
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling stable MnO2 matrix for aqueous zinc-ion battery cathodes†
Yiding
Jiao
 a,
Liqun
Kang
b,
Jasper
Berry-Gair
a,
Kit
McColl
a,
Jianwei
Li
a,
Haobo
Dong
a,
Hao
Jiang
c,
Ryan
Wang
b,
Furio
Corà
a,
Dan J. L.
Brett
b,
Guanjie
He
*abd and
Ivan P.
Parkin
*a
a,
Liqun
Kang
b,
Jasper
Berry-Gair
a,
Kit
McColl
a,
Jianwei
Li
a,
Haobo
Dong
a,
Hao
Jiang
c,
Ryan
Wang
b,
Furio
Corà
a,
Dan J. L.
Brett
b,
Guanjie
He
*abd and
Ivan P.
Parkin
*a
aChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk; i.p.parkin@ucl.ac.uk
bDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
cKey Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science & Technology, 130 Meilong Road, Shanghai 200237, China
dJoseph Banks Laboratories, School of Chemistry, University of Lincoln, Green Lane, Lincoln, LN6 7DL, UK
First published on 20th October 2020
Abstract
The primary issue faced by MnO2 cathode materials for aqueous Zn-ion batteries (AZIBs) is the occurrence of structural transformations during cycling, resulting in unstable capacity output. Pre-intercalating closely bonded ions into the MnO2 structures has been demonstrated as an effective approach to combat this. However, mechanisms of the pre-intercalation remain unclear. Herein, two distinct δ-MnO2 (K0.28MnO2·0.1H2O and K0.21MnO2·0.1H2O) are prepared with varying amounts of pre-intercalated K+ and applied as cathodes for AZIBs. The as-prepared K0.28MnO2·0.1H2O cathodes exhibit relatively high specific capacity (300 mA h g−1 at 100 mA g−1), satisfactory rate performance (35% capacity recovery at 5 A g−1) and competent cyclability (ca. 95% capacity retention after 1000 cycles at 2 A g−1), while inferior cyclability and rate performance are observed in K0.21MnO2·0.1H2O. A stable δ-MnO2 phase is observed upon cycling, with the reversible deposition of Zn4SO4(OH)6·5H2O (ZSH), ion migration between electrodes and synchronous transition of Mn valence states. This work firstly and systematically reveals the role of the pre-intercalated ions via density functional theory simulations and show that above a threshold K/Mn ratio of ca. 0.26, the K ions suppress structural transformations by stabilizing the δ phase. To demonstrate its commercial potential, AZIBs with high-loading active materials are fabricated, which deliver adequate energy and power densities compared with most commercial devices.
Introduction
Issues of energy shortage and environmental pollution need to be addressed for human survival and social development. Thus, the development of renewable energy sources such as solar, wind and hydropower has become a global imperative.1,2 To effectively deliver energy from renewable sources and decarbonize transport systems, energy storage devices with desirable capacity, rate performance and cyclability are required. Li-ion batteries (LIBs) have been commercialized for portable devices and electric vehicles due to their wide operating voltage window and competitive energy/power densities.3 However, safety issues, expensive configurations, and concerns about critical materials availability are barriers to future development and applications.4–6 Therefore, cost-effective, safe, and environmentally-friendly energy storage devices as alternatives to LIBs are in urgent demand. Among other candidates, such as technologies based on alternate charge-carrying cations (e.g. K+, Na+, Al3+ and Ca2+),7–9 aqueous Zn-ion batteries (AZIBs) are drawing significant attention due to their low redox potential (−0.76 V vs. standard hydrogen electrode), outstanding theoretical capacity (820 mA h g−1), abundant resources, high safety and ambient fabrication.10–12Although extensive research focuses on Zn anodes and electrolyte modifications, a reliable and high-performance cathode has always been pursued to commercialize AZIBs. Among the candidates, vanadium-based cathodes exhibit the highest specific capacity (>300 mA h g−1) and prolonged cyclability, but their working voltage window is relatively low (<1.0 V vs. Zn/Zn2+), which hinders their practical use for high energy density applications.13–17 Prussian blue analogs (PBAs) operate at a high working voltage (1.5–1.7 V vs. Zn/Zn2+) but suffer from the relatively low specific capacity (50–120 mA h g−1).18,19 Manganese-based cathodes not only share outstanding specific capacity (200–300 mA h g−1) and high working voltage range (1.3–1.35 V vs. Zn/Zn2+)20–24 but also exhibit the advantages of abundant reserves, simple fabrication, low cost and environmental friendliness, and have therefore become a research hotspot.25–27 However, the application of manganese-based cathodes is severely hindered by substantial intrinsic issues. The soluble Mn2+, derived from the disproportionation reaction of Mn3+ and structural transformation, causes severe capacity decay upon cycling.28–34 Structural transformations are likely to occur between the tunnel and layered structures.30 According to previous reports, α-, β-, γ-, and λ-MnO2 usually experience phase transition into Zn-buserite, during which the volumetric change associated with this process causes significant residual stress and contributes to the amorphized manganese-based cathodes, ultimately leading to capacity loss during cycling.35 Several strategies have been proposed to improve performance. As an electrolyte additive, MnSO4 has been proven to suppress excessive Zn2+ insertion by limiting Mn vacancies upon cycling and maintaining structural integrity.36 The hybridization with carbon-based materials, such as graphene and carbon nanotubes, has been demonstrated as an efficient method to provide a protective host structure and prolongs the cyclability.20,37–43
Pre-intercalation has been widely applied in other layered materials such as vanadium oxides to enhance stability upon cycling; choice of the interlayer guest species acting as structure stabilizing “pillars” allows to tune lattice spacing, enhance ion mobility, confer inherent conductivity via shallow donor levels associated with reduced V ions.38,44–48 Besides, the presence of interlayer water in aqueous batteries screens the interaction between intercalated ion and cathode, leading to faster intercalation processes. Likewise, pre-intercalation of closely bonded ions has also been preliminarily studied to improve the performance of Mn-based cathodes.20 The effect of pre-intercalated ions is concluded as the electrostatic force between per-intercalated ions and O and the enhanced structural stability. However, such a conclusion is too vague and omits the possible structural transformation induced by the pre-intercalation, which leaves the working mechanism of pre-intercalation an unexplored area. The structure–performance relationship between pre-intercalated cations and electrochemical behavior needs to be considered and discussed.
