
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNa2Fe2OS2, a new earth abundant oxysulphide cathode material for Na-ion batteries†
Jacinthe
Gamon‡
 a,
Arnaud J.
Perez‡
a,
Leanne A. H.
Jones
bc,
Marco
Zanella
a,
Luke M.
Daniels
a,
Rhun E.
Morris
a,
Chiu C.
Tang
d,
Tim D.
Veal
bc,
Laurence J.
Hardwick
ab,
Matthew S.
Dyer
a,
John B.
Claridge
a and
Matthew J.
Rosseinsky
*a
a,
Arnaud J.
Perez‡
a,
Leanne A. H.
Jones
bc,
Marco
Zanella
a,
Luke M.
Daniels
a,
Rhun E.
Morris
a,
Chiu C.
Tang
d,
Tim D.
Veal
bc,
Laurence J.
Hardwick
ab,
Matthew S.
Dyer
a,
John B.
Claridge
a and
Matthew J.
Rosseinsky
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: rossein@liverpool.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, Chadwick Building, Peach Street, Liverpool, L69 7ZF, UK
cDepartment of Physics, University of Liverpool, Oliver Lodge Laboratory, Oxford Street, Liverpool, L69 7ZE, UK
dDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
First published on 21st September 2020
Abstract
Multiple anion materials are of particular interest for the discovery of new crystal structures and offer an original way to modulate physical properties, including energy storage materials with enhanced performances. Through careful synthesis optimization, a new Na2Fe2OS2 phase was prepared by two different routes: high temperature solid-state synthesis and simple mechanochemical synthesis. The long-range and local structure of Na2Fe2OS2 was studied by Rietveld refinement of neutron and X-ray diffraction data combined with EXAFS data refinement. The phase comprises an amorphous and a crystalline part which has an anti-K2NiF4 structure, corresponding to the n = 1 member of the homologous anti-Ruddlesden–Popper [AX][ABX3]n series. Its electrochemical properties as a cathode material were studied in Na half cells and Na-ion full cells, revealing that the material becomes fully amorphous upon initial desodiation to Na0.5Fe2OS2, but maintains a reversible capacity of 135 mA h g−1 in full cells where up to 1.2 Na+ can be reversibly extracted and reinserted when compensating for the Na lost in SEI formation. The stability of the pristine material and its structural evolution upon charging are discussed, paving the way for further optimization of this material. Being composed exclusively of earth-abundant elements and stable under dry air, Na2Fe2OS2 perfectly illustrates the great opportunity of multiple anion chemistry to explore new structure types and develop better energy storage systems.
1. Introduction
In the 1970s, research on intercalation materials to store energy was motivated by the energy crisis from increasing oil prices. Pioneering research in that field led to the worldwide development of Li-ion batteries, the transformation of consumer electronics and a revolution in our access to information. With the acceleration of climate change due to fossil fuels consumption, the need for cleaner energy sources and energy storage systems becomes critical to lead a technological transition towards electricity-powered transportation and large-scale energy storage capabilities. It remains unclear whether the cost of Li-ion batteries will meet this grand challenge as the technology relies heavily on non-abundant/unevenly distributed elements such as lithium, nickel or cobalt, with prices sensitive to market and political decisions. The development of Na-ion batteries, whose upper continental crust abundance (ucca) is significantly higher than that of Li (2.567 wt% vs. 0.0022 wt%, respectively),1 as a cheaper alternative is therefore a promising route to diversify technology for grid or local storage applications that do not require high energy density. Current efforts to prepare Na-ion batteries using Na3V2(PO4)2F3 or layered P2-Nax(M/M′)O2 (with M, M′ = Ni, Co, Cr, V) as the cathode materials and hard carbon as the anode material focus on competing with Li-ion battery systems in terms of performance. However, these do not provide a significant difference in terms of price and abundance of materials, because of the presence of rare transition metals (ucca of V, Cr, Co, Ni are 0.0053, 0.0035, 0.0012, 0.0019 wt%, respectively).1,2 The search for new sodium cathode materials which contain highly abundant transition metals such as Mn (ucca of 0.0527 wt%) or more so Fe (ucca of 3.089 wt%) is therefore strongly needed to reduce the cost and environmental impact of batteries.While many studies focus on the substitution of cations within an oxide lattice, the combination of two anions in one structure type offers alternative ways to modulate structure and properties.3 Hetero-anionic materials containing anions of different sizes and electronegativity tend to form layered structures, favorable for chemical intercalation, in which each cation bonds to one of the anions according to their preferred chemical interactions.4–8 The use of oxyfluorides as cathode materials is an active field of research,9,10 as the higher electronegativity of fluoride ions leads to high voltage electrode materials. Less electronegative anions, such as chalcogenides (Ch = S2−, Se2−), are more easily oxidized than oxides, favoring Ch2− anionic redox activity11 and higher capacities.12 The presence of highly polarizable chalcogenide anions is also a strong advantage to increase electronic conductivity, which often limits the electrochemical properties in oxide systems.
In that matter, oxysulphides represent a promising family of materials, which is relatively unexplored, mostly due to their lower voltage compared to oxides. However, recent results on a new anti-perovskite Li2FeOS have shown that the low voltage can be compensated by large capacities alongside the advantage of using abundant elements.13
Reported for its interesting electronic and magnetic properties,14 Na2Fe2OSe2 is a good candidate for Na-ion batteries with a reasonable amount of Na that gives a theoretical capacity of 162 mA h g−1. Its layered structure should lead to good Na-ion conductivity and good electronic conductivity in the Fe2O layer. The sulphide equivalent Na2Fe2OS2, which has not been reported, has an even higher theoretical capacity of 225 mA h g−1, while being composed of elements that are among the most abundant on our planet.1
Due to its reactivity against oxidation with oxygen and hydrolysis with water, synthesis and processing of sulphide materials is expensive and often requires the use of toxic gases and/or solvents which generate safety and environmental issues at the industrial level. Developing low temperature and solvent-free synthesis routes of sulphide materials has therefore become an important research topic.15–17 We report here on the synthesis of the promising oxysulphide Na2Fe2OS2 through mechanochemistry, a scalable, room temperature route, with thorough characterization of its structural features, and unravel its electrochemical properties as a cathode material for Na-ion batteries through a combination of advanced characterization techniques.
2. Results
2.1 Synthesis and structure determination
Na2Fe2OS2 was first prepared through a solid-state synthesis route (sample denoted as Na2Fe2OS2-SS), by reproducing the method used for the synthesis of the analogue compound Na2Fe2OSe2 described by He et al.14 After indexing the pattern to the I4/mmm space group (a = 4.04222(1) Å, c = 14.07319(9) Å), a Rietveld refinement was performed, starting from the structural model of Na2Fe2OSe2 and replacing selenium with sulphur atoms. A good fit is obtained with this model (part 1.1 in the ESI, Fig. S1, Tables S1 and S2†). Sample purity was assessed through the quantitative phase analysis method implemented in FullProf,18 which indicated weight fractions of 89.3(4)%, 7.37(7)%, 0.07(2)% and 3.26(3)% of the main phase Na2Fe2OS2, Na3Fe2S4, NaFe2O3 and FeO respectively. To reduce the amount of impurities, different synthesis conditions were evaluated (part 1.2 in ESI, Fig. S2–S4†). Unfortunately, none of these conditions improved sample purity, with longer reaction times leading to decomposition of the phase, suggesting that Na2Fe2OS2 is metastable.Mechanochemical synthesis has been shown to be a powerful tool for the synthesis of metastable and soft materials such as chalcogenides,19–21 while avoiding severe limitations on the use of toxic sulphurizing agents and therefore gaining substantial interest from industry.22,23 It additionally has the benefit of producing sub-micron particles with appreciable amount of structural disorder and improved ionic conductivity,24–26 leading scientists to adopt this method for battery material processing.27–30 Mechanochemical synthesis was thus used for the preparation of Na2Fe2OS2. Details on the optimization procedure for the mechanochemical synthesis can be found in the Methods section and in part 2.1 of the ESI.† By adding an excess of Na2S to the reaction media (1Na2O + 2FeS + 0.75Na2S), we were able to isolate the target Na2Fe2OS2 phase. During high energy ball milling, high temperatures in excess of 700 °C can be reached through friction at the contact point of colliding balls and over a very short period of time (10−4 to 10−3 s). These high temperatures have been described by the plasma-magma and impulse models,31 and are due to the large amount of energy transferred to the reagents, estimated as the kinetic energy of the ball before collision32 (of the order of ∼100 J).33 The Na2Sx polysulphides,34 with melting points ranging from 490 °C for Na2S2 to 252 °C for Na2S6, are therefore likely to act as a flux, known to favor the crystallization of sulphide materials.35 The excess, unreacted Na2S was removed through washing the powder in anhydrous methanol, resulting in a powder free from any Na3Fe2S4 and FeO crystalline impurities (cf. ESI, part 2.1†). In-depth structural and electrochemical properties characterization were then performed on this mechanosynthesized and methanol-washed sample, denoted Na2Fe2OS2-MW.
2.2 Composition and microstructure
The X-ray diffraction (XRD) pattern indicates the presence of crystalline Na2Fe2OS2 resulting from mechanosynthesis. A Le Bail fit against synchrotron X-ray diffraction (SXRD) data shows that the pattern can be indexed using a similar cell to that of Na2Fe2OS2-SS (I4/mmm, a = 4.0325(1) Å and c = 14.077(1) Å) with contributions from an Fe metal crystalline impurity and from ZrO2 at Q = 2.1 Å−1 (introduced from the ball milling media) (Fig. S10†).The micro-structural analysis (implemented in Fullprof, cf. Methods)36 revealed an average coherent scattering domain size of 22(2) nm (based on the Debye–Scherrer model) with no particular size anisotropy, and an average maximum strain of 31.98(5)‰ (upper limit of the apparent strain defined by Stokes and Wilson)37 compared to 136.8(2) nm and 5.51(1)‰, respectively for Na2Fe2OS2-SS. The SXRD pattern shows a broad contribution between 1–2 Å−1, which can be partially attributed to the capillary used for the measurement (Fig. S10†). However, the significant contribution of the background to the SXRD pattern indicates the potential presence of an additional amorphous phase which does not contribute to the Bragg scattering, and is not unheard of through mechanosynthesis methods.38
The relative proportions of crystalline and amorphous components in the material were assessed using the Rietveld quantitative amorphous content analysis method by adding crystalline Na3Fe2S4 as a suitable internal standard (cf. ESI part 2.2.2†). This analysis revealed that 39(5) wt% of the phase is crystalline, i.e. 61(5) wt% of the material is not observed using diffraction experiments. This result highlights the importance of using the quantitative phase analysis method with an internal standard to study mechanosynthesized materials. Attempts to increase the extent of crystallization in the compound by annealing at 400 °C or 600 °C, or by extended ball milling resulted in the decomposition of the phase, further confirming the metastability of Na2Fe2OS2.
The elemental analysis performed by Inductively Coupled Plasma Optical Emission Spectrometry (ICP/OES), CHNS, together with Scanning Electron Microscopy coupled with Wavelength-Dispersive X-ray spectroscopy (SEM-WDX) revealed an overall composition: Na2.2(1)Fe2.00(1)OS2.29(1) (Table S4 and Fig. S11†) close to the expected composition Na2Fe2OS2. The excess Na and S content can be attributed to some remaining sodium sulphide species (2.5(5)% according to the mass balance from the washing procedure).
Transmission Electron Microscopy (TEM) imaging and Energy Dispersive X-ray spectroscopy (EDX) mapping were performed to gain more information about the microstructure of the sample. The imaging revealed the presence of large micrometric particles composed of agglomerated nanograins (Fig. 1a). The elementary ∼20 nm coherent scattering domains revealed by XRD could not be isolated. EDX mapping revealed a homogeneous distribution of the four elements over the whole particle, aside from the 70 nm wide Fe metal grain impurity (Fig. 1b). These results, added to the estimated lateral resolution from the EDX probe of ∼2–5 nm,39 rule out the presence of any other impurity with grains larger than 7 nm and show that the Na2Fe2OS2 composition is homogeneous on the nanometer scale.
 | ||
| Fig. 1 Characterisation of Na2Fe2OS2-MW. (a) TEM imaging of a particle of Na2Fe2OS2-MW and (b) the corresponding EDX mapping images of the elemental distribution. (c) Infrared spectra of Na2Fe2OS2-SS, Na2Fe2OS2-MW and of a Na3Fe2S4 reference prepared by solid-state synthesis. Rietveld refinement of Na2Fe2OS2-MW against (d) synchrotron X-ray diffraction and neutron powder diffraction data from (e) bank 2 (2θ = 168.330°) and (f) bank 5 (2θ = 30.000°) of Polaris instrument, with Iobs (red dots), Icalc (black line), Iobs–Icalc (blue line), and Bragg reflections (black tick marks for Na2Fe2OS2, blue tick marks for Fe, and pink tick marks for ZrO2). Contribution of the capillary to the background of the pattern is shown in Fig. S10.† | ||
Fourier Transform Infrared spectroscopy (FTIR) was used to further probe the compositional homogeneity on the local scale and discriminate the presence of amorphous impurities. Fig. 1c shows the spectra of the crystalline and mechanosynthesized samples along with a Na3Fe2S4 reference material. Both Na2Fe2OS2-MW and Na2Fe2OS2-SS samples show similar features, with one group of broad and low intensity peaks between 700 and 550 cm−1, a broad and intense signal between 550 and 400 cm−1, and another group of different intensity peaks between 400 and 280 cm−1. Additionally, Na2Fe2OS2-SS shows a signal with increasing intensity below 280 cm−1, which is not observed in Na2Fe2OS2-MW. This signal can be attributed to the Na3Fe2S4 impurity (Fig. 1c and Table S7†).
