
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scalable upcycling of thermoplastic polyolefins into vitrimers through transesterification†
Goutam Prasanna
Kar
 ,
Mohand Osman
Saed
and
Eugene Michael
Terentjev
*
,
Mohand Osman
Saed
and
Eugene Michael
Terentjev
*
Cavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 0HE, UK. E-mail: emt1000@cam.ac.uk
First published on 27th October 2020
Abstract
About 150 million tons of disposed plastic is accumulated each year globally. A massive challenge will be addressed even if a fraction of this amount is reclaimed as relevant feedstock for innovative materials, provided this transformation is accomplished through an affordable process with minimal resources in a high-throughput manner. Vitrimers, the dynamic networks enabled by an associative covalent bond exchange, are an emerging class of materials that combine the best of thermoplastic and thermoset characteristics. Here we report that high performance vitrimers can be produced through chemical transformation of commodity thermoplastic polyolefins (TPOs) in a simple and economical way. Polypropylene (PP) and polyethylene (PE) retrieved from recycling have been converted into permanently crosslinked networks that are rubber-elastic above the melting point, and are capable of bond exchange at a further elevated temperature. We find that the dynamically crosslinked network shows thermally triggered shape-memory behaviour with 90% recovery after multiple fixity–recovery cycles. With superior mechanical stability compared to the precursor TPO, dynamic networks can establish interfacial covalent bonding to assemble objects of complex shapes through welding. The developed method can be applied to a wide range of TPOs without prior knowledge of their precise composition. It suggests a new direction towards recovery of ‘smart’ materials for sustainable and affordable technologies from plastic recycling, using conventionally operated instruments, without the need to upgrade the infrastructure of the polymer processing industry.
Introduction
It is estimated that by 2050 the accumulated disposed plastic in the ocean will surpass marine life.1 Worldwide, about 6300 million metric tons of disposed plastic has been generated, about 9% of which was recycled.2 Although today's world seems unimaginable without the use of plastic materials, disposed plastic is a serious concern because of the environmental impact and global health threats.3–6 It is hence imperative to chemically transform these disposed plastics into functional materials with high technological relevance.7–9The two classes of polymeric materials, thermoplastics and thermosets, are abundant in modern technology. Thermoplastics are lightweight and tough, withstand harsh solvents, and are used for structurally less-demanding applications. Thermoplastics can be melted and re-processed, although the issue of contamination by unwanted additives arises in their recycling. In contrast, the crosslinked thermosets are insoluble, and are often used in applications demanding high strength (the famous example being the fiber-reinforced epoxy composite for making plastic airframes10,11). However, thermosets cannot be recycled or re-processed, which is a major drawback in modern sustainable society.
A new class of ‘smart’ plastic materials, named vitrimers, has emerged recently to achieve a trade-off between thermoplastics and thermosets.12 Vitrimers are permanently crosslinked polymer networks that are insoluble in solvents, but unlike thermosets they can plastically flow at high temperatures, allowing their reprocessing and making a true multi-use plastic.13–15 Importantly, due to the associative nature of their bond exchange, vitrimers retain the same degree of crosslinking even in the stress-induced plastic-flow regime, so the total connectivity remains constant. This means that insolubility remains in force, and contamination on reprocessing is not such a problem as with molten thermoplastics. At an elevated temperature, the bond exchange is activated with a single characteristic rate16 of the bond-exchange reaction (and the associated single value of its activation energy ΔG), which is in stark contrast with a broad distribution of relaxation times (and energy barriers) in physically crosslinked thermoplastic networks. On cooling the material to ‘operating temperature’ the bond exchange is effectively frozen, thereby fixing the network topology and the reference shape of the crosslinked plastic. Thus, vitrimers can be recycled and re-designed for desired purposes, but only via high-stress moulding at high temperatures, while still retaining the physical characteristics of a solid thermoset. In contrast, classical thermoplastic networks would always retain a degree of residual creep at operating temperatures on the tail of the distribution of relaxation rates, and also flow as ordinary viscous liquids at high temperature.
A range of promising technologies are possible with vitrimers, which offer superior mechanical strength, creep resistance, weldability and shape memory, while maintaining the ease of processability.14,17 Textiles, flexible electronics, soft robotics, and aerostructural and automotive parts are ubiquitous with such polymer systems, driving the need for upcycling commodity plastics via an economical industrial process to meet the unprecedented demand.7,18
Thermoplastic polyolefins, such as PP and PE, are dominant among the synthetic polymers produced globally. The relatively low cost of production from inexpensive natural gas, and the resulting high usage, makes TPOs the most abundant plastic waste in today's world.2 Several challenges must be overcome to develop an industrially viable process to chemically transform recycled thermoplastics into vitrimers: (1) the method of introduction of dynamically exchangeable crosslinks should be ‘general’, so that it can be applied to a wide variety of polymers, in particular, TPOs with their inert C–C backbone. (2) The chemistry of transformation from a thermoplastic to a vitrimer should be robust to circumvent the expensive and energy demanding purification process of removing additives/contaminants embedded in unsorted plastics. (3) Finally, the process should be scalable, i.e. the protocol should offer the flexibility of upscaling the whole process for on-demand high-throughput processing of industrial amounts of recycled plastic.