In this work, two K+ pre-intercalated birnessite MnO2 materials with varied amounts of K+ were prepared via sol–gel and hydrothermal methods, respectively. Detailed physical and electrochemical characterizations were executed to disclose their dissimilarities in composition and the impact on electrochemical behavior. The AZIBs fabricated with K0.28MnO2·0.1H2O (K0.28MO) deliver a relatively high specific capacity of 300 mA h g−1 at 100 mA g−1. Even at a high current density of 2 A g−1, the AZIBs exhibit a sufficient specific capacity of 100 mA h g−1 and maintain >95% of their capacity over 1000 cycles, which is at the top level of relevant materials.26,27 In contrast, the AZIBs fabricated with K0.21MnO2·0.1H2O (K0.21MO) exhibit inferior performance. The energy storage mechanisms are thoroughly investigated with systematic ex situ analysis. A stable δ-MnO2 primitive phase was observed throughout the cycling process, along with the reversible deposition/dissolution of Zn4SO4(OH)6·5H2O (ZSH) phase, ion migration and simultaneous change of Mn valence state. The pre-intercalated K ions's underlying function was further scrutinized via density functional theory (DFT) simulations, demonstrating the phase selection role played by the K/Mn ratio and thus the structural stabilization for the birnessite phase achieved via the pre-intercalation of K ions at higher concentration. Moreover, to demonstrate practical applications, batteries were assembled with high loading (ca. 10 mg cm−2) of active materials, which show outstanding energy and power densities compared to current commercial energy storage systems.
Results and discussion
Materials and characterization
Birnessite K0.28MO was synthesized with a sol–gel method, while birnessite K0.21MO was obtained via a one-step hydrothermal process (detailed methods are described in ESI†). Transmission electron microscopy (TEM), X-ray fluorescence analysis (XRF), energy-dispersive X-ray spectroscopy (EDX) and coupled elemental mappings were used to characterize the morphology and elemental distribution.K0.28MO exhibits nanoplates's shape with a width and length of 50–100 nm (Fig. S1A†). The morphology of K0.28MO and K0.21MO are similar and further supported by the results shown in Fig. S2A, S1B, C and D.† Furthermore, the crystallographic structures of the two materials were studied by X-ray diffraction (XRD, Fig. 1A). All X-ray diffraction characteristic peaks of the two materials are perfectly indexed to the birnessite K0.28MnO2·0.54H2O phase (hexagonal, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (166), JCPDS 86-0666).49,50 The magnified image of the (003) diffraction peak shows that in K0.28MO, it shifts towards higher diffraction angles compared with K0.21MO.28,51 The enlarged TEM image further reveals that the lattice spacing of K0.28MO is 7.04 Å, which corresponds to the (003) lattice plane (Fig. S1B†). The homogenous elemental distribution of K, Mn and O is observed in K0.28MO (Fig. S3†). XRF analysis proves that the molar K/Mn ratio of K0.28MO is 0.28, while for K0.21MO, the ratio is 0.21 (Fig. S2E and S1F†). The water contents of both materials were further investigated by thermogravimetric analysis (TGA), as shown in Fig. S4.† Most of the physically absorbed water content evaporates from room temperature to 150 °C. Complete loss of structural water content occurred between 150 °C and 500 °C. The removal of the structural water content corresponds to a 2 wt% loss of total mass, corresponding to 0.1 moles of H2O per mole of K0.28MnO2 or K0.21MnO2.
m (166), JCPDS 86-0666).49,50 The magnified image of the (003) diffraction peak shows that in K0.28MO, it shifts towards higher diffraction angles compared with K0.21MO.28,51 The enlarged TEM image further reveals that the lattice spacing of K0.28MO is 7.04 Å, which corresponds to the (003) lattice plane (Fig. S1B†). The homogenous elemental distribution of K, Mn and O is observed in K0.28MO (Fig. S3†). XRF analysis proves that the molar K/Mn ratio of K0.28MO is 0.28, while for K0.21MO, the ratio is 0.21 (Fig. S2E and S1F†). The water contents of both materials were further investigated by thermogravimetric analysis (TGA), as shown in Fig. S4.† Most of the physically absorbed water content evaporates from room temperature to 150 °C. Complete loss of structural water content occurred between 150 °C and 500 °C. The removal of the structural water content corresponds to a 2 wt% loss of total mass, corresponding to 0.1 moles of H2O per mole of K0.28MnO2 or K0.21MnO2.
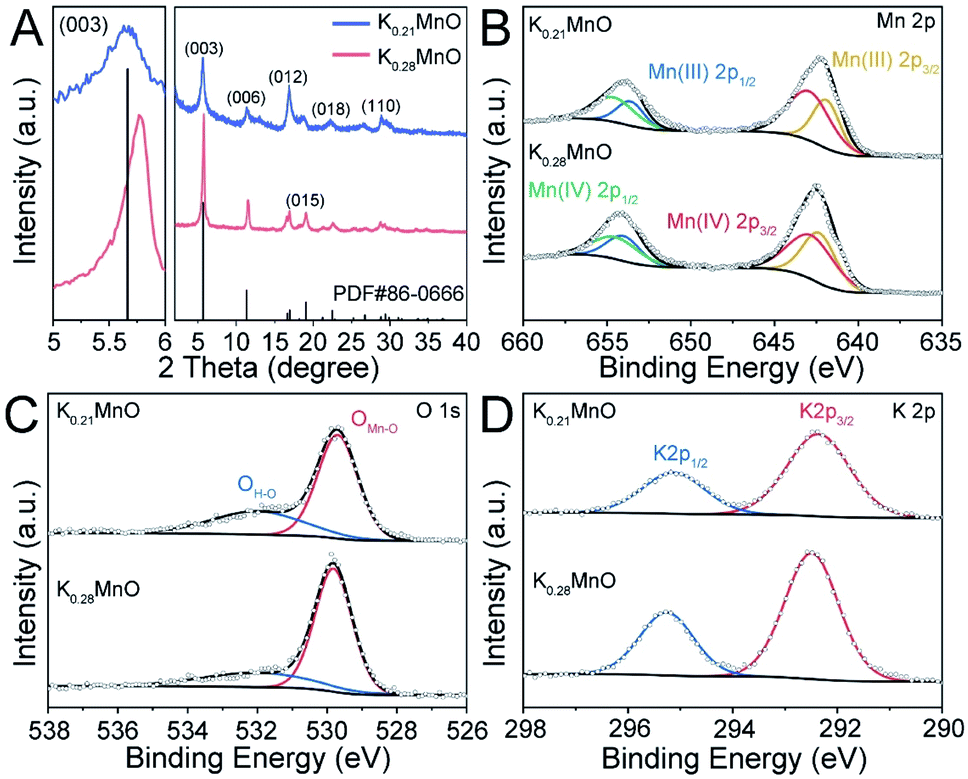 | ||
| Fig. 1 Material Characterization. (A) XRD patterns of K0.21MO and K0.28MO. (B) Mn 2p XPS spectra of K0.21MO and K0.28MO. (C) O 1s XPS spectra of K0.21MO and K0.28MO. (D) K 2p XPS spectra of K0.21MO and K0.28MO. | ||
To validate the counter-intuitive result that increased interlayer concentration of K causes a contraction of the interlayer separation, we performed a series of DFT calculations examining the equilibrium structure of KxMnO2 at varying K content x between 0 and 0.5. The results concerning the c lattice parameter and snapshots of the interlayer structures are reported in Fig. S5.† We observe an increase of interlayer spacing at low K content, followed by a contraction beyond a threshold value of ca. 0.3, which results from the stronger electrostatic interaction between the intercalated K+ ions and the reduced MnO2 layers. A contraction of interlayer separation, as observed experimentally, is therefore consistent with the atomic information provided by the computational study. The DFT-optimized lattice parameter for intercalant free δ-MnO2 is 13.05 Å, which expands to 20.32 Å upon intercalation of water at an H2O/Mn ratio of 0.25. Both values are in good agreement with experiment and previous reports.52,53