Peaks were fitted using the Gaussian function in order to compare positions and full width at half maximum (FWHM). Fig. S15† shows the result of the fit and fitting parameters are reported in Table S8.† Vibration modes with wavenumbers between 400 and 280 cm−1, which are also observed for Na3Fe2S4 (Fig. 1c), can be attributed to Fe–S stretching modes.40–42 For Na2Fe2OS2-SS this region consists of 3 distinct peaks of FWHM ranging from 10 to 24 cm−1 whereas for Na2Fe2OS2-MW it consists of a much broader signal. This could be fitted by three overlapping contributions and the two main peaks at 292(2) and 310(2) cm−1 show large FWHM of 109(3) and 50(2) cm−1, respectively. The comparison between FWHM of Na2Fe2OS2-SS and Na2Fe2OS2-MW reflects the high amount of structural disorder as well as the large strain present in the mechanosynthesized sample, therefore susceptible to show a broader range of Fe–S distances. Vibrations at higher wavenumbers, between 550 and 400 cm−1, can be attributed to metal–oxygen stretching bonds.43 Interestingly, these peaks have comparable FWHM for Na2Fe2OS2-MW (52(3) and 40(2) cm−1) and Na2Fe2OS2-SS (66(7) and 64(3) cm−1), which is consistent with the rigid nature of the short Fe–O bonds compared to Fe–S bonds.
Overall, no additional peaks could be identified for Na2Fe2OS2-MW. This observation suggests that the intimately combined coherent and incoherent scattering domains have similar local structure with a broader range of interatomic distances compared to the purely crystalline phase (Na2Fe2OS2-SS).
2.3 Structural refinement and structure description
The structure of the crystalline part of Na2Fe2OS2-MW was first determined through Rietveld analysis by refining the model obtained for Na2Fe2OS2-SS against the SXRD data of Na2Fe2OS2-MW. Neutron data was then used to obtain the atomic displacement parameters (adp) and site occupancy factors (sof) of light elements (Na and O), which were then included in the refinement of the SXRD data. This model gave a good fit of all data banks, with SXRD and neutron banks 2 and 5 shown in Fig. 1d, e and f, respectively (all banks in Fig. S12a–d in ESI†), and outcome of the refinement presented in Tables S5 and S6.† The quantitative phase analysis performed with the Rietveld method revealed a crystalline phase composed of 98.2(1) wt% Na2Fe2OS2 and 1.8(2) wt% Fe metal impurity. A decrease of the a lattice parameter was observed compared to Na2Fe2OS2-SS (Table 1). Interestingly, the values for the anisotropic and isotropic adp of all atoms were much larger than those obtained for Na2Fe2OS2-SS, and are represented as displacement ellipsoid around atoms in Fig. 2d–g.| a (Å) | 4.0325(1) (Na2Fe2OS2-MW); 4.04222(1) (Na2Fe2OS2-SS) | |||
| c (Å) | 14.077(1) (Na2Fe2OS2-MW); 14.07319(9) (Na2Fe2OS2-SS) | |||
| V (Å3) | 228.5(2) (Na2Fe2OS2-MW); 229.950(2) (Na2Fe2OS2-SS) | |||
| Site | Na | Fe | O | S |
| Wyckoff position | 4e | 4c | 2a | 4e |
| Site symmetry | 4mm | mmm | 4/mmm | 4mm |
| (x, y, z) | (0, 0, 0.176(2)) | (0, 0.5, 0) | (0, 0, 0) | (0, 0, 0.376(3)) |
| sof | 1 | 0.99(2) | 1 | 0.96(4) |
| B iso/B11, B22, B33, Beq (Å2) | 1.6(3) | 2.0(4), 1.0(4), 1.8(4), 2.1(5) | 0.1(2), 0.1(2), 1.6(9), 1.2(5) | 2.3(5) |
| Coordination | 6 | 6 | 6 | 9 |
| BVS | 1.06(2) | 1.80(1) | 2.14(1) | 1.84(1) |
| Distance (Å) | d Na–S: 2.808(3) | d Fe–S: 2.670(2) × 4 | — | — |
| d Na–S: 2.9410(8) × 4 | d Fe–O: 2.0152(1) × 2 | — | — | |
| d Na–O: 2.478(2) | d Fe–Fe: 2.8500(2) | — | — | |
| d Na–Fe: 3.1936(17) | — | — | ||
| Angles (°) | S–Na–S: 104.30(10) × 4 | S–Fe–S: 98.02(4) × 2 | — | — |
| S–Na–S: 86.50(2) × 4 | S–Fe–S: 81.98(9) × 2 | — | — | |
| S–Na–S: 151.41(2) × 2 | S–Fe–S: 180.00(9) × 2 | — | — | |
| S–Na–O: 75.70(6) × 4 | S–Fe–O: 90.00(5) × 8 | — | — | |
| S–Na–O: 179.97(12) | O–Fe–O: 180 | — | — | |
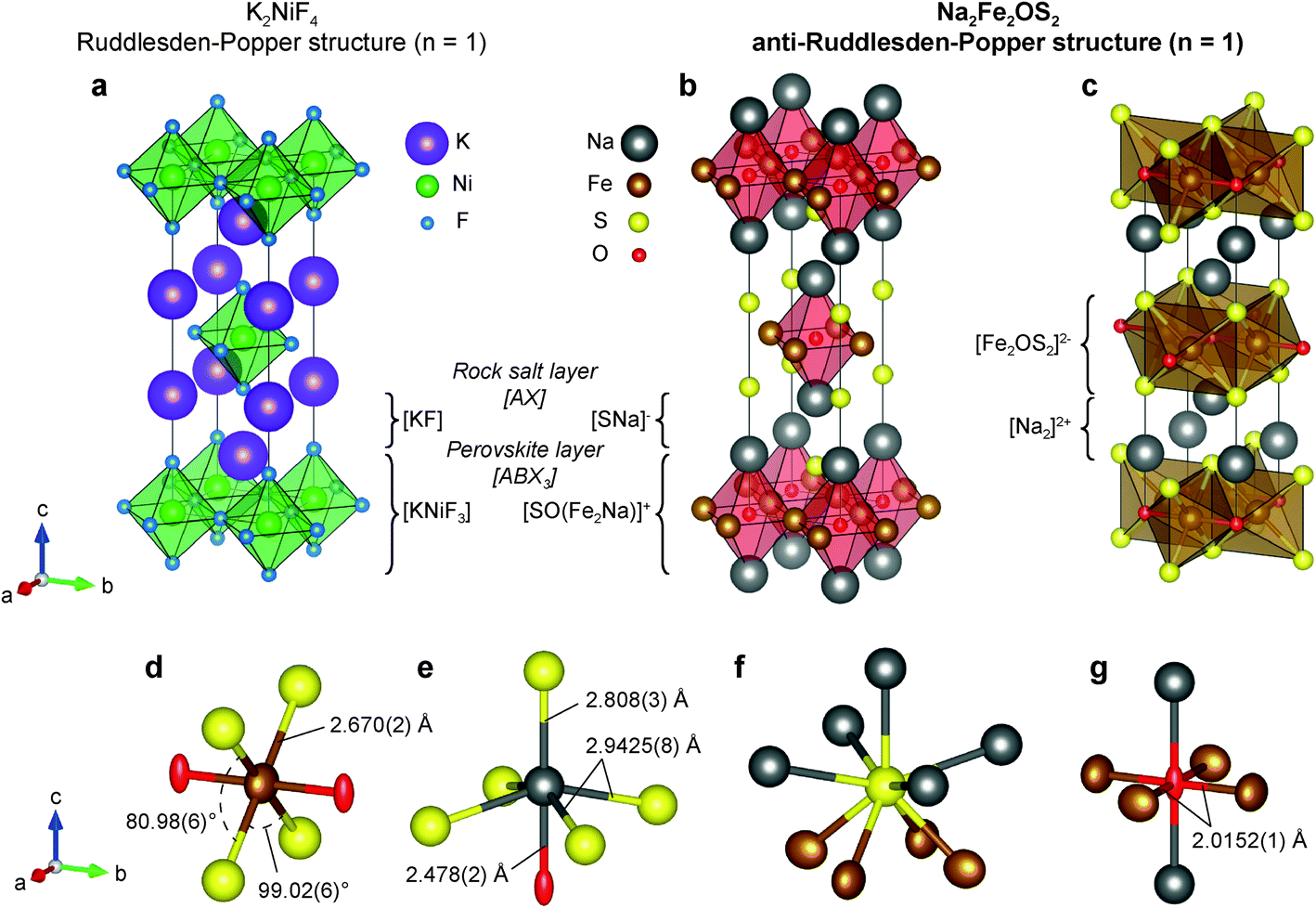 | ||
| Fig. 2 Structure of Na2Fe2OS2 from the refinement obtained against ND and SXRD data of the mechanosynthesized sample (Na2Fe2OS2-MW). (a) The K2NiF4 (n = 1 Ruddlesden–Popper) structure is compared to (b) the anti-K2NiF4 structure (n = 1 anti-RP) of Na2Fe2OS2. (c) The layered structure and heteroleptic environment of Fe in the Na2Fe2OS2 structure are also highlighted. Individual atomic environments and the corresponding atomic displacement parameters obtained from Rietveld refinement are represented with atoms shown as displacement ellipsoid with a probability of 99% for (d) Fe (brown ellipsoid), (e) Na (grey sphere), (f) S (yellow sphere) and (g) O (red ellipsoid). | ||
There was no indication of anisotropic displacements of S and Na from the refinement, but anisotropic displacement parameters were found for Fe and O (Table 1), suggesting possible displacement of these atoms. However, a model with Fe atoms displaced from the 4c (0, 0.5, 0) onto a 16n (x, 0.5, z) site did not improve the fit (Fig. S13†) and was therefore discarded. During independent refinement, sof of Na refined to a value higher than 1, and was therefore fixed to 1, while the sof of Fe, S and O refined to 0.99(2), 0.96(4) and 0.98(1) respectively. In contrast to Na2Fe2OS2-SS for which the refinement highlighted an Fe deficiency (Table S2†), the refined composition of Na2Fe2OS2-MW, Na2Fe1.98(2)S1.96(4)O0.98(1), is closer to the ideal stoichiometry and could explain why this sample has a smaller a lattice parameter.
Na2Fe2OS2 was found to be isotypic with its selenium analogues.14 The phase crystallizes in an anti-K2NiF4 type structure (Fig. 2a). K2NiF4 corresponds to the n = 1 member of the Ruddlesden–Popper (RP) homologous series of general formula [AX][ABX3]n, where A is a large cation, B a smaller cation and X an anion (Fig. 2b). In the anti-structure of Na2Fe2OS2, the [ABX3] layer is an anti-perovskite slab which can be written as [SO(Fe2Na)]+ and the rocksalt [AX] layer as [SNa]−, with large sulphur anions on the A site, small oxygen anions on the B site, and iron and sodium cations ordered on the two inequivalent X sites. As expected from the size and electronegativity difference of O2− and S2−, anion ordering is clearly evidenced in the structure. Interestingly, the layers of the anti-RP phases are charged, which is in contrast with the regular n = 1 RP phase.
