef CAS.
- Z. Dai, X. Shi, H. Liu, H. Li, Y. Han and J. Zhou, RSC Adv., 2018, 8, 1218–1224 RSC.
- M. Cho, M. Karaaslan, S. Chowdhury, F. Ko and S. Renneckar, ACS Sustain. Chem. Eng., 2018, 6, 6434–6444 CrossRef CAS.
- O. Hosseinaei, M. Åkerström, A. Uhlin, P. Tomani and S. Danielsson, in NWBC 2018 Proceedings of the 8th Nordic Wood Biorefinery Conference, ed. E. Hytönen and J. Vepsäläinen, 2018, pp. 157–163 Search PubMed.
- M. Cho, F. K. Ko and S. Renneckar, ACS Omega, 2019, 4, 5345–5355 CrossRef CAS.
- P. Schlee, O. Hosseinaei, D. Baker, A. Landmér, P. Tomani, M. J. Mostazo-López, D. Cazorla-Amorós, S. Herou and M. M. Titirici, Carbon, 2019, 145, 470–480 CrossRef CAS.
- P. Schlee, S. Herou, R. Jervis, P. R. Shearing, D. J. L. Brett, D. Baker, O. Hosseinaei, P. Tomani, M. M. Murshed, Y. Li, M. J. Mostazo-López, D. Cazorla-Amorós, A. B. Jorge Sobrido and M. M. Titirici, Chem. Sci., 2019, 10, 2980–2988 RSC.
- F. J. García-Mateos, R. Berenguer, M. J. Valero-Romero, J. Rodríguez-Mirasol and T. Cordero, J. Mater. Chem. A, 2018, 6, 1219–1233 RSC.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X.-F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134–141 CrossRef CAS.
- J. L. Espinoza-Acosta, P. I. Torres-Chávez, J. L. Olmedo-Martínez, A. Vega-Rios, S. Flores-Gallardo and E. A. Zaragoza-Contreras, J. Energy Chem., 2018, 27, 1422–1438 CrossRef.
- J.-W. Jeon, L. Zhang, J. L. Lutkenhaus, D. D. Laskar, J. P. Lemmon, D. Choi, M. I. Nandasiri, A. Hashmi, J. Xu, R. K. Motkuri, C. A. Fernandez, J. Liu, M. P. Tucker, P. B. McGrail, B. Yang and S. K. Nune, ChemSusChem, 2015, 8, 428–432 CrossRef CAS.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS.
- J. R. Dillen, S. Dillén and M. F. Hamza, Pulp and Paper: Wood Sources, in Reference Module in Materials Science and Materials Engineering, ed. S. Hashmi, Elsevier, Oxford, 2016, pp. 1–6 Search PubMed.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- D. Baker and O. Hosseinaei, High Glass Transition Lignins and Lignin Derivatives for the Manufacture of Carbon and Graphite Fibers, US Pat. 20140271443A1, 2014.
- J. Rencoret, A. Gutiérrez, L. Nieto, J. Jiménez-Barbero, C. B. Faulds, H. Kim, J. Ralph, Á. T. Martínez and J. C. del Río, Plant Physiol., 2011, 155, 667–682 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- J. Rönnols, H. Schweinebarth, A. Jacobs, J. S. Stevanic, A.-M. Olsson, A. Reimann and F. Aldaeus, Nord. Pulp Pap. Res. J., 2015, 30, 550–561 Search PubMed.
- R. Mörck, H. Yoshida and K. P. Kringstad, Holzforschung, 1986, 40, 51–60 CrossRef.
- H. Yoshida, R. Mörck, K. P. Kringstad and H. Hatakeyama, Holzforschung, 1987, 41, 171–176 CrossRef CAS.
- R. Katahira, T. J. Elder and G. T. Beckham, A Brief Introduction to Lignin Structure, in Lignin Valorization: Emerging Approaches, The Royal Society of Chemistry, 2018, pp. 1–9 Search PubMed.
- J. Ralph, G. Brunow and W. Boerjan, Lignins, in Encyclopedia Of Life Sciences, John Wiley & Sons, 2007, pp. 1–10 Search PubMed.
- O. Hosseinaei, D. P. Harper, J. J. Bozell and T. G. Rials, ACS Sustain. Chem. Eng., 2016, 4, 5785–5798 CrossRef CAS.
- S. Kubo and J. F. Kadla, Biomacromolecules, 2005, 6, 2815–2821 CrossRef CAS.
- Y. Uraki, Y. Sugiyama, K. Koda, S. Kubo, T. Kishimoto and J. F. Kadla, Biomacromolecules, 2012, 13, 867–872 CrossRef CAS.
- O. Hosseinaei, D. P. Harper, J. J. Bozell and T. G. Rials, Int. J. Mol. Sci., 2017, 18, 1410 CrossRef.
- T. Faravelli, A. Frassoldati, G. Migliavacca and E. Ranzi, Biomass Bioenergy, 2010, 34, 290–301 CrossRef CAS.
- T. Elder, Energy Fuels, 2014, 28, 1175–1182 CrossRef CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- C. Lian, H. Liu, D. Henderson and J. Wu, J. Phys.: Condens. Matter, 2016, 28, 414005 CrossRef.
- N. B. Mckeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS.
- J. Weber, Q. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- M. J. Bleda-Martínez, J. A. Maciá-Agulló, D. Lozano-Castelló, E. Morallón, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2005, 43, 2677–2684 CrossRef.
- L. Guan, L. Yu and G. Z. Chen, Electrochim. Acta, 2016, 206, 464–478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta09093j |
| This journal is © The Royal Society of Chemistry 2020 |