X-ray photoelectron spectroscopy (XPS) was applied to analyze the chemical composition and electronic structure of the two materials. As shown in Fig. 1B, both materials deliver similar Mn 2p shoulder peaks, which can be attributed to Mn 2p1/2 (654.4 eV) and Mn 2p3/2 (642.7 eV) with a spin-orbital splitting of 11.7 eV. A slight difference in the Mn(IV) and Mn(III) ratio is observed, deriving from different pre-intercalated K+ concentrations. The presence of O and K can also be confirmed by O 1s and K 2p spectra. As shown in Fig. 1C, two characteristic peaks in the O 1s spectra can be identified as OMn–O (529.7 eV) and OH–O (532.0 eV). Two shoulder peaks are also noted in the K 2p scan, which can be ascribed to K 2p1/2 (295.2 eV) and K 2p3/2 (292.5 eV) with the spin-orbital splitting of 2.77 eV (Fig. 1D).
Raman spectra are further applied to study the inter-layer and intra-layer bond strength (Fig. S6†). The Raman shift at ca. 640 cm−1 is assigned to the inter-layer Mn–O bonds, while the Raman shift at ca. 570 cm−1 is assigned to the intra-layer Mn–O bonds. It is indicated that the inter-layer Mn–O bonds have been strengthened in K0.28MnO, while the intra-layer Mn–O bonds are weakened.
Further proof of the difference in Mn valence states in the bulk structures of these two materials can be found in the Mn L3,2-edge near-edge X-ray absorption fine structure (NEXAFS spectra). As shown in Fig. S7A,† Mn L3,2-edge absorption features for both materials are very similar but different from c-Mn2O3 and c-MnO standard reference samples. The intensity ratio of L3/L2 absorption of both materials shows similarities with β-MnO2, which confirms high-spin states and similar oxidation states of Mn(IV). However, the first absorption peak at 641 eV is distinguished from the prominent absorption peak at 643.5 eV compared to β-MnO2, which results from Mn's different chemical environments in the framework. In the magnified L3-edge features, a slight shift (∼0.2 eV) towards lower energy of both the leading-edge position and the gravity center of the L3-edge is observed (Fig. S7B†), while similar shifts also exist in the L2-edge (Fig. S7C†), which both indicate a lower Mn oxidation state in K0.28MO compared to K0.21MO.
Electrochemical performance
The as-synthesized materials were assembled as cathodes in AZIB coin cell configurations using 3 M ZnSO4 and 0.2 M MnSO4 aqueous solution as the electrolyte for electrochemical tests. Fig. 2A illustrates the CV curves of K0.28MO at different scan rates from 0.1 to 1.0 mV s−1. Two couples of redox peaks can be observed, indicating a two-step electrochemical process. With the increase of the scan rate, CV curves present minor peak shifts, representing dependable reversibility and stability during the polarization processes. A linear fitting was further carried out between peak current densities (i) and the square root of the scan rate (v1/2) to evaluate the diffusion-controlled and capacitive contributions. It was calculated that the capacitive contribution is 53.7% of the total specific capacity at 0.1 mV s−1, and it gradually increases to 78.6% at 1.0 mV s−1 (Fig. 2B). CV curves are also measured with K0.21MO, which shows a similar two redox couple system and demonstrates similar electrochemical processes (Fig. S8A†). The capacitive contribution of K0.21MO is 34.7% at 0.1 mV s−1 and 62.7% at 1.0 mV s−1, much lower than that of the K0.28MO cathode, which can be explained by more active surface charge storage of K0.28MO (Fig. S8B†). To further understand the inherent resistive behavior of both materials, EIS was performed before and after the cell was charged and discharged for 200 cycles at 2 A g−1. We observed no change in charge-transfer resistance (R3) of K0.28MO, but there is an increase of R3 for K0.21MO (Fig. S8C and D†).2,54 It is reported that the dissolution of Mn is the primary reason for the capacity loss and impedance rise.55 Therefore, the results indicate that a higher amount of pre-intercalated K+ ions effectively suppress the Mn dissolution and subsequent impedance rise.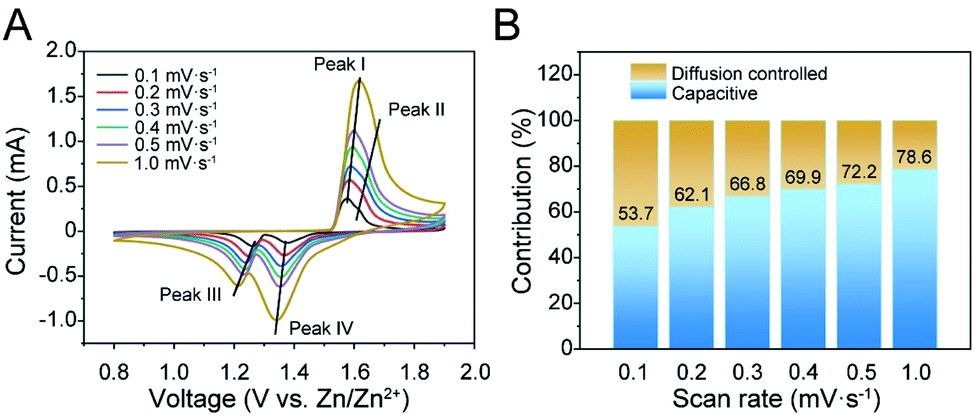 | ||
| Fig. 2 Electrochemical behavior of K0.28MO in 3 M ZnSO4 + 0.2 M MnSO4 electrolyte. (A) Cyclic voltammetry (CV) curves of K0.28MO material at different scan rates from 0.1–1.0 mV s−1. (B) The corresponding capacitive and diffusion-controlled contribution from the CV curves. | ||
To further elucidate the dissimilarity in electrochemical performance resulting from pre-intercalation, cycling performance and charge/discharge profiles were measured in AZIB coin cells. As shown in Fig. 3A, at 100 mA g−1, K0.28MO exhibited a lower initial capacity of 150 mA h g−1 with a gradual rise to 300 mA h g−1 at the 50th cycle. By contrast, K0.21MO exhibited a higher initial capacity of 230 mA h g−1, while the capacity reached only 260 mA h g−1 at the 50th cycle. The comparison of low rate performance confirms that higher amounts of pre-intercalated K+ help maintain structural integrity, ensuring a higher cycling capacity. However, the pre-intercalated K+ ions occupy the active sites for Zn intercalation and thus hinder the intercalation/deintercalation of other cations in the first few cycles, sacrificing a part of the initial capacity. The cycle number was further extended to 60 to prove the cycling stability of K0.28MO at the low rate. The battery can maintain a specific capacity of >300 mA h g−1 after the 50th cycle (Fig. S9†).