The structure can also be described as alternating layers of [Fe2OS2]2− and sodium double layers [Na2]2+ (Fig. 2c). Other phases containing this isostructural [M2OQ2]2− motif (M = Fe, Ti; Q = S, Se, As, Sb) have been reported in the literature. Among them, Na2Ti2Pn2O (Pn = As, Sb)44 is isostructural to Na2Fe2OS2 and Na2Fe2OSe2 (with Ti in the Fe site and Pn in the S or Se site); BaTi2Pn2O shows a barium single layer (Fig. S14b†), and La2O2Fe2OSe2 a fluorite type [La2O2]2+ layer (Fig. S14c†)45,46 instead of a sodium double layer [Na2]2+. Several phases containing the anti-[CuO2] square planar configuration within the [M2OQ2]2− layer have been studied as potential superconductors,47,48 as the square planar lattice is an important feature of the well-known high temperature cuprate superconductors.49,50 They revealed interesting physical properties such as anti-ferromagnetic ordering in the 50–100 K range14,51 or spin-density-wave/charge-density-wave instabilities.48,52,53
Looking more closely at individual atomic environments in Na2Fe2OS2, Fe is coordinated by two oxygen atoms in the anti-[CuO2] type square planar layer, and four S atoms above and below the plane (Fig. 2d). Fe therefore sits in an octahedral site compressed along the axial direction (Fe–O), with the four S atoms in the equatorial plane forming a rectangle with two obtuse (99.02(6)°) and two acute (80.98(6)°) S–Fe–S angles (Fig. 2d). The Fe–O distance (dFe–O = 2.0152(1) Å, interatomic distances are reported in Table 1) is shorter than that observed in FeO (dFe–O = 2.16885 Å) where Fe2+ is also 6-fold coordinated. On the contrary, the Fe–S distance (dFe–S = 2.670(2) Å) is longer than in most iron sulphide compounds, yielding a bond valence sum (BVS) of 1.80(1) on the Fe atom, close to the expected +2 theoretical value. This geometry around the Fe site is reflected in the values of the adp: the displacement ellipsoid of Fe shows a marked elongation along the a and c axis, revealing a disk-shaped displacement in the equatorial plane of the FeO2S4 octahedra (Fig. 2d). This is also consistent with the higher polarizability of the S2− anions in comparison to O2− which yields more rigid bonds. The rigidity of the Fe–O bond is further featured in the comparison with the structure of other compounds presenting the Fe2OCh2 (Ch = S, Se) layer. For instance, the a lattice parameter (twice the Fe–O distance) in Na2Fe2OSe2 (4.107(8) Å)45 and La2O2Fe2O2Se2 (4.084466(9) Å)14 is very close to that of Na2Fe2OS2 (1.8 and 1.2% difference, respectively). On the contrary, the longer c lattice parameter (14.641(8) and 18.59798(7) Å) accommodates the larger Se2− ions and [La2O2]2+ slabs (3.9 and 24% difference, respectively). Oxygen anions are coordinated by four iron atoms in the ab plane and two sodium atoms along c (Fig. 2g). The adp of oxygen highlight a large elongation of the displacement ellipsoid of oxygen along the c axis. This has been observed previously in compounds with similar structures and was attributed to the preferred five-fold coordination of O over the six-fold coordination.54 Sulphur atoms are slightly off plane with the sodium atoms of the same layer (z(Na)–z(S) = 0.052(5)), such that S is displaced towards the closest Fe2O layer (Fig. 2f). The [SNa]− layer therefore differs from a pure [AX] rocksalt layer in which Na and S atoms occupy the same plane. Finally, sodium atoms lie in a distorted NaOS5 octahedral site (Fig. 2e), with one short, four long Na–S distances, and one short Na–O distance.
2.4 Stability
Remarkably, it is possible to synthesize Na2Fe2OS2 by mechanosynthesis under dry air (see Methods). The diffraction pattern of the obtained material (blue line, Fig. 3a) is similar to that of the sample synthesized under argon (black line, Fig. 3a), with lattice parameters: a = 4.0417(2) and 4.0436(3) Å for Na2Fe2OS2OS synthesized under Ar and dry air, respectively and c = 14.078(2) and 14.084(2) Å. The stability of the phase under dry air is very important for future applications, as dry rooms are routinely used in the industry for battery assembly.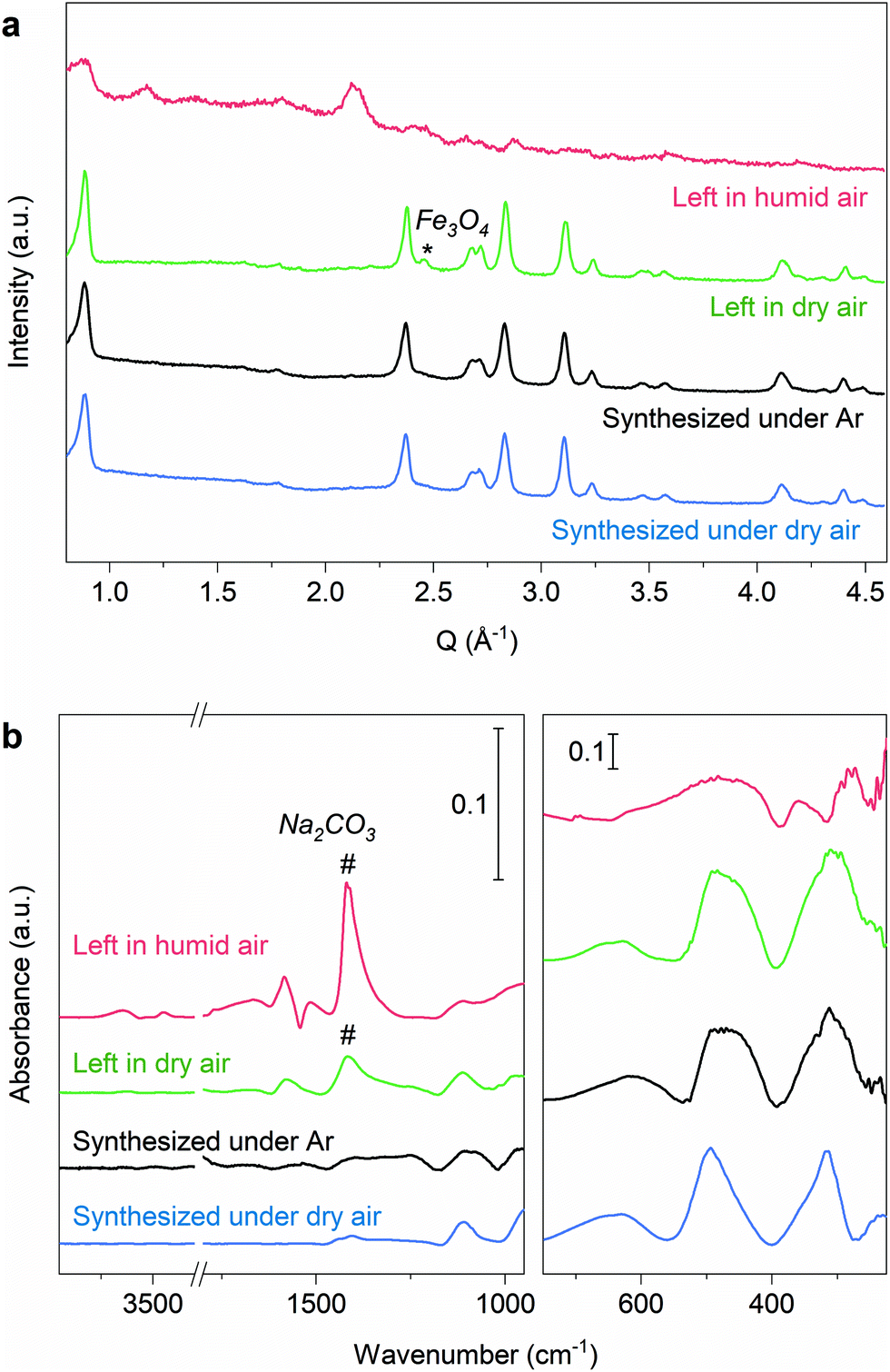 | ||
| Fig. 3 Stability of Na2Fe2OS2. (a) XRD diagram and (b) infrared spectra of the Na2Fe2OS2 powder synthesized by mechanosynthesis under Ar (black line) and under dry air (blue line) before methanol washing showing similar results. Leaving the mechanosynthesized sample under flowing dry air for 20 hours shows that some of the Fe metal impurity is oxidized into Fe3O4 (asterisk) and the formation of some Na2CO3, without any change of the Na2Fe2OS2 phase (green line). The same sample left under humid atmosphere for 20 hours (RH ∼ 40%, red line) shows the decomposition of the phase and a large increase of Na2CO3. | ||
The stability of the phase under different atmospheres was further assessed by first synthesizing a sample under Ar and then storing it under dry air or under ambient atmosphere (relative humidity ∼40%) for 20 hours. Within seconds, the powder left under ambient atmosphere changed texture (formation of agglomerates) and became amorphous according to X-ray diffraction (red line, Fig. 3a). In contrast, no loss in crystallinity was observed for the sample left under dry air as attested by the similarity of the XRD pattern with that of the sample stored under Ar (green line, Fig. 3a). This suggests that Na2Fe2OS2 decomposes in the presence of humidity.
The infrared spectra of the three samples are shown on Fig. 3b, with assignment of the peaks in Table S7.† The spectra of the sample synthesized or stored under dry air are similar to that of the pristine material, whereas that of the sample left under humid atmosphere shows features from carbonate and hydroxide decomposition products, further demonstrating the stability of both the crystalline and amorphous content of Na2Fe2OS2 under dry air.
2.5 Electrochemical properties as a cathode material
Swagelok-type three-electrode cells were assembled using Na2Fe2OS2-MW with metallic Na as the counter/reference electrodes, 1 M NaPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (ethylene carbonate:dimethyl carbonate) with 2 wt% FEC (fluoroethylene carbonate) as an electrolyte, and were cycled between 1.5 and 3 V vs. Na+/Na. The initial charge leads to the removal of approximately 1.5 Na+ ions per formula unit (f.u.) from the material to reach the composition Na0.5Fe2OS2 (Fig. 4a), giving a capacity of 170 mA h g−1. The voltage increases up to 3 V, with discrete features that can be better observed on the differential capacity curve (Fig. 4b) at 1.90, 2.07 and 2.17 V versus Na+/Na. These processes are not observed upon discharge, whereas the capacity is almost fully recovered upon reduction of the material to 1.5 V, giving the composition Na1.95Fe2OS2 after the first cycle. Subsequent cycles show a smooth voltage curve, with broad features at 2.15 and 2.45 V on oxidation in the differential capacity curve, suggesting that the material undergoes some activation during the initial desodiation process. The practical reversible capacity of the material is therefore limited to 162 mA h g−1, i.e., 68% of the theoretical capacity, with an average voltage of 1.9 V versus Na+/Na. We compared the cycling properties using 1 M NaPF6 in EC:DMC electrolyte with and without FEC as an additive (Fig. 4c), with the rate capability curve for the former in Fig. S16.† In the electrolyte without additive, the coulombic efficiency (CE) improves from 87% on the first cycle to a maximum of 98% on the 4th cycle (Fig. 4d) and then continuously decreases with further cycling. The low CE suggests that electrolyte degradation products are continuously formed on the Na anode and oxidized at the cathode. Using FEC as an additive has been shown to slow the degradation of carbonate based electrolytes at the Na anode by forming a passivating layer,55 with the appearance of a signature polarization step measured on the anode potential upon Na stripping (Fig. 4a, bottom). This is consistent with the higher CE on the first cycle (96%). However, FEC does not completely prevent the formation of degradation products, as some Na is freshly plated at each cycle. More surprising is the CE value larger than 100% on the next cycles, while the capacity is still slowly decreasing. At this point, it is clear that side reactions play a significant role in the chemistry of the Na cell. But it remains difficult to understand if the cycling properties of Na2Fe2OS2 are affected by intrinsic degradation of the material itself or by other factors, such as poisoning by electrolyte degradation products formed at the Na electrode. Moreover, the difference between the charge and discharge curves suggests that processes with different kinetics are happening on charge and discharge. We therefore used several analytical techniques to understand the evolution of the material upon sodiation/desodiation.