After the conception of vitrimers12 in 2011 in the seminal work of Leibler et al., dioxaborolane metathesis19,20 and exchange of vinylogous urethanes21 have been employed to introduce adaptable networks into TPOs. In one of the earlier studies by Leibler et al. it was demonstrated that the network dynamics is mediated by the exchange between B–O bonds and boronic ester groups.19 However, it turns out that the dioxaborolane metathesis has quite a low activation energy: Ea ≈ 16 kJ mol−1 as quoted by Leibler et al.19 In fact, there was a suggestion that this exchange reaction has a rate faster than the internal network relaxation dynamics and so the Arrhenius plot does not give an adequate thermal activation behaviour.22 This means that the mechanical stability of the resulting vitrimer at operating temperatures might not be sufficient, as some residual creep could be induced by stress due to this ‘easy’ bond-exchange. A more stable and mechanically robust system is required, also including a simplified chemical approach for industrial implementation.
Here we address the challenges mentioned above. First, we demonstrate a reactive extrusion process to impart a dynamic covalent network onto the originally inert C–C backbone of polypropylene (PP) with well characterized physical properties, acquired from a standard laboratory supplier. We then replicate this method on the PP and PE collected from a recycling pile without having prior knowledge of the physical characteristics and embedded additives/contaminants. To maintain a congenial economic environment, our method of chemical transformation of TPOs relies on a melt compounder, a widely used piece of equipment in industrial production.
Two distinct mechanisms of dynamic networks have been identified in vitrimer chemistry – dissociative and associative cross-link exchange.23 In dissociative exchange, chemical bonds are broken before the new crosslinks form, and the intermediate state has a significant lifetime. During the bond cleavage, the effective viscosity of the material drops abruptly.24 In contrast, associative exchange happens through synergistic bond cleavage and new bond formation, thereby always maintaining a fixed crosslink density, and following the Arrhenius thermal activation law.23,25 Among associative bond exchange mechanisms, transesterification between ester and hydroxyl groups has been demonstrated to be highly efficient12,26–28 (see Fig. 1). In addition, the catalytic control in the transesterification reaction with a number of thermally stable catalysts, such as triazobicyclodecene (TBD), triphenylphosphine (PPh3), and zinc(II) acetylacetonate hydrate (Zn(acac)2), offers tremendous flexibility in tuning the exchange kinetics and relaxation of dynamic covalent bonds.29,30 In this paper, we want to explore the opportunities offered by transesterification bond exchange between ester and hydroxyl groups in functionalized TPOs.
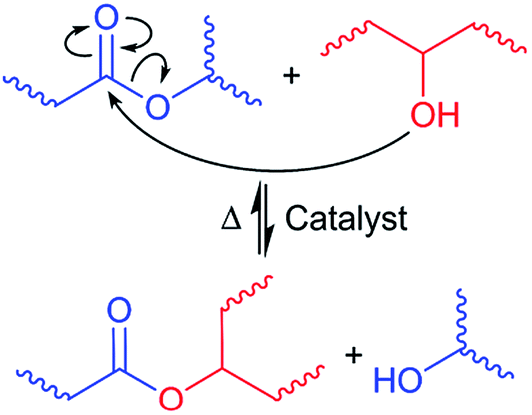 | ||
| Fig. 1 Thermo-reversible bond exchange between ester and hydroxyl moieties through transesterification. | ||
Materials and methods
Materials
PP (for details see Table S1†) was provided by INEOS Olefins & Polymers USA. Polypropylene recycled bottles (PPb) and HDPE disposed packaging (PEp) retrieved from a waste bin at the Cavendish Laboratory in Cambridge were washed thoroughly with hot water to remove visible dirt, and vacuum-dried at 80 °C for 6 h before undergoing reactive extrusion. Maleic anhydride (MA), dicumyl peroxide (DCP), bisphenol A diglycidyl ether (DGEBA), zinc acetylacetonate hydrate (Zn(acac)2), xylene, dimethyl formamide, KOH, ethanol, and bromothymol blue were procured from Sigma Aldrich.Reactive extrusion
Thermoplastic polyolefins (TPOs, i.e. PP, PPb and PEp) were functionalized with MA through reactive extrusion in a 7 cm3 conical twin-screw compounder, HAAKE MiniLab 3 (Thermo Fisher), with an integrated recirculation channel under a continuous nitrogen flow. Required amounts of vacuum-