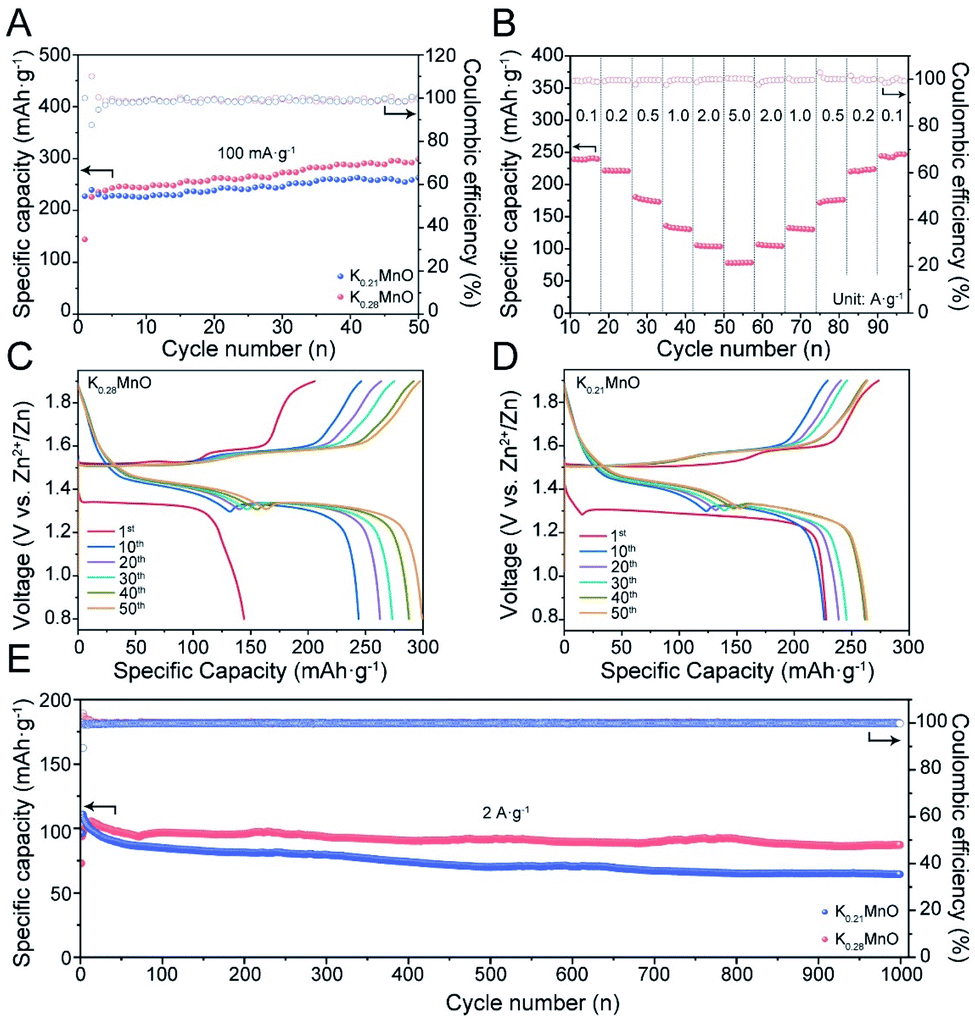 | ||
| Fig. 3 Electrochemical performance of electrodes tested in coin cells. (A) Cycling performance of K0.28MO and K0.21MO materials at a current density of 100 mA g−1. (B) Rate performance of K0.28MO. (C and D) Charge/discharge profiles of K0.28MO and K0.21MO cells at 100 mA g−1, respectively. (E) Cycling performance of K0.28MO and K0.21MO materials at a current density of 2 A g−1. | ||
The rate performance of both materials was further evaluated. Both materials exhibit good rate performance after 10 cycles of activation. The capacity retention at 5 A g−1 for the K0.28MO cathode is 35% of the value measured at 100 mA g−1 (Fig. 3B), while for K0.21MO, the capacity retention is 28% (Fig. S10†). This can be explained by the better structural integrity upon cycling achieved in K0.28MO. The charge/discharge profiles at the 1st, 10th, 20th, 30th, 40th and 50th cycles at the current density of 100 mA g−1 are reported in Fig. 3C and D. The typical two-stage voltage plateaus are fully retained after 50 cycles, illustrating the steady reaction kinetics.
To further confirm the cyclability at a high rate, both materials are cycled at 2 A g−1 for 1000 cycles (Fig. 3E). K0.28MO exhibited a high specific capacity of 100 mA h g−1 and high capacity retention of 95%, while for K0.21MO, the specific capacity dropped rapidly with 30% decay. It is inferred that the higher concentration of K+ ions helps to stabilize the layer structure and thus prevent the cathode from self-dissolution and amorphization upon cycling, which significantly prolongs the AZIB cycle life.
To further confirm the influence of K pre-intercalation. K0.28MO was further compared with pristine δ-MnO2 with no pre-intercalated cations. As depicted in Fig. S11A,† the (003) interlayer spacing of δ-MnO2 is larger than both K0.28MO and K0.21MO, which derives from the repulsive force between the interlayer O atoms. EDX analysis further confirms pure Mn and O elemental distribution with no pre-intercalation (Fig. S11B†).
The CV analysis also shows two couples of redox peaks and a two-step reaction process. However, the peak current and position slightly differ from K0.28MO, which results from the difference in the chemical environment and the subsequent difference in reaction kinetics (Fig. S12†).