:DMC (ethylene carbonate:dimethyl carbonate) with 2 wt% FEC (fluoroethylene carbonate) as an electrolyte, and were cycled between 1.5 and 3 V vs. Na+/Na. The initial charge leads to the removal of approximately 1.5 Na+ ions per formula unit (f.u.) from the material to reach the composition Na0.5Fe2OS2 (Fig. 4a), giving a capacity of 170 mA h g−1. The voltage increases up to 3 V, with discrete features that can be better observed on the differential capacity curve (Fig. 4b) at 1.90, 2.07 and 2.17 V versus Na+/Na. These processes are not observed upon discharge, whereas the capacity is almost fully recovered upon reduction of the material to 1.5 V, giving the composition Na1.95Fe2OS2 after the first cycle. Subsequent cycles show a smooth voltage curve, with broad features at 2.15 and 2.45 V on oxidation in the differential capacity curve, suggesting that the material undergoes some activation during the initial desodiation process. The practical reversible capacity of the material is therefore limited to 162 mA h g−1, i.e., 68% of the theoretical capacity, with an average voltage of 1.9 V versus Na+/Na. We compared the cycling properties using 1 M NaPF6 in EC:DMC electrolyte with and without FEC as an additive (Fig. 4c), with the rate capability curve for the former in Fig. S16.† In the electrolyte without additive, the coulombic efficiency (CE) improves from 87% on the first cycle to a maximum of 98% on the 4th cycle (Fig. 4d) and then continuously decreases with further cycling. The low CE suggests that electrolyte degradation products are continuously formed on the Na anode and oxidized at the cathode. Using FEC as an additive has been shown to slow the degradation of carbonate based electrolytes at the Na anode by forming a passivating layer,55 with the appearance of a signature polarization step measured on the anode potential upon Na stripping (Fig. 4a, bottom). This is consistent with the higher CE on the first cycle (96%). However, FEC does not completely prevent the formation of degradation products, as some Na is freshly plated at each cycle. More surprising is the CE value larger than 100% on the next cycles, while the capacity is still slowly decreasing. At this point, it is clear that side reactions play a significant role in the chemistry of the Na cell. But it remains difficult to understand if the cycling properties of Na2Fe2OS2 are affected by intrinsic degradation of the material itself or by other factors, such as poisoning by electrolyte degradation products formed at the Na electrode. Moreover, the difference between the charge and discharge curves suggests that processes with different kinetics are happening on charge and discharge. We therefore used several analytical techniques to understand the evolution of the material upon sodiation/desodiation.
 | ||
| Fig. 4 Electrochemical properties of Na2Fe2OS2 in Na cells. (a) Voltage curve and (b) differential capacity curve measured in a three-electrode Swagelok cell configuration with Na as counter and reference electrodes, and 1 M NaPF6 in EC:DMC + 2 wt% FEC as an electrolyte. The contribution of the Na counter electrode to the overall polarization of the cell (bottom curve in (a)) can be ignored in this configuration. The differential capacity curve shows the disappearance of well-defined redox process at the end of the first charge. Capacity retention (c) and coulombic efficiency (d) of the material cycled between 1.5 and 3 V vs. Na+/Na with and without FEC as an additive. | ||
2.6 Structural evolution upon cycling
In situ X-ray diffraction was performed to understand the structural evolution of the crystalline part of the material during the first charge/discharge cycle. An electrode with an approximate loading of 40 mg was cycled up to 3 V at a C/60 rate (1 Na+/f.u. exchanged in 60 h) and one-hour long diffraction patterns were repeatedly measured during cycling. The patterns are plotted as a contour plot in Fig. 5a with the voltage curve, showing first a biphasic transition with an intensity decrease of the peaks from the pristine phase and the appearance of a new set of peaks after the removal of approximately 0.3 Na+/f.u. (Fig. 5b). This biphasic process corresponds to the sharp redox process observed in the differential capacity curve at 1.9 V (Fig. 4b). Upon oxidation from 2.05 V to 3 V, corresponding to a Na content varying from x = 1.7 to x = 0.5 in NaxFe2OS2, the Bragg reflections corresponding to this new phase disappear (Fig. 5c), leaving only the reflections from Be and Al that belong to the electrochemical cell. This amorphization is irreversible as the Bragg reflections do not return when discharging the cell to 1.5 V (x ≈ 1.95) after a full charge to 3 V. Close examination of the background shows some change upon cycling, suggesting the reversible evolution of the amorphous phase on the next cycles, but it is difficult to discuss from diffraction data. A second in situ experiment with a fresh electrode was then performed to probe the structural reversibility of the first biphasic process taking place at 1.9 V. The cell was cycled at C/80 and limited to 2.05 V on charge, then discharged at C/40. The reflections corresponding to the pristine material first disappear to be replaced by the new set of reflections on charge (Fig. S17†), and they reappear again on discharge, showing the reversibility of this biphasic phase transition. | ||
| Fig. 5 Structural evolution of Na2Fe2OS2-MW upon charging. (a) In situ XRD experiment and corresponding voltage curve, showing a new structure for the composition Na1.7Fe2OS2 (indicated by a dashed line) followed by a complete amorphisation of the material. The data is shown as a colormap, dash signs indicate peaks from the Be/Al belonging to the in situ setup. (b) Selected diffraction patterns highlighting the biphasic process from Na2Fe2OS2 to Na1.7Fe2OS2 and (c) the amorphisation from Na1.7Fe2OS2 to Na0.5Fe2OS2. (d) Fourier difference map around the Fe site from the refinement of Na1.7Fe2OS2 SXRD data using the same model as Na2Fe2OS2-MW and an sof for Na of 0.85. (e) Final Rietveld refinement of the SXRD pattern of the partially charged phase with refined composition Na1.6(1)Fe2OS2, with Iobs (red dots), Icalc (black line), Iobs–Icalc (blue line), and Bragg reflections (black tick marks for Na1.6(1)Fe2OS2, blue for Fe, orange for Fe3O4 and pink for ZrO2) and comparison with the initial model (inset). (f) Plausible Fe migration pathways highlighted with a yellow arrow through the face sharing of Fe and Na octahedra and with blue arrows through the vacant tetrahedral site (in blue) leading to the final disordered structural model for Na1.6(1)Fe2OS2. | ||
2.7 Structure of the intermediate NaxFe2OS2 phase (x ∼ 1.7)
High quality synchrotron X-ray data were collected ex situ on the new phase NaxFe2OS2 charged at 2.05 V vs. Na+/Na showing the same set of Bragg reflections as observed in the in situ cell. The new peaks can be indexed using the same space group as the pristine material, with a decrease and increase of a and c lattice parameters, respectively (a = 3.8814(7) Å and c = 14.258(3) Å) as shown by the Le Bail fit on Fig. S18.† Assuming that the removal of 0.3 Na+/f.u. is homogeneous between both the crystalline and amorphous part of the pristine sample, the composition of the new crystalline structure should be approximately Na1.7Fe2OS2. Using the structural model of Na2Fe2OS2, from which 0.3 Na atoms are removed per formula unit, does not yield a good Rietveld fit (Fig. S19a†). The actual structure was determined through successive analysis of the electronic density in Fourier difference maps (Fig. S17–S19 in part 3.2 of ESI† for details) revealing, in particular, a strong negative electron density on the Fe site (Fig. 5d). A good fit to the synchrotron X-ray data was finally obtained by introducing anti-site disorder between the Fe and Na sites and displacing Fe atoms from the initial 4c position (0, 0.5, 0) onto a 16n (x, 0.5, z) position determined from the Fourier difference map (white arrows on Fig. 5d). Fig. 5e shows the final fit of the SXRD data and the inset highlights the improvement of the fit compared to the ordered model without anti-site defects and the outcome of the refinement is presented in Tables S8–S10.† Two possible cationic migration pathways can favor the formation of anti-site disorder once some Na sites are left vacant by the desodiation process: (i) through faces shared by Fe and vacant (Na) octahedral sites (yellow arrow on Fig. 5f) and (ii) through an interstitial tetrahedral site (blue arrows on Fig. 5f). This interstitial site is actually believed to be responsible for the high ionic conductivity in the oxide ion conductor La2NiO4+δ Ruddlesden–Popper phase, with excess O2− anions occupying this site,56 and hence is a candidate for Na mobility in the anti-structure studied here.The final structural model defined for the intermediate disordered phase is presented in Fig. 5f. The refined composition, Na1.6(1)Fe2.0(1)OS1.98(10), is close to the expected Na1.7Fe2OS2 composition, suggesting that sodium is homogeneously removed from the amorphous and crystalline parts of the pristine sample. The 16n site is occupied by 62(12)% Fe and 38(12)% Na, while the 4e site contains 41(3)% Na and 38(4)% Fe. This partial occupation represents a high degree of cation disorder, with almost random occupation of Fe and Na on both sites. The BVS value of the 16n site (2.28(7)) is slightly higher than expected for Fe2+ cations, suggesting that potentially oxidized Fe3+ species are most probably located in these positions rather than the 4e site. The larger size of the 4e site and smaller BVS value (1.08(8)), should be more favourable to host Fe2+, which is also more mobile than Fe3+ cations. Assuming that Fe3+ remains in the 16n site, the composition of the initial [Fe2OS2]2− layer becomes [(Fe1.2(2)Na0.8(2))OS2]2.4−, whereas that of the former sodium double layer becomes [Na0.76(8)Fe2+0.82(6)]2.4+.
The decrease of the a lattice parameter reflects the presence of Fe3+, which affects the average Fe–O distance in the ab plane. By considering the rms displacements of both Fe, S and O atoms from the value of their adp, the Fe–O distances range between 1.84(13) and 2.18(13) Å and the Fe–S distances range from 2.080(5) and 3.416(6) Å. It is probable that, on a local scale, S and Fe or Na will arrange within their potential sites so as to prevent the smallest distance which is not favourable for Fe or Na and S bonding. In turn, the increase in the c lattice parameter counterbalances this effect and acts to maintain satisfactory Na–Na and Na–S distance. Overall, a unit cell volume shrinkage is observed (214.8(1) Å3 compared to 228.91(3) Å3 for the pristine sample).
Rietveld analysis was used to calculate the phase fraction of the crystalline material compared to the Fe impurity, expected to be insensitive to cycling, and thus check whether part of the phase becomes amorphous during the phase transition from Na2Fe2OS2 to Na1.7Fe2OS2. The amount of crystalline phase decreases slightly from 98.2(1) wt% for Na2Fe2OS2 to 96.1(3) wt% for Na1.7Fe2OS2, suggesting that only a small part of the crystalline Na2Fe2OS2 phase (2.1(3) wt%) loses crystallinity during the charge to 2.05 V. This shows that the amorphous and crystalline parts of the starting material behave similarly during the electrochemical process and is reinforced by the comparison of the apparent size of the crystallites. Microstructural analysis of Na1.7Fe2OS2, performed in the same way as for Na2Fe2OS2-MW, did not reveal any particular size anisotropy, with a volume-averaged apparent size of the crystallites of 24(5) nm and an average maximum strain of 106.4(6)‰. The apparent particle size did not decrease during the charge to 2.05 V, which indicates that little side amorphization occurs during this first process. However, the anomalously large strain could find its origin through increased structural disorder. This seems consistent with the wide range of bond lengths which indicates significant local relaxation from the average refined cell metrics.
The successive electrochemical reactions happening in Na2Fe2OS2 can be summarized as follow:
| Na2Fe2OS2 (crystalline) → Na1.6Fe2OS2 (crystalline) + 0.4Na+ + 0.4e− | (1) |
| Na1.6Fe2OS2 (crystalline) → Na0.5Fe2OS2 (amorphous) + 1.1Na+ + 1.1e− | (2) |
| Na0.5Fe2OS2 (amorphous) ↔ Na1.95Fe2OS2 (amorphous) + 1.45Na+ + 1.45e− | (3) |
Limiting the cycling between 1.5 and 2.05 V to retain the crystallinity of the material only gives a capacity of 34 mA h g−1 (0.3 Na+/e− exchanged), which is not large enough to represent a good compromise between capacity and structural stability. Despite the material's loss of crystallinity, an initial charge capacity of 170 mA h g−1 (1.5 Na+/e−) and a discharge capacity of 162 mA h g−1 are obtained when cycling between 1.5 and 3 V. This is a significant capacity and it is therefore important to obtain some insight on the local structure evolution of the amorphous compounds upon cycling of the material.
2.8 EXAFS analysis of iron local environment
X-ray absorption spectroscopy at the Fe K-edge was used to obtain information on the local structure of the pristine and ex situ samples at different states of charge/discharge on the first cycle. This technique has the advantage of probing both the crystalline and amorphous parts of the sample. The extended X-ray absorption fine structure (EXAFS) data gives information on the first few coordination shells around the absorbing Fe atoms, which will be designated as Fe* for clarity. A simple comparison of the magnitude of the Fourier transform of EXAFS data for the pristine and the cycled materials (charged at 2.05 V, 3 V and discharged at 1.5 V after a complete cycle) clearly highlights the loss of order beyond the first coordination shell of atoms around Fe* upon oxidation (Fig. 6a): the magnitude of the signal in the 1–2.2 Å R-range, corresponding to the first shell of atoms around Fe*, is not significantly modified, whereas the signal becomes very weak above the R value of 2.2 Å. This is consistent with the loss of crystallinity and long-range order of the material upon oxidation as observed by in situ XRD. | ||
| Fig. 6 Local environment around Fe atoms from EXAFS at the Fe K-edge. (a) Magnitude of the Fourier transform of EXAFS data for the pristine Na2Fe2OS2 and selected ex situ samples charged to various voltages, showing the loss of correlation beyond the first atomic shell around absorbing Fe atoms when the material is oxidized. (b) Detail of the contributions used to fit the data for the pristine sample in the 1–3 Å R-range. A short Fe–S distance (dark green line) is required to fit the data properly in addition to the scattering of the crystalline phase (dashed lines). Experimental data is shown as blue circle markers and the fit as a black line. | ||
The EXAFS data were fitted to obtain more precise information on the bond distances in the different samples. In the pristine material, structural information for the crystalline part of the sample is available up to R-values of 5–6 Å, and bond distances can be constrained based on the crystallographic parameters obtained from diffraction data (a, c, zNa, zS, see Fig. S22 and Table S11 in ESI† for more detailed information). This model gives a good fit of the data beyond the first shell, but a poor result in the 1–2.4 Å R-range. This suggests that additional contributions in the first shell of atoms around the Fe* absorbing atom, i.e., Fe*–O and/or Fe*–S distances, have to be included to model the data. A good fit could be obtained by adding a short Fe*–S distance (2.290(6) Å) in the model (Fig. 6b), whereas the addition of a Fe*–O distance gave large bond distance values (>2.3 Å), highly correlated to the Fe*–S scattering path, and did not improve the fit significantly. This contribution was therefore not included in the final model. The final fit of the EXAFS data is shown in Fig. S23a and b, and fitting parameters in Table S12.† Interestingly, the short Fe–S distance (2.290(6) Å) is much shorter than the average Fe–S distance of the crystallographic model (2.615(10) Å). This distance could reflect local distortions in the crystalline part of the sample, which can be inferred from the large atomic displacement parameters on Fe and S atoms. The root mean square (rms) displacements of both atoms along the Fe–S vector were calculated from the outcome of the refinement of the diffraction data (detail in part 2.4 of ESI and Table S8†). It shows that the Fe–S distance in the pristine material can be as short as 2.34(15) Å if the rms displacement is considered (green line in Fig. S24†), therefore matching, within error, the distance obtained with EXAFS. Alternatively, the short Fe–S distance could reflect the environment of Fe in the amorphous part of the sample. Future work will consist of an in-depth structural and compositional characterization of this amorphous content produced by ball milling.
The phase charged to 2.05 V, which retains crystallinity according to X-ray diffraction, shows a similar EXAFS signal compared to the fully amorphous samples, whereas one would expect some signal to persist beyond the first shell of atoms. Simulating EXAFS data for different models considering the multiple Fe–Fe distances in Na1.7Fe2OS2, due to antisite defect (38% of Fe on the 4e site) and/or displacement of Fe from the 4c to 16n site, results in a significant reduction of the intensity beyond the first shell (Fig. S25 in ESI†). This explains why the EXAFS data of the Na1.7Fe2OS2 material resembles that of the amorphous samples (3 V and 1.5 V), despite being crystalline. Almost no information beyond the first shell (Fe*–O and Fe*–S) can be obtained because of the increased disorder leading to multiple Fe–Fe distances in Na1.7Fe2OS2.
For the ex situ cycled samples charged at 3 V (Na0.5Fe2OS2) and discharged at 1.5 V after a complete cycle (Na1.95Fe2OS2), the EXAFS fit was restrained to the first two shells, Fe*–O, Fe*–S and Fe*–Fe. A comparison of different fitting models gives good results for mixed oxide/sulphide coordination environments (Rf < 0.1%, see Fig. S27†), whereas pure oxide and sulphide models result in poor fits (Rf > 2.5%). The result of the final fits is shown in Fig. S23† with refined parameters in Table S12.† This analysis suggests that iron in the amorphous material formed upon cycling is still coordinated by oxygen and sulphur atoms, although it is difficult to discriminate between heteroleptic Fe environments (FeOxSy), as in the crystalline Na2Fe2OS2 compound, and/or an oxysulphide with a distribution of homoleptic polyhedra (FeOx and FeSy). The Fe–S distances obtained for the cycled samples (between 2.256(3) and 2.296(5) Å) are close to the short Fe–S distance found for the pristine sample (2.290(6) Å). These distances are typical of Fe–S bonds in tetrahedral FeS4 environments, and the Fe–O distances in the amorphous samples (between 1.878(6) and 1.927(19) Å) are typical of tetrahedral FeO4 environments, which suggests that the amorphisation of the material is associated with a large reorganization of cations/anions in the material. Both Fe*–O and Fe*–S distances decrease upon oxidation of the material to 2.05 V (1.917(8) and 2.268(2) Å, respectively) and further to 3 V (1.878(6) and 2.256(3) Å), and increase again on reduction to 1.5 V (1.927(19) and 2.296(5) Å), suggesting some degree of Fe oxidation/reduction upon cycling.
2.9 Cycling properties in Na-ion full cells
As the material becomes fully amorphous during the initial charge, we need to verify that the reversible capacity of the material is real, and not simply an artefact due to the presence of excess Na in the metallic sodium anode. We therefore assembled Na-ion full cells with hard carbon (HC) as an anode so that the only source of Na in the cell is that present in Na2Fe2OS2 at the beginning of the experiment. This also brings the advantage of preventing electrolyte degradation reactions at the more reactive Na metal anode, so that FEC is no longer needed as an additive in the electrolyte. The lower and upper voltage limits for cycling in full cell were fixed to 0 and 3 V. This way, the potential of the Na2Fe2OS2 electrode varies between 1.5 and 3 V vs. Na+/Na (thus reaching the composition Na0.5Fe2OS2) while the hard carbon electrode potential varies between 1.5 and 0 V vs. Na+/Na on discharge and charge, respectively. The charge/discharge capacities and coulombic efficiency (CE) of the Na2Fe2OS2/HC full cell are compared in Fig. 7 with that of the Na2Fe2OS2/Na half cell, with 1 M NaPF6 in EC:DMC as electrolyte for both cells. The low CE of 58.5% on the first cycle (Fig. 7b) reflects the loss of Na during the formation of the solid electrolyte interphase (SEI) on the hard carbon anode, meaning that further cycling of NaxFe2OS2 is limited to the 1.36 ≤ x ≤ 0.47 composition range. This corresponds to a discharge capacity of 100 mA h g−1 and a specific energy of 160 W h kg−1. Longer cycling in the full cell shows a constant CE of approximately 98% and slow capacity loss over time. The excess sodium in half cells compensates for the loss of compositional range, giving 86.8% efficiency on the first cycle, while also allowing for mitigated capacity loss with cycling (Fig. 7a). This shows that the Na half cell system adds artefacts in the determination of cathode properties and is unreliable to study long-term cycling.
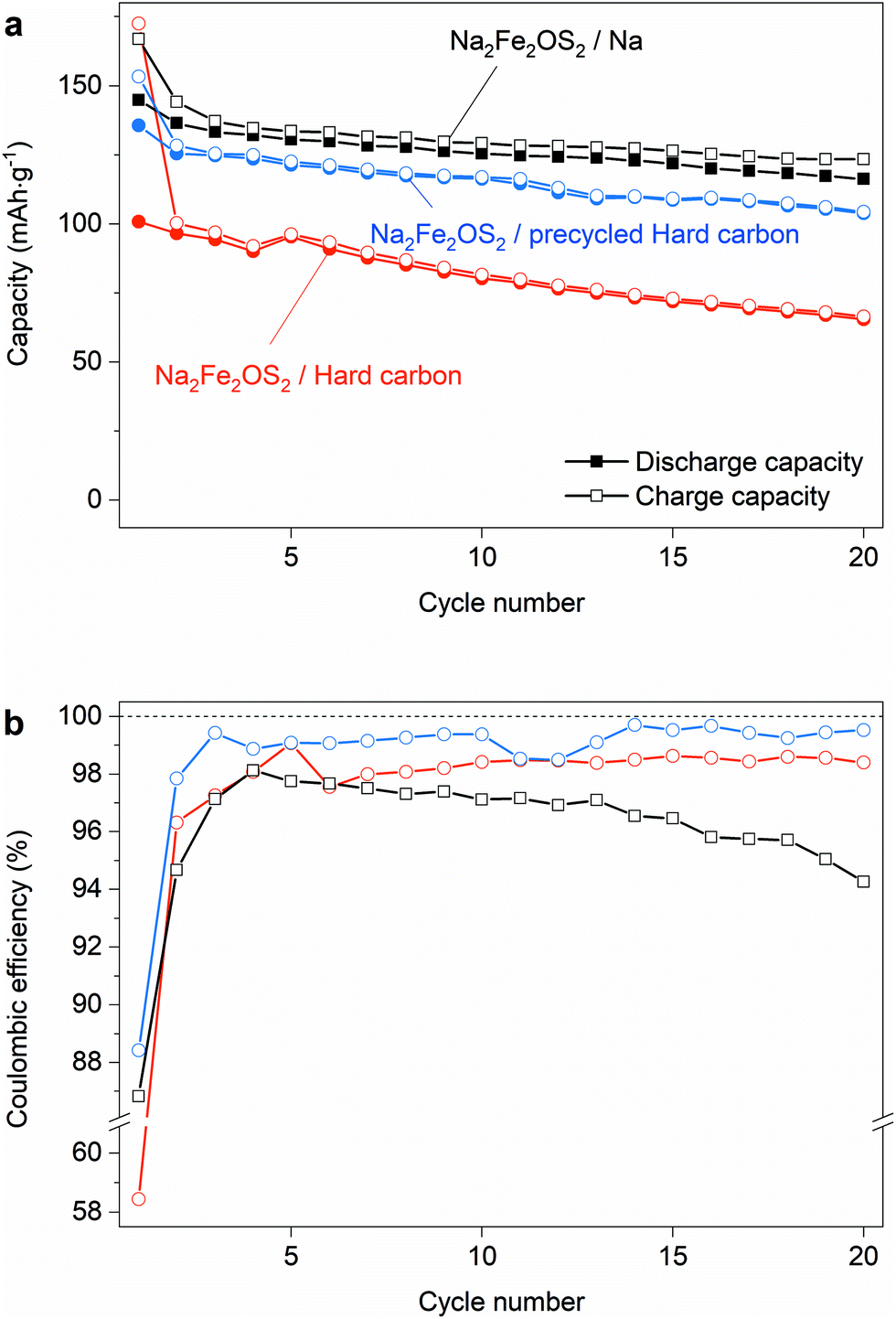 | ||
| Fig. 7 Performances of Na2Fe2OS2 in Na-ion full cells. (a) Charge/discharge capacity retention and (b) coulombic efficiency comparison between half cell (black) and full cells using pristine (red) and precycled (blue) hard carbon anodes and 1 M NaPF6 in EC:DMC electrolyte. All cells are cycled at C/10. | ||
To remove the effect of SEI formation on the hard carbon and access more reversible capacity from Na2Fe2OS2, we precycled a hard carbon electrode in a Na half cell to form the SEI, recovered it after 5 cycles, washed it in DMC and assembled a full cell with that electrode which was again cycled between 0 and 3 V. This time, the CE on the first cycle reaches 88.4%, suggesting that Na2Fe2OS2 also contributes to the irreversible capacity during the first cycle, and the specific energy of the cell reaches 215 W h kg−1. The CE then settles around 99% for the next 20 cycles, and the capacity further decreases with cycling (77% capacity retention after 20 cycles), although more slowly than with the pristine hard carbon anode (65% capacity retention). Cells opened after 10 and 50 cycles were first compared visually, showing very different results. After 10 cycles, no obvious degradation was observed, but after 50 cycles the glass fiber separator clearly appeared dark where the cathode was placed (Fig. S27a†). XPS measurements on the hard carbon anode after 50 cycles (Fig. S27c and d†) show signals corresponding to Fe 2p and S 2p peaks, thus proving that long term cycling results in iron and sulphur dissolution, migration and deposition on the anode side. This explains, at least in part, the low CE efficiency and the capacity loss with cycling.
To further explore Fe dissolution in this material, a 3-electrode cell (hard carbon counter electrode, Na reference electrode) was used to charge the material above 3 V vs. Na+/Na and extract the remaining 0.5 Na+ from Na0.5Fe2OS2 (Fig. S27b†). The voltage stopped increasing at 3.2 V vs. Na+/Na, dropped to 3.15 V, then remained at a flat potential of 3.05 V. This gives an apparently unlimited capacity, well above the theoretical capacity of Na2Fe2OS2 or the hard carbon electrode, and leads to a complete loss of cycling performances afterwards. The color of the separator recovered from the cell appeared dark (Fig. S27a†), and XPS analysis of the hard carbon anode (Fig. S27c and d†) recovered from a coin cell after cycling in equivalent conditions, shows that the Fe/S dissolution can also be triggered by overcharging the cathode above 3.2 V vs. Na+/Na. Soluble species in the electrolyte can potentially act as redox shuttles, as can be the case in Na–S systems,57 thus providing such large capacity.