As a consequence, the cycling stability of δ-MnO2 demonstrates results consistent with expectations. Specifically, as shown in Fig. S13,† the maximum capacity reaches 150 mA h g−1, which is much higher than K0.28MO, demonstrating outstanding energy storage capability of the δ-MnO2 phase. However, the specific capacity of δ-MnO2 experienced a fast decline of more than 1/3 of its maximum capacity. The cycling instability comes from the unsupported unstable δ-MnO2 and subsequent dissolution of the cathode material.
Energy storage mechanism
To confirm the maintained structural integrity and explore the underlying energy storage mechanism of K0.28MO, combined ex situ XRD, XPS, High Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM), and NEXAFS measurements were carried out to determine if phase transitions of the pristine material and microstructural evolution are involved upon cycling. Fig. 4A provides the specific voltage points at which the coin cells were disassembled to obtain the cathodes for subsequent testing. As shown in Fig. 4B, the δ-MnO2 phase with a characteristic peak at ca. 5.6° was observed throughout the cycling process, confirming that the primitive structure is retained upon cycling. This result contrasts with previous reports of phase transitions occurring after cycling.29,32,56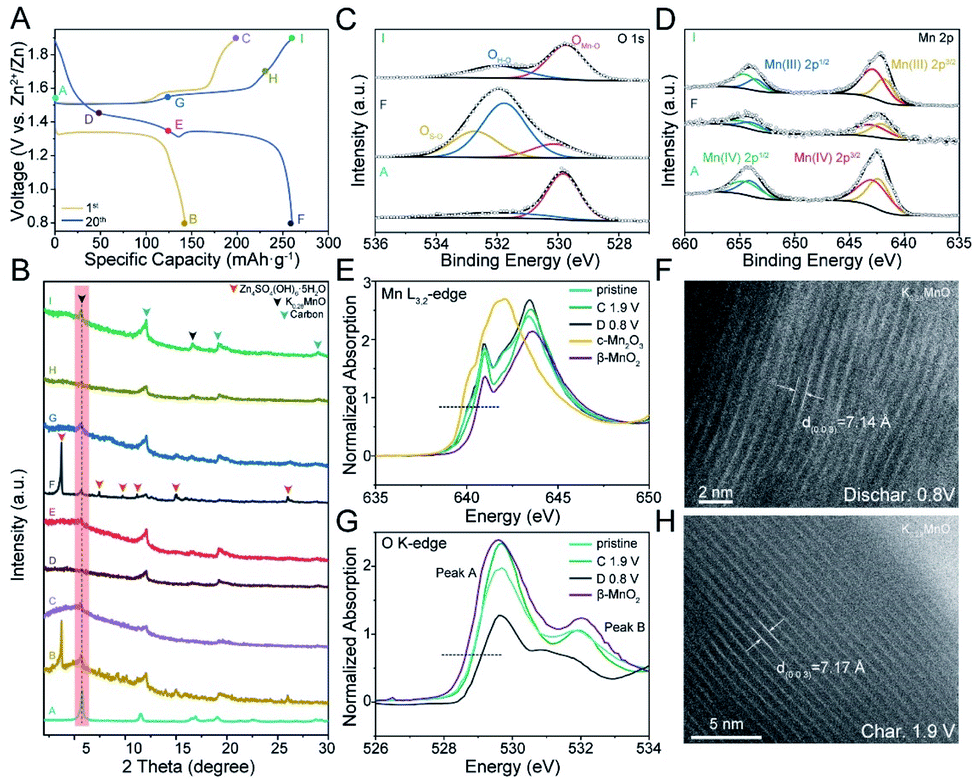 | ||
| Fig. 4 Mechanism study of K0.28MO electrode. (A) Charge/discharge curves of K0.28MO at the 1st and 20th cycle. (B) Ex situ XRD pattern of rainsed cathode collected at various states marked in (A). (C and D) Ex situ XPS spectra of O 1s (C) and Mn 2p (D) at different discharge/charge states of the K0.28MO electrode. (E) Ex situ near-edge X-ray absorption fine structure (NEXAFS) spectra at Mn L3,2-edge of pristine, charged, discharged K0.28MO and reference materials. (F) Ex situ High Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM) image of K0.28MO at a fully discharged state. (G) Ex situ NEXAFS spectra at O K-edge of pristine, charged, discharged K0.28MO and reference materials. (H) Ex situ HAADF-STEM image of K0.28MO at a fully charged state. | ||
Also, as shown in Fig. 4B, a new phase with a strong characteristic diffraction peak (2θ) at 8° was discovered in the initial discharge process (A → B) and the later discharge process from 1.35 V to 0.8 V (E → F), which can be assigned to ZSH.20 The change in O 1s XPS curves also reveals the emergence of S–O species at 532.8 eV and an enhanced peak at 531.9 eV corresponding to O–H species (Fig. 4C). The ZSH phase shows a unique two-dimensional layered structure formed by stacking of Zn(OH)2 with ZnSO4 from the electrolyte while absorbing water molecules between the layers. ZSH is formed on the surface of the cathode material (Fig. S14†). During both the initial charge process (B → C) and later charge process from 0.8 V to 1.55 V (F → G), the characteristic peaks of ZSH disappear. With increasing discharge depth, the ZSH layers dissolve back to the electrode, indicating the reversible formation of the ZSH phase (Fig. S15†). The results confirm the reversible deposition/dissolution of the ZSH phase on the cathode material's surface.20,23,57 It has been reported that based on the Zn2+/H+ intercalation mechanism in AZIBs, with the intercalation of H+, OH− is reversibly released to react with excessive Zn2+ and SO42− in the electrolyte to deposit the ZSH layers.23,57 Thus, the discovery of ZSH in this work confirms the dual ion intercalation mechanism for birnessite MnO2 cathodes.
Considering the faster migration and smaller hydrated radius, the first discharge plateau shown in Fig. 4A is assigned as the intercalation of H+. Due to the decrease of the H+ around the cathodes and a further increase of the polarization, the intercalation of Zn2+ soon becomes dominant. The slower intercalation kinetics means that the intercalation of Zn2+ requires a higher overpotential, and thus the discharge plateau is lower.
The change of the Mn oxidation state on both surface and bulk structures was further studied. During the discharge process, the Mn(III) content increases while Mn(IV) content reduces; In the charging process, Mn(IV) content reaches a maximum value, indicating the highest oxidation state at fully charged state (Fig. 4D).