Overall, the material itself shows an intrinsic instability when charged above 3.2 V vs. Na+/Na, which cannot be prevented. However, cation dissolution over long term cycling has been mitigated by using surface coatings or adding electrolyte additives for other cathode materials in the past, such as LiFePO4,58,59 and we expect that further studies will be successful in preventing this issue and improving the long term cycling properties of Na2Fe2OS2.
3. Discussion
We report the use of an oxysulphide as a cathode material in a Na-ion battery. Very few transition metal oxysulphide materials containing alkali metals have been reported in the literature, and we have been unable to locate previous Na cathode applications of these materials (cf. part 4 in ESI† for details on literature search). Previous studies focused mostly on conversion reaction and topotactic chemical intercalation of alkali metal cations into originally alkali metal-free transition metal oxysulphides, such as copper,23 vanadium,60,61 titanium,62 tungsten,63 and molybdenum64,65 oxysulphides, the Ruddlesden Popper phases Ln2Ti2O5S2 (Ln = Y, Nd),5,66 or the layered Sr2MnO2Cu2m−0.5Sm+1 (m = 1, 2, and 3) phases.67 Some of these materials show good reversibility for electrochemical insertion of lithium and sodium, but none of them contain alkali metals in their as-prepared form,60,68 limiting their use to anode materials for Li or Na-ion batteries only. The only example of a sodium-containing oxysulphide for Na-ion battery is the Na2Cu2.09O0.5S2 phase which has been investigated as a conversion-type anode material.69The Na2Fe2OS2 material gives a theoretical capacity of 225 mA h g−1, 75% of which is obtained on the first charge to 3 V. The reversible capacity is 162 mA h g−1, which corresponds to an approximated volumetric capacity of 552 mA h cm−3 (considering the density of the crystallized Na2Fe2OS2 phase). For comparison, materials considered as potential cathode material for Na-ion batteries such as Na3V2(PO4)2F3 and Na2/3(Fe1/2Mn1/2)O2 have volumetric capacities of 349 and 570 mA h cm−3, respectively.2 In that regard, the performances of Na2Fe2OS2 fall within the right range for commercial application (cf. Table S15†).
In contrast to the parent material Na2Fe2OSe2, which can easily be prepared by a solid-state route between 600 and 700 °C,14 Na2Fe2OS2 is metastable and decomposes at high temperature. An interesting point of comparison is the metastable anti-perovskite phase Li2FeOS, whose synthesis requires quenching from 750 °C to room temperature.13 A lower temperature synthesis route, via mechanochemistry, was developed to obtain the kinetically-stabilized Na2Fe2OS2 phase. Mechanosynthesis has been shown to be a powerful tool for the synthesis of sulphide and oxysulphide materials in the past.70–72 This method has the advantage of operating at room temperature and ambient pressure, which leads to easy processing/up-scaling and enables the formation of metastable phases with high doping concentration for instance.25,45 The stability and conceivable synthesis of Na2Fe2OS2 under dry air will also facilitate its handling for any future application. The study of the microstructure of the material showed that it is composed of an amorphous part and a crystalline part, having coherently scattering domains of 20 nm, both of composition Na2Fe2OS2.
Using X-ray and neutron diffraction data, the crystalline component was fully characterized. Na2Fe2OS2 is the n = 1 member of the anti-Ruddlesden–Popper [SNa][SO(Fe2Na)]n homologous series, with divalent iron and two sodium ions. The structural evolution of the “anti” structure Na2Fe2OS2 upon desodiation can be compared to the topochemical oxygen deintercalation from the “normal” Ruddlesden–Popper phase Ca2MnO4 to form the oxygen deficient Ca2MnO3.5 phase under reducing conditions (Fig. 8).73,74 In the oxygen deficient RP phase, oxygen vacancies are preferentially found in the [MnO2] square plane of the RP phase, with an ordering that depends on the nature of the A cation. In the sodium deficient anti-RP structure, with refined composition Na1.6(1)Fe2.0(1)OS1.98(10), the sites left vacant by the deintercalated Na atoms are filled by the migration of Fe atoms, so that the vacancies preferentially sit in the [Fe2O] plane. Interestingly, the number of vacancies in Na1.6(1)Fe2.0(1)OS1.98(10) is close to that found in the Ca2MnO3.5 phase, but they are disordered over all the cation sites, a difference that could simply arise from the presence of two different cations Fe2+ and Na+ in the anti-RP phase, as opposed to only one anion O2− in the RP phase. Furthermore, the displacement of Fe atoms from the 4c to 16n position takes after the displacement of some O atoms into tetrahedral OCa2Mn2 sites for the oxygen deficient RP phase, in order to compensate for the loss of oxygen anions and reduce the repulsion between cations. Further reduction of the RP phase leads to a loss of cationic order to form the disordered rocksalt Ca2MnO3, whereas it is possible to oxidize the anti-RP phase NaxFe2OS2 to x = 0.5, which leads to the complete amorphisation of the material. Fe atoms in the amorphous phase formed are coordinated by O and/or S atoms, with Fe–O/Fe–S distances consistent with a tetrahedral environment, e.g. FeO2S2. As the loss of crystallinity of the material is triggered by the favorable vacancy formation on the Fe site of the [Fe2O] square plane, the question arises how to stabilize this layer and avoid or delay the amorphisation of the material. A possible answer could come from the study of the n = 2 members of the “normal” Ruddlesden–Popper series. By moving from Y3+ cations in YSr2Mn2O7 to a larger Nd3+ cation in NdSr2Mn2O7, the oxygen vacancies in the corresponding (Y/Nd)Sr2Mn2O5.5 phases are displaced from the [MnO2] planes to the apical oxygen sites corresponding to the Na sites in the anti-structure of Na2Fe2OS2. Following this reasoning, using larger Se2−/Te2− anions instead of S2− could stabilize the vacant sodium sites, thus preventing the migration of Fe atoms and the loss of crystalline order, or at least delay it to higher desodiation levels.
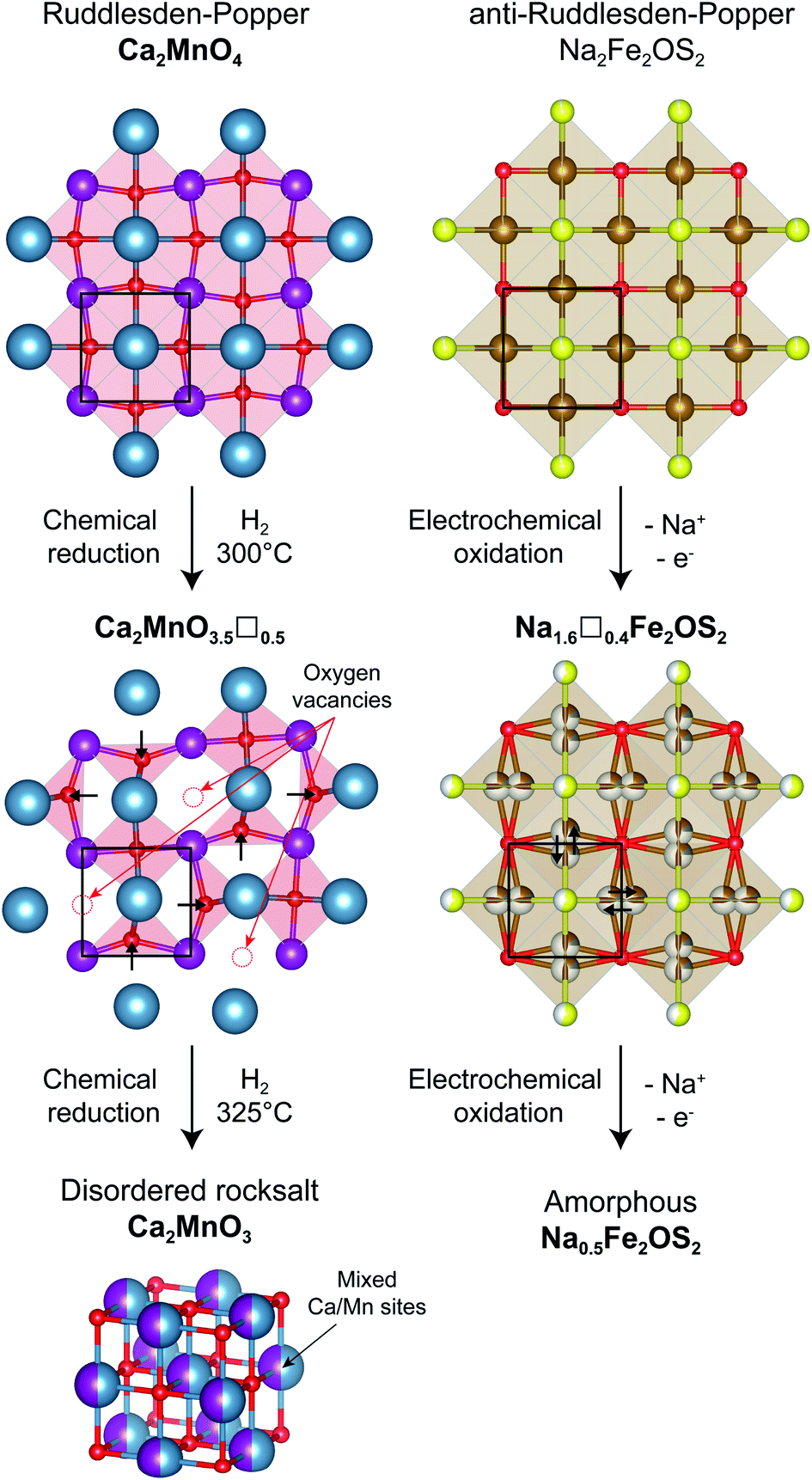 | ||
| Fig. 8 Comparison between the topochemical oxygen deintercalation from the Ruddlesden–Popper phase Ca2MnO4 (left) and the electrochemical sodium deintercalation from the anti-Ruddlesden–Popper phase Na2Fe2OS2 (right). Only the [MnO2] and [Fe2O] square lattices of the two structures are represented for clarity, highlighted with a black square, with out-of-plane Ca/S atoms. Oxygen vacancies (red circles) in the reduced Ca2MnO3.5 phase are ordered in the plane, affecting the position of other oxygen atoms (black arrows). A similar displacement of cations is observed in the oxidized Na1.6Fe2OS2 phase, with the difference that vacancies are disordered. Further reduction/oxidation of these materials leads to the formation of a disordered Ca2MnO3 phase and an amorphous Na0.5Fe2OS2 phase, respectively. | ||
This new structure type among cathode materials paves the way for the exploration of new phases with other transition metal cations and/or higher order members of the series. A few n = 1 compounds have been reported in the anti-RP series,14,44 but the only higher n member reported is the n = 3 ((Ba,Sr)3n+1ONn−1)Bin+1 anti-RP compound.54 The theoretical capacity is expected to decrease with increasing values of n (191 mA h g−1 for n = 2, 178 mA h g−1 for n = 3 and converging to 150 mA h g−1 for n = ∞, corresponding to the anti-perovskite NaFe2OS) but these values remain attractive and higher order structures could present improved structural stability with cycling or other more valuable features compared to Na2Fe2OS2. It is interesting to note that the previously reported anti-perovskite Li2FeOS, which has a much higher theoretical capacity (455 mA h g−1), is the end member of another anti-RP series [FeS][Li2FeOS]n, and shows more promise to increase the capacity. We were not able to stabilize the Na equivalent Na2FeOS phase, or alternatively the anti-RP Li2Fe2OS2 phase. The relative size of cations and anions seems to play a role in the stabilization and local structural distortions of anti-perovskite and anti-Ruddlesden–Popper phases, in the same way it does for perovskite and Ruddlesden–Popper phases, which are prone to distortions and octahedral tilting that have been extensively studied.75 The larger difference in ionic radii between sodium (r(Na+, [VI]) = 1.16 Å) and iron (r(Fe2+, [VI]) = 0.78 Å) cations is well suited to the anti-RP phases with two distinct cations sites corresponding to the axial and equatorial anion sites in the RP structure itself, whereas iron and lithium (r(Li+, [VI]) = 0.76 Å) have similar radii that favor mixed occupancy on the unique cation site of the anti-perovskite structure. Substituting iron with earlier transition metals (Ti, V, Cr, Mn), which have larger ionic radii, could help in stabilizing both Na anti-perovskite and Li anti-RP structures by decreasing/increasing the difference in ionic radii, respectively.
Despite the full amorphization of the phase, a good capacity is obtained and maintained throughout cycling. The charge compensation mechanism is expected to involve both Fe and S species and remains to be unraveled. In that regard, the disentanglement of redox contributions will represent a fascinating challenge. Analysis of EXAFS data at the Fe K-edge enabled us to understand the local structure surrounding Fe. It showed that the oxysulphide phase is maintained both in the amorphous part of the pristine sample and throughout the charge–discharge process during cycling, with Fe being coordinated to both S2− and O2− anions. This feature could help maintain the electrochemical properties of the material upon long-term cycling and shows that the crystallinity is not a prerequisite for good cyclability in this material. Amorphization has been shown to play a crucial role in promoting the electrochemical performance of Na–Fe–P–O based cathodes,76,77 and, in this regard, it would also be interesting to further study sodium oxysulphide glass materials for Na-ion batteries. Oxysulphides represent a very promising opportunity as cathode materials for sodium-ion batteries, with the possibility for both transition metal cations and sulphur anions to participate in the redox process to enhance overall capacity.
4. Conclusion
Na2Fe2OS2 is the first example of a cathode material for Na-ion batteries with an anti-Ruddlesden–Popper structure. A specific energy of 160 W h kg−1 is obtained between 0 and 3 V in a full cell configuration using a hard carbon anode, and can be increased above 200 W h kg−1 if the loss of Na during SEI formation on the hard carbon is compensated. This system is therefore interesting for large scale storage applications particularly as it consists only of elements amongst the most abundant on the planet. We developed a simple mechanosynthesis approach that not only works under inert atmosphere but also under dry air, so that preparation and processing of the material can take place in dry rooms which are already commonly used to assemble commercial Li-ion cells. The promising properties of Na2Fe2OS2 highlight the potential of new oxysulphides, and more generally new multiple anion materials, as cathode materials for metal-ion batteries. The anti-Ruddlesden–Popper structure itself presents a large chemical flexibility for the discovery of new phases, with the potentiality to replace/combine iron with other transition metal cations, use larger anions than sulphur and to extend the homologous series [SNa]−([SO(Fe2Na)]+)n beyond n = 1, by compensating charges of both layers through the substitution with lower valence cations or lower oxidation state anions. These routes could help to stabilize the material upon desodiation and increase the capacity retention, together with classic optimization methods such as electrode coatings and electrolyte engineering. As an example of material entangling complex structural and redox processes, this work calls for further studies on the transport, electronic and magnetic properties of multiple anion materials. Controlling the physical properties of materials based on anion substitution instead of, or together with, cation substitution can help to drastically reduce the environmental impact of technologically relevant materials in all domains of materials science.5. Methods
5.1 Materials
Na (ACS reagent, 99.99%, dry), Na2S (97.3%, anhydrous) and S (99.998%, trace metal basis) were purchased from Sigma Aldrich. FeS (99.98%, metals basis), Fe2O3 (99.9%, metal basis) were obtained from Alfa Aesar. Na2O (98%) from abcr GmbH and methanol (99.9%, Extra Dry, AcroSeal™, ACROS Organics™) from Thermo Fisher.5.2 Synthesis
:1/3:4/3:2/3 ratio, transferred into an alumina crucible and placed in a quartz tube before sealing under vacuum (10−4 mbar). The tube containing the sample was heated to 800 °C at a ramp rate of 5 °C min−1, held at 500 °C for 20 hours, and cooled down at a ramp rate of 5 °C min−1. The resulting powder was ground in an agate mortar for 15 min before transferring in an alumina crucible and re-annealing under vacuum at 600 °C for 20 hours using the same heating and cooling ramp rates. The resulting powder was then manually ground in order to obtain a fine powder.
5.3 Stability test
To test the stability of Na2Fe2OS2 under different atmospheres, ∼200 mg of the sample prepared by mecanosynthesis before methanol washing (Na2Fe2OS2-M) was maintained under ambient atmosphere for 20 hours on the bench, before performing an XRD scan of the powder. In parallel, ∼200 mg of the same sample was loaded in a glass tube which was then connected to a Swagelok® tube fitting and closed with valves in an argon filled glovebox. Outside the glovebox, a compressed air supply was linked to a molecular sieve desiccant the output of which was connected to the tube containing the sample. A flow of dry air was thus maintained inside the glass tube for 20 hours. Another experiment consisted of synthesizing the Na2Fe2OS2-M sample under a dry air atmosphere, following the procedure described above. Prior loading in the planetary mill, the bowl was filled with dry air by flowing synthetic air through the gassing valves of the gassing lid for ∼5 min.5.4 Elemental analysis
5.5 Electron microscopy
Wavelength Dispersive X-ray spectroscopy (WDX) was performed using a Tescan S8000 scanning electron microscope (SEM) equipped with a WDX detector from Oxford Instruments. The detector was calibrated with appropriate standards for each chemical element. Data acquisition and analysis was performed using INCA software. Powder sample was sprinkled on a carbon tape and coated with a thin layer of carbon with a Quorum sputter coater in order to avoid charging.Transmission Electron Microscopy (TEM) and energy dispersive X-ray spectroscopy (EDX) mapping was performed with a JEOL 2100+ equipped with an EDX detector from Oxford instruments. Powder sample was ball milled in methanol under inert atmosphere to reduce particles size. Few drops of the methanol suspension were deposited on a holey carbon film Ni TEM grid. The grid was loaded into a beryllium TEM holder and inserted in the JEOL 2100+. In order to avoid exposing the material to air the sample preparation and loading was perform under inert atmosphere.
5.6 Infrared spectroscopy
Infrared spectroscopy measurements were done in ATR mode using a Nicolet iS50 FTIR spectrometer with CsI beamsplitters (ThermoFisher Scientific) inside a nitrogen containing glovebox (O2, H2O < 0.1 ppm).5.7 Diffraction
Routine analysis of phase purity and lattice parameters were performed on a Bruker D8 Advance diffractometer with a monochromated Cu source (Kα1, λ = 1.54418 Å) in powder transmission Debye Scherrer geometry (capillary) with sample rotation. Synchrotron X-ray diffraction (SXRD) was performed on pristine and ex situ samples at the I11 beamline at Diamond Light Source (Oxfordshire, UK), with an incident wavelength of 0.826540(1) Å using a wide-angle position sensitive detector, and samples sealed in Ø = 0.3 mm glass capillaries to prevent air exposure. Time-of-flight (ToF) neutron powder diffraction (ND) data was collected for the pristine material at room temperature using the Polaris instrument at ISIS neutron source (Oxfordshire, UK). Samples were loaded in Ø = 6 mm vanadium cylindrical cans and sealed in an argon-filled glovebox. The structural models were refined by the Rietveld method78,79 as implemented in the Fullprof suite.80In situ XRD was performed using an electrochemical cell equipped with a Be window (250 μm thick) and an Al current collector (8 μm thick) on a Rigaku SmartLab diffractometer with a 9 kW rotating anode providing a parallel beam of Mo Kα1 radiation (λKα1 = 0.709032 Å).The program FullProf was used to obtain information about the microstructure, following the method described by Rodriguez-Carjaval et al. This model uses the Scherrer formula, which considers that the size broadening can be written as a linear combination of spherical harmonics. Peak shapes were modelled using the spherical harmonics expansions36 in a tetragonal material with Laue class 4/m. The microstructural information given in the output files were then treated using GFOURIER program81 to visualize the particle shapes.
The amount of amorphous content was determined by using the quantitative phase analysis method described in the literature.82–84 This method consists of mixing a known amount of the Na2Fe2OS2 material with an internal standard of similar absorption coefficient, Na3Fe2S4, and to compare the relative weight percent calculated by the program with the relative weight percent determined experimentally, which includes the contribution of amorphous phases. This method is described in the ESI, part 2.3.4.†
5.8 Electrochemical characterization
Electrochemical characterization was performed in 3-electrode Swagelok cells and 2025-type coin cells. The positive electrode consisted of a laminated mixture of active material (Na2Fe2OS2), conductive carbon (C65 from Timcal) and binder (polytetrafluoroethylene, PTFE dried from a 60% aqueous suspension from Sigma-Aldrich) in proportions 85:10:5 by weight for measurement in Na cells. This ratio was changed to 55:30:15 for electrodes used in full cells in order to balance cathode/anode capacities. For ex situ characterization by XRD, the active material was simply mixed with 10 wt% C65 conductive carbon and used as a powder. Active material loadings were typically between 5 and 10 mg and electrode thickness was approximately 100 μm. Metallic Na was used as an anode for half cells and hard carbon (HC) electrodes for full cells. Hard carbon electrodes were prepared by ball-milling hard carbon with C65 conductive carbon for 20 min, then dispersing the powder in a 1% aqueous solution of carboxymethylcellulose (CMC) to obtain a 81:9:10 ratio of HC:C65:CMC, and casted over an Al foil. Electrodes were punched after solvent evaporation and further dried under vacuum at 100 °C for 12 h before use. 1 M NaPF6 in EC:DMC (1:1) with or without 2 wt% fluoroethylenecarbonate (FEC) additive was used for the electrolyte. The positive and negative electrodes were separated by Whatman GF/D borosilicate glass fiber membranes soaked with the electrolyte. All parts were assembled in an Ar-filled glovebox. Galvanostatic cycling was performed at a C/10 rate (defined as 1 Na+ exchanged in 10 h, considering the chemical formula Na2Fe2OS2) between 1.5 and 3 V versus Na+/Na. After cycling, samples for ex situ characterization were recovered inside the glovebox, washed three times in anhydrous DMC and dried under vacuum. For the study of capacity retention, the number of cycles was limited to 20 cycles due to the identification of Fe and S dissolution as the main reason for capacity loss over cycling.
5.9 X-ray absorption spectroscopy
Self-standing electrodes, with an optimized density for X-ray absorption measurements at the Fe K-edge, were prepared and cycled at different charge/discharge states, washed in DMC, and sealed in a pouch bag to prevent air exposure of the samples. X-ray absorption spectra were measured at the B18 beamline at Diamond Light Source (Oxfordshire, UK) in transmission mode. The data were calibrated by fixing at 7112 eV the maximum of the derivative of an Fe metal foil reference placed after the samples, and normalized with the Athena software.85 Fitting of the EXAFS data was done using the Artemis software of the same suite, starting from the crystallographic model obtained from diffraction experiment to build the initial paths used for fitting. The data were fitted in R space using k, k2 and k3-weighed data together, with 2.5–12.5 Å−1k-range/1–6 Å R-range for the pristine material, and 3–12.6 Å−1k-range/1–3 Å R-range for the cycled samples. E0 and S02 were first fixed at values obtained from fitting reference materials (FeO, Fe2O3, FeS, FeS2, NaFeO2, Na3Fe2S4) and let to refine only at the end of the procedure when a satisfactory fit was obtained.5.10 X-ray photoemission spectroscopy
Measurements were performed on a Thermo Fisher Scientific NEXSA spectrometer using a micro-focused monochromatic Al Kα source, with an X-ray source power of 150 W. Low energy electrons and Ar ions were used for charge neutralization. Survey spectra were run at a pass energy of 200 eV and high-resolution spectra at a pass energy of 40 eV. All samples for XPS analysis were prepared in a glovebox and loaded in the spectrometer using an air-tight sample transfer chamber to avoid any exposure to air.Conflicts of interest
The authors declare no competing financial interest. Underlying data is available at http://datacat.liverpool.ac.uk/id/eprint/1043.Acknowledgements
We thank EPSRC for funding under EP/N004884. We thank Diamond Light Source for access to beamlines I11 and I18, Dr Sarah Day, Prof. Alan Chadwick and Dr Giannantonio Cibin for assistance on the beamlines. We thank STFC for access to Polaris (Xpress proposal 1990220) and Dr Ron Smith for running the measurements. XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’), operated by Cardiff University and UCL, under contract No. PR16195. Dr Mark Isaacs is greatly acknowledged for his support during the measurement. MJR thanks the Royal Society for the award of a Research Professor position. We acknowledge Dr Konstantin Luzyanin, Mr Stephen Moss and Mrs Jean Ellis (University of Liverpool) for ICP-OES and CHNS measurements, and Dr Quinn Gibson and Dr Wesley Surta for helpful discussions.References
- A. A. Yaroshevsky, Geochem. Int., 2006, 44, 48 CrossRef.
- R. Dugas, B. Zhang, P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A867 CrossRef CAS.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef.
- A. M. Kusainova, P. S. Berdonosov, L. G. Akselrud, L. N. Kholodkovskaya, V. A. Dolgikh and B. A. Popovkin, J. Solid State Chem., 1994, 112, 189 CrossRef CAS.