Reliable proof of the reversible change of the Mn oxidation state is provided by NEXAFS (Fig. 4E). Mn L3,2-edge NEXAFS could directly probe the electron dipole-allowed transition from Mn 2p to unoccupied valence 3d states, strongly correlated with the electronic configuration and oxidation state. The systematic shift of L3 leading edge position and gravity center of L3/L2 edge towards lower energies could indicate the decrease of average oxidation states. Compared to commercial β-MnO2 and Mn2O3 electrodes, the average oxidation state at fully charged state is very close to Mn4+, while at fully discharged state, the curve shows more resemblance with Mn2O3 with a negative energy shift (0.2 eV), indicating the presence of its lowest oxidation state (Mn3+). The sharp absorption peaks at 641 eV at both charged and discharged states are distinguished from the main absorption edge, which is considered characteristic features of birnessite structure.55
Besides, as shown in Fig. 4G, the pre-edge feature in O K-edge NEXAFS spectra originates from electron transitions from ligand O 1s orbitals to unoccupied Mn 3d states due to the hybridization of O 2p and Mn 3d (t2g and eg) states. For Mn(IV) with 3d3 (t32ge0g) configuration, two absorption peaks could be identified: the lower energy peak at 528.8 eV (Peak A) is attributed to spin-down t2g and spin-up eg transitions (nearly equivalent crystal field splitting and exchange splitting) and the higher energy peak at 532.0 eV (Peak B) arise from the spin-down eg transitions. For Mn(III) with 3d4 (t32ge1g) configuration, three absorption peaks could be observed: spin-up eg, spin-down t2g and spin-down eg transitions, from low energies to high energies, respectively. As shown in Fig. 4G, for the charged state, the pre-edge feature is almost identical to β-MnO2, while the discharged state presents a broad absorption peak (530.8 to 531.8 eV), which is contributed from Mn(III) content.
At the fully discharged state, the characteristic peak of K0.28MO slightly shifts towards higher angles, which can be ascribed to the intercalation of H+/Zn2+ ions and enhanced ionic interaction. The corresponding (003) lattice spacing of K0.28MO at a full discharged state was observed as 7.14 Å (Fig. 4F), while the lattice spacing increased to 7.17 Å at full charged state, which is enlarged due to the deintercalation of H+/Zn2+ and reduced ionic interaction (Fig. 4H).49
HAADF-STEM of K0.28MO is also applied to detect the orientation of (012) lattice plane. As shown in Fig. S16,† the lattice space remained stable during both the charge and discharge process. Moreover, the orientation of (012) lattice plane is disordered, which suggests possible lattice defects. Lattice defects provide pathways for ion intercalation/deintercalation, enabling higher capacity and fast ion diffusion kinetics.
Structural stabilization effect of K ions via DFT simulation
The structural details of MnO2, therefore, deserve a more in-depth analysis. The rich polymorphic chemistry of MnO2 comprises dense, tunnel and layered structures. The stable phase in the absence of intercalated cations is the rutile β-MnO2, followed by the tunnel α-MnO2 structure. The relative polymorph stability is modified by the presence and concentration of different cations,6 with the α-MnO2 phase becoming stable at low intercalation levels. Only at high intercalation does the layered birnessite δ-MnO2 structure become the stable polymorph. Therefore, it is reasonable to expect a structural strain upon electrochemical cycling in energy storage applications that varies the concentration of intercalated species.The two materials investigated in the present work contain different amounts of pre-intercalated K, which may affect the phase stability; to understand whether this is the case, we have studied through DFT calculations the structure and stability of KxMnO2 in α and δ phases as a function of K content in the relevant range of 0 ≤ x ≤ 0.5. Our calculations confirm that indeed the layered birnessite phase is unstable compared to α-MnO2 in the absence of K. At low K content, up to x = 0.125, intercalation energy is higher in the tunnels of α-MnO2 than in between the layers of δ-MnO2 and the energy difference between the polymorphs increases (Fig. 5). However, further K intercalation above x = 0.125 is more stable in the δ-MnO2 phase, where the 2-D layered structure provides a larger intercalation volume. At x = 0.25, all interstices of α-MnO2 are fully occupied; the presence of any K above x = 0.25 requires split-interstitials in the tunnel α-MnO2 phase, associated with strong destabilization energy, calculated as 3.7 eV per K ion. The difference in successive intercalation energies between alpha and delta is provided in Fig. S17.† Above ca. 0.26, the layered δ-MnO2 polymorph becomes the thermodynamic ground state in the KxMnO2 chemistry. Enforcing an even higher K concentration of x = 0.5 in α-MnO2 in the DFT calculations yields a layered structure resembling the δ phase upon geometry optimization (Fig. S17†), emphasizing the intercalation-induced structural instability. It is interesting to observe that the two materials studied experimentally have K content on opposite sides of the α–δ phase transition. The best performing K0.28MO has a K content that positions it in the phase space where the layered polymorph is stable, which substantiates the experimental observation of its phase stability upon cycling. In contrast, the K0.21MO material with lower K content will repeatedly cross the α–δ phase transition boundary upon further intercalation of Zn during cycling, explaining its structural instability.
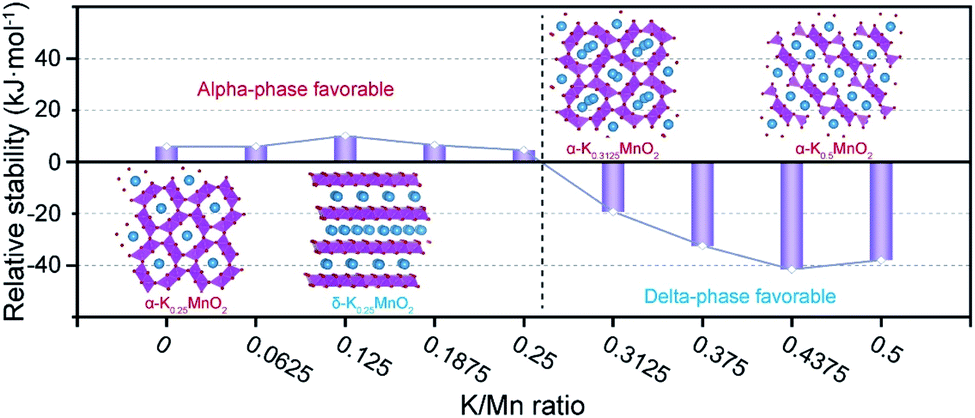 | ||
| Fig. 5 The relative stability of delta phase to alpha phase as a function of K/Mn ratio via DFT simulation. | ||
In summary, the overall electrochemical reaction of K0.28MO can be illustrated as in Fig. 6A. The primitive birnessite phase is well maintained throughout the cycling process, while ZSH is deposited reversibly simultaneous H+/Zn2+ intercalation/deintercalation, contributing to the overall capacity.
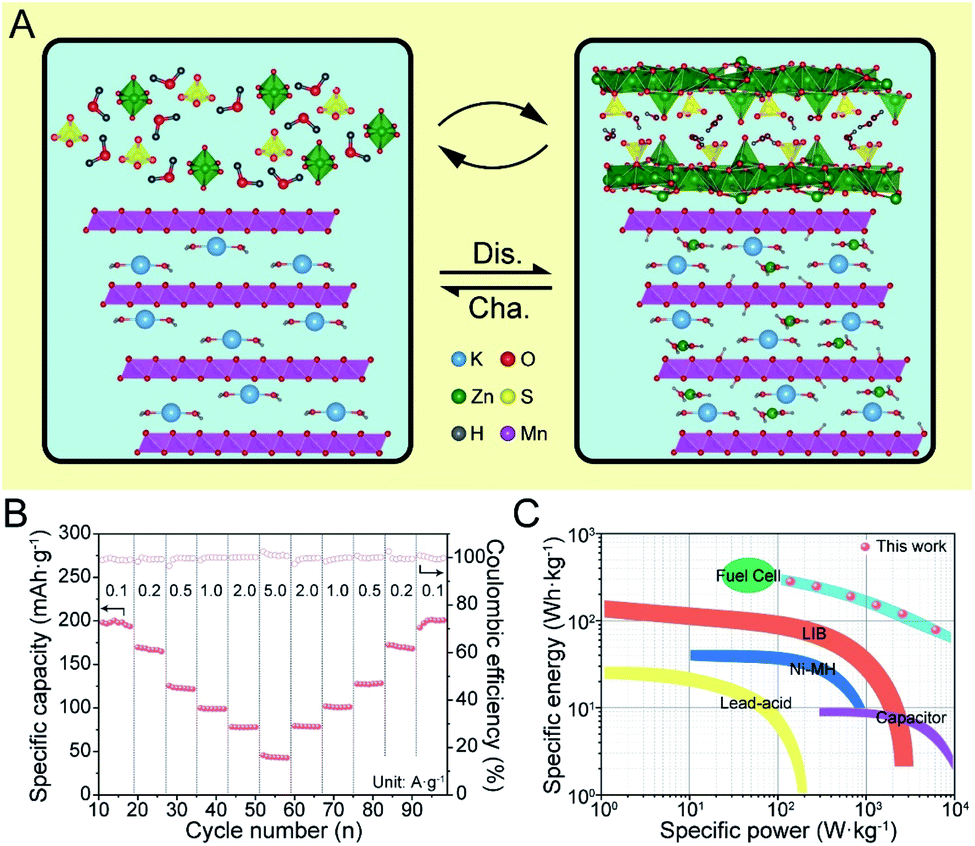 | ||
| Fig. 6 Energy storage mechanism and potential for industrialization. (A) Schematic illustration of the electrochemical reaction of K0.28MO in aqueous AZIBs. (B) Rate performance of K0.28MO cell with high mass loading (ca. 10 mg cm−2). (C) Comparison of the Ragone plot (based on the weight of cathode materials) of the K0.28MO cell with typical commercial energy storage systems. | ||
Commercialization potential
To further demonstrate the potential commercial applications of the as-designed materials, the battery was assembled with typical high mass loading used in commercial lithium-ion batteries (ca. 8–10 mg cm−2). As shown in Fig. 6B, the AZIBs with high loading mass shows competent rate performance close to normal loading ones, which maintains ca. 200 mA h g−1 at 100 mA g−1 and keeps ca. 50 mA h g−1 at a high current density of 5 A g−1. Besides, there is almost no capacity loss after stepwise current density changes. To compare with current commercial energy storage systems, a Ragone plot is constructed, and it is worth mentioning that the K0.28MO cell shows adequate energy and power densities, which indicates its potential for commercialization (Fig. 6C).Conclusions
In summary, the relationship between phase stability, structural integrity and the proportion of pre-intercalated ions was established for K pre-intercalated MnO2 cathodes. The underlying mechanisms responsible for the phase instability of pristine birnessite MnO2 upon cycling are identified through a combined experimental and computational approach. K concentration above a threshold value, as realized in K0.28MO, is critical for phase stability. Zn intercalation in K0.28MO has been characterized and quantified under different electrochemical conditions. We further demonstrated that Zn–K0.28MO cells achieved a high capacity of 300 mA h g−1 and long-term cycle life of up to 1000 cycles without any significant decrease in the specific capacity. The enhanced rate performance also confirmed the fast kinetics enabled by K+ supported pathways. These findings signal reasonable potential and stimulate future research via intercalation engineering for improving the performance of AZIBs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge Diamond Light Source and electron Physical Science Imaging Centre for the allocated beamtime at E01 (session ID: MG24450). The authors acknowledge Helmholtz-Zentrum Berlin for the allocated beamtime at the ISISS beamline of BESSY II (proposal ID: 19108389-ST). The authors would like to thank the Engineering and Physical Sciences Research Council (EPSRC, EP/L015862/1, EP/R023581/1), STFC Batteries Network (ST/R006873/1), RSC Mobility Grant (M19-7656) for funding support. Via the authors' membership of the UK's HEC Materials Chemistry Consortium funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). The authors are grateful to the UK Materials and Molecular Modelling Hub for computational resources, partially funded by EPSRC (EP/P0202194/1). The authors acknowledge the use of the UCL Kathleen High Performance Computing Facility (Kathleen@UCL) and associated support services to complete this work. Y. Jiao thanks the funding support from China Scholarship Council/University College London for the joint PhD scholarship. Y. J. thanks the support from his colleagues and families. In particular, Y. J. wants to thank H. X. for her invaluable support and encouragement over the years. Will you marry me?Notes and references
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320 CrossRef CAS.
- M. Armand, Nature, 2001, 414, 359–367 CrossRef.
- J. Hassoun, K.-S. Lee, Y.-K. Sun and B. Scrosati, J. Am. Chem. Soc., 2011, 133, 3139–3143 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- J.-M. Tarascon and M. Armand, in Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 171–179 Search PubMed.
- Z. Liu, J. Wang, H. Ding, S. Chen, X. Yu and B. Lu, ACS Nano, 2018, 12, 8456–8466 CrossRef CAS.
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 CrossRef CAS.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, ACS Nano, 2016, 10, 9738–9744 CrossRef CAS.
- L. Ma, N. Li, C. Long, B. Dong, D. Fang, Z. Liu, Y. Zhao, X. Li, J. Fan and S. Chen, Adv. Funct. Mater., 2019, 29, 1906142 CrossRef CAS.
- F. Wan and Z. Q. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS.
- F. Wan, X. Y. Wang, S. S. Bi, Z. Q. Niu and J. Chen, Sci. China: Chem., 2019, 62, 609–615 CrossRef CAS.
- Y. Liu, P. Hu, H. Liu, X. Wu and C. Zhi, Materials Today Energy, 2020, 17, 100431 CrossRef.
- S. Liu, H. Zhu, B. Zhang, G. Li, H. Zhu, Y. Ren, H. Geng, Y. Yang, Q. Liu and C. C. Li, Adv. Mater., 2020, 32, 2001113 CrossRef CAS.
- K. Lu, B. Song, Y. X. Zhang, H. Y. Ma and J. T. Zhang, J. Mater. Chem. A, 2017, 5, 23628–23633 RSC.
- L. Y. Zhang, L. Chen, X. F. Zhou and Z. P. Liu, Sci. Rep., 2015, 5, 18263 CrossRef CAS.
- M. Tian, C. Liu, J. Zheng, X. Jia, E. P. Jahrman, G. T. Seidler, D. Long, M. Atif, M. Alsalhi and G. Cao, Energy Storage Materials, 2020, 29, 9–16 CrossRef.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- Z. Chen, P. Wang, Z. Ji, H. Wang, J. Liu, J. Wang, M. Hu and Y. Huang, Nano-Micro Lett., 2020, 12, 75 CrossRef.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- J. H. Huang, Z. Wang, M. Y. Hou, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef.
- S. Khamsanga, R. Pornprasertsuk, T. Yonezawa, A. A. Mohamad and S. Kheawhom, Sci. Rep., 2019, 9, 8441 CrossRef.
- J. J. Wang, J. G. Wang, H. Y. Liu, C. G. Wei and F. Y. Kang, J. Mater. Chem. A, 2019, 7, 13727–13735 RSC.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y. W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121–125 CrossRef CAS.
- S.-D. Han, S. Kim, D. Li, V. Petkov, H. D. Yoo, P. J. Phillips, H. Wang, J. J. Kim, K. L. More and B. Key, Chem. Mater., 2017, 29, 4874–4884 CrossRef CAS.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemComm, 2015, 51, 9265–9268 RSC.
- B. Lee, C. S. Yoon, H. R. Lee, K. Y. Chung, B. W. Cho and S. H. Oh, Sci. Rep., 2014, 4, 6066 CrossRef CAS.
- X. W. Wu, Y. H. Xiang, Q. J. Peng, X. S. Wu, Y. H. Li, F. Tang, R. C. Song, Z. X. Liu, Z. Q. He and X. M. Wu, J. Mater. Chem. A, 2017, 5, 17990–17997 RSC.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef.
- C. Y. Zhu, G. Z. Fang, J. Zhou, J. H. Guo, Z. Q. Wang, C. Wang, J. Y. Li, Y. Tang and S. Q. Liang, J. Mater. Chem. A, 2018, 6, 9677–9683 RSC.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li and P. Bhattacharya, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- M. Chamoun, W. R. Brant, C.-W. Tai, G. Karlsson and D. Noréus, Energy Storage Materials, 2018, 15, 351–360 CrossRef.
- M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Islam, D. T. Pham, J. Jo, S. Kim, J. P. Baboo, Z. Xiu, K. S. Lee, Y. K. Sun and J. Kim, Chem. Mater., 2017, 29, 1684–1694 CrossRef CAS.
- J. Li, K. McColl, X. Lu, S. Sathasivam, H. Dong, L. Kang, Z. Li, S. Zhao, R. Wang, D. J. L. Brett, P. R. Shering, F. Cora, G. He, A. Kafizas, C. J. Carmalt and I. P. Parkin, Adv. Energy Mater., 2020, 10, 2000058 CrossRef CAS.
- F. W. Ming, H. F. Liang, Y. J. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef.
- Y. X. Zeng, X. Y. Zhang, R. F. Qin, X. Q. Liu, P. P. Fang, D. Z. Zheng, Y. X. Tong and X. H. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef.
- Q. C. Zhang, C. W. Li, Q. L. Li, Z. H. Pan, J. Sun, Z. Y. Zhou, B. He, P. Man, L. Y. Xie, L. X. Kang, X. N. Wang, J. Yang, T. Zhang, P. P. Shum, Q. W. Li, Y. G. Yao and L. Wei, Nano Lett., 2019, 19, 4035–4042 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS.
- X. Deng, Y. Xu, Q. An, F. Xiong, S. Tan, L. Wu and L. Mai, J. Mater. Chem. A, 2019, 7, 10644–10650 RSC.
- P. He, G. B. Zhang, X. B. Liao, M. Y. Yan, X. Xu, Q. Y. An, J. Liu and L. Q. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang and Y. Shao, Adv. Mater., 2018, 30, 1703725 CrossRef.
- X. Yao, Y. Zhao, F. A. Castro and L. Mai, ACS Energy Lett., 2019, 4, 771–778 CrossRef CAS.
- T. Sun, Q. Nian, S. Zheng, J. Shi and Z. Tao, Small, 2020, 16, 2000597 CrossRef CAS.
- J. Xiao, L. Wan, X. Wang, Q. Kuang, S. Dong, F. Xiao and S. Wang, J. Mater. Chem. A, 2014, 2, 3794–3800 RSC.
- H. He, H. Tong, X. Song, X. Song and J. Liu, J. Mater. Chem. A, 2020, 8, 7836–7846 RSC.
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, Small, 2020, 16, 2001228 CrossRef CAS.
- O. Ghodbane, F. Ataherian, N. L. Wu and F. Favier, J. Power Sources, 2012, 206, 454–462 CrossRef CAS.
- Y. Yang, X. Su, L. Zhang, P. Kerns, L. Achola, V. Hayes, R. Quardokus, S. L. Suib and J. He, ChemCatChem, 2019, 11, 1689 CrossRef CAS.
- C. Zhan, J. Lu, A. J. Kropf, T. Wu, A. N. Jansen, Y.-K. Sun, X. Qiu and K. Amine, Nat. Commun., 2013, 4, 2437 CrossRef.
- R. Qiao, T. Chin, S. J. Harris, S. Yan and W. Yang, Current Applied Physics, 2013, 13, 544–548 CrossRef.
- M. H. Alfaruqi, S. Islam, D. Y. Putro, V. Mathew, S. Kim, J. Jo, S. Kim, Y. K. Sun, K. Kim and J. Kim, Electrochim. Acta, 2018, 276, 1–11 CrossRef CAS.
- L. Chen, Z. Yang, H. Qin, X. Zeng and J. Meng, J. Power Sources, 2019, 425, 162–169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta08638j |
| This journal is © The Royal Society of Chemistry 2020 |