- S. J. Clarke, S. G. Denis, O. J. Rutt, T. L. Hill, M. A. Hayward, G. Hyett and Z. A. Gál, Chem. Mater., 2003, 15, 5065 CrossRef CAS.
- M. O. Figueiredo, Inorg. Chim. Acta, 1987, 140, 161 CrossRef CAS.
- N. Charles, R. J. Saballos and J. M. Rondinelli, Chem. Mater., 2018, 30, 3528 CrossRef CAS.
- S. J. Clarke, P. Adamson, S. J. C. Herkelrath, O. J. Rutt, D. R. Parker, M. J. Pitcher and C. F. Smura, Inorg. Chem., 2008, 47, 8473 CrossRef CAS.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Förstermann, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Energy Environ. Sci., 2018, 11, 926 RSC.
- D. Deng, ChemNanoMat, 2017, 3, 146 CrossRef CAS.
- A. Dugast, R. Brec, G. Ouvrard and J. Rouxel, Solid State Ionics, 1981, 5, 375 CrossRef CAS.
- S. Saha, G. Assat, M. T. Sougrati, D. Foix, H. Li, J. Vergnet, S. Turi, Y. Ha, W. Yang, J. Cabana, G. Rousse, A. M. Abakumov and J.-M. Tarascon, Nat. Energy, 2019, 4, 977 CrossRef CAS.
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, J. Am. Chem. Soc., 2017, 139, 9645 CrossRef CAS.
- J. B. He, D. M. Wang, H. L. Shi, H. X. Yang, J. Q. Li and G. F. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205212 CrossRef.
- W. Liu, B. Guo, C. Mak, A. Li, X. Wu and F. Zhang, Thin Solid Films, 2013, 535, 39 CrossRef CAS.
- P. G. Mccormick, J. Ding, W.-F. Miao and R. Street, Process for the Production of Ultrafine Particles, CA2230443C, 2009.
- F. M. Michel, M. A. A. Schoonen, X. V. Zhang, S. T. Martin and J. B. Parise, Chem. Mater., 2006, 18, 1726 CrossRef CAS.
- R. J. Hill and C. J. Howard, J. Appl. Crystallogr., 1987, 20, 467 CrossRef CAS.
- P. Baláž, M. Baláž, M. Achimovičová, Z. Bujňáková and E. Dutková, J. Mater. Sci., 2017, 52, 11851 CrossRef.
- K. Kanazawa, S. Yubuchi, C. Hotehama, M. Otoyama, S. Shimono, H. Ishibashi, Y. Kubota, A. Sakuda, A. Hayashi and M. Tatsumisago, Inorg. Chem., 2018, 57, 9925 CrossRef CAS.
- V. V. Zyryanov and N. F. Uvarov, Inorg. Mater., 2005, 41, 281 CrossRef CAS.
- P. Baláž, M. Hegedüs, M. Achimovičová, M. Baláž, M. Tešinský, E. Dutková, M. Kaňuchová and J. Briančin, ACS Sustainable Chem. Eng., 2018, 6, 2132 CrossRef.
- L. D'alencon, T. Le Mercier, M.-D. Braida, J. Gamon and P. Barboux, Mechanochemical Synthesis of Rare Earth Sulfides, WO/2019/121819, 2019.
- R. Schlem, S. Muy, N. Prinz, A. Banik, Y. Shao-Horn, M. Zobel and W. G. Zeier, Adv. Energy Mater., 2019, 1903719 Search PubMed.
- A. Düvel, M. Wilkening, R. Uecker, S. Wegner, V. Šepelák and P. Heitjans, Phys. Chem. Chem. Phys., 2010, 12, 11251 RSC.
- J. Mbah, B. Krakow, E. Stefanakos and J. Wolan, Electrochem. Solid-State Lett., 2009, 12, E12 CrossRef CAS.
- N. V. Kosova, in High-Energy Ball Milling, ed. M. Sopicka-Lizer, Woodhead Publishing, 2010, pp. 331–360 Search PubMed.
- S. Soiron, A. Rougier, L. Aymard and J.-M. Tarascon, J. Power Sources, 2001, 97–98, 402 CrossRef CAS.
- C. Yang, Y. Li, Y. Chen, Q. Li, L. Wu and X. Cui, Small, 2019, 15, 1970044 CrossRef.
- N. Muralidharan, C. N. Brock, A. P. Cohn, D. Schauben, R. E. Carter, L. Oakes, D. G. Walker and C. L. Pint, ACS Nano, 2017, 11, 6243 CrossRef CAS.
- P. Baláž, in Mechanochemistry Nanosci. Miner. Eng., ed. P. Baláž, Springer, Berlin, Heidelberg, 2008, pp. 1–102 Search PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC.
- Y. Bai, F. He, B. Fu and X. Han, International Journal of Innovative Computing, Information and Control, 2014, 10, 1715 Search PubMed.
- M. G. Kanatzidis, Chem. Mater., 1990, 2, 353 CrossRef CAS.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571 RSC.
- J. Rodríguez-Carvajal, Study of Micro-Structural Effects by Powder Diffraction Using the Program FULLPROF, http://www.cdifx.univ-rennes1.fr/fps/Microstructural_effects.pdf Search PubMed.
- A. R. Stokes and A. J. C. Wilson, Proc. Phys. Soc., 1944, 56, 174 CrossRef CAS.
- V. V. Boldyrev, Thermochim. Acta, 1987, 110, 303 CrossRef.
- J. J. Friel and C. E. Lyman, Microsc. Microanal., 2006, 12, 2 CrossRef CAS.
- D. Mitra, S. J. George, Y. Guo, S. Kamali, S. Keable, J. W. Peters, V. Pelmenschikov, D. A. Case and S. P. Cramer, J. Am. Chem. Soc., 2013, 135, 2530 CrossRef CAS.
- L. Zhou, J. Liu and F. Dong, Spectrochim. Acta, Part A, 2017, 173, 544 CrossRef CAS.
- L. Lauterbach, H. Wang, M. Horch, L. B. Gee, Y. Yoda, Y. Tanaka, I. Zebger, O. Lenz and S. P. Cramer, Chem. Sci., 2015, 6, 1055 RSC.
- T. Zhang, N. Zhao, J. Li, H. Gong, T. An, F. Zhao and H. Ma, RSC Adv., 2017, 7, 23583 RSC.
- A. Adam and H.-U. Schuster, Z. Anorg. Allg. Chem., 1990, 584, 150 CrossRef CAS.
- D. G. Free, N. D. Withers, P. J. Hickey and J. S. O. Evans, Chem. Mater., 2011, 23, 1625 CrossRef CAS.
- J. M. Mayer, L. F. Schneemeyer, T. Siegrist, J. V. Waszczak and B. V. Dover, Angew. Chem., Int. Ed. Engl., 1992, 31, 1645 CrossRef.
- T. C. Ozawa and S. M. Kauzlarich, Sci. Technol. Adv. Mater., 2008, 9, 033003 CrossRef.
- T. Yajima, K. Nakano, F. Takeiri, T. Ono, Y. Hosokoshi, Y. Matsushita, J. Hester, Y. Kobayashi and H. Kageyama, J. Phys. Soc. Jpn., 2012, 81, 103706 CrossRef.
- S. K. Gupta, H. Jangam and N. Sharma, Res. Dev. Mater. Sci., 2018, 7, 1 Search PubMed.
- J. G. Bednorz and K. A. Müller, Z. Phys. B Condens. Matter, 1986, 64, 189 CrossRef CAS.
- E. E. McCabe, C. Stock, E. E. Rodriguez, A. S. Wills, J. W. Taylor and J. S. O. Evans, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 100402 CrossRef.
- R. H. Liu, D. Tan, Y. A. Song, Q. J. Li, Y. J. Yan, J. J. Ying, Y. L. Xie, X. F. Wang and X. H. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 144516 CrossRef.
- D. J. Singh, New J. Phys., 2012, 14, 123003 CrossRef.
- F. Gäbler, Y. Prots and R. Niewa, Z. Anorg. Allg. Chem., 2007, 633, 93 CrossRef.
- R. Dugas, A. Ponrouch, G. Gachot, R. David, M. R. Palacin and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A2333 CrossRef CAS.
- A. Demourgues, F. Weill, B. Darriet, A. Wattiaux, J. C. Grenier, P. Gravereau and M. Pouchard, J. Solid State Chem., 1993, 106, 317 CrossRef CAS.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 1 Search PubMed.
- K. Amine, J. Liu and I. Belharouak, Electrochem. Commun., 2005, 7, 669 CrossRef CAS.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243 RSC.
- G. Tchangbedji, D. A. Odink and G. Ouvrard, J. Power Sources, 1993, 44, 577 CrossRef CAS.
- G. Ouvrard, G. Tchangbedji, P. Deniard and E. Prouzet, J. Power Sources, 1995, 54, 246 CrossRef CAS.
- H. Martinez, A. Benayad, D. Gonbeau, P. Vinatier, B. Pecquenard and A. Levasseur, Appl. Surf. Sci., 2004, 236, 377 CrossRef CAS.
- I. Martin, P. Vinatier, A. Levasseur, J. C. Dupin and D. Gonbeau, J. Power Sources, 1999, 81–82, 306 CrossRef CAS.
- K. M. Abraham and D. M. Pasquariello, Chem. Mater., 1993, 5, 1233 CrossRef CAS.
- L. Benoist, D. Gonbeau, G. Pfister-Guillouzo, E. Schmidt, G. Meunier and A. Levasseur, Surf. Interface Anal., 1994, 22, 206 CrossRef CAS.
- S. G. Denis and S. J. Clarke, Chem. Commun., 2001, 2356 RSC.
- O. J. Rutt, G. R. Williams and S. J. Clarke, Chem. Commun., 2006, 2869 RSC.
- K. M. Abraham and D. M. Pasquariello, Chem. Mater., 1993, 5, 1233 CrossRef CAS.
- J. Xu, J. He, W. Ding, Z. Hong and F. Huang, Adv. Energy Mater., 2019, 9, 1900170 CrossRef.
- J. Gamon, S. Haller, E. Guilmeau, A. Maignan, T. Le Mercier and P. Barboux, J. Solid State Chem., 2018, 263, 157 CrossRef CAS.
- X. Zhu, Z. Wen, Z. Gu and S. Huang, J. Electrochem. Soc., 2006, 153, A504 CrossRef CAS.
- C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1 CrossRef CAS.
- M. E. Leonowicz, K. R. Poeppelmeier and J. M. Longo, J. Solid State Chem., 1985, 59, 71 CrossRef CAS.
- K. R. Poeppelmeier, M. E. Leonowicz and J. M. Longo, J. Solid State Chem., 1982, 44, 89 CrossRef CAS.
- B. V. Beznosikov and K. S. Aleksandrov, Crystallogr. Rep., 2000, 45, 792 CrossRef.
- F. Xiong, H. Tao and Y. Yue, Front. Mater., 2020, 6 DOI:10.3389/fmats.2019.00328.
- S. Nakata, T. Togashi, T. Honma and T. Komatsu, J. Non-Cryst. Solids, 2016, 450, 109 CrossRef CAS.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 192, 55 CrossRef.
- J. Gonzalez-Platas and J. Rodriguez-Carjaval, GFOURIER: a Windows/Linux program to calculate and display Fourier maps, Program available with the FullProf suite Search PubMed.
- T. Suzuki-Muresan, P. Deniard, E. Gautron, V. Petříček, S. Jobic and B. Grambow, J. Appl. Crystallogr., 2010, 43, 1092 CrossRef CAS.
- A. G. De La Torre, S. Bruque and M. a. G. Aranda, J. Appl. Crystallogr., 2001, 34, 196 CrossRef CAS.
- N. Henry, P. Deniard, S. Jobic, R. Brec, C. Fillet, F. Bart, A. Grandjean and O. Pinet, J. Non-Cryst. Solids, 2004, 333, 199 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis details for the solid state synthesis and mecanosynthesis of Na2Fe2OS2, details of the different synthesis attempts and of the optimisation and washing procedure, morphology of the sample prepared by mechanosynthesis, result of the elemental analysis, details of the Rietveld refinement and structure outcomes of the solid state and mechanosynthesis samples before and after washing, bond distance and angles in Na2Fe2OS2-MW, XRD results of the stability test, in situ XRD of Na2Fe2OS2-MW, EXAFS fits of the ex situ samples and outcome of the refinement, details of the structure determination and outcome of the Rietveld refinement of Na1.7Fe2OS2. CIF file for Na2Fe2OS2. Associated structural information of Na2Fe2OS2. CCDC 1990654. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ta07966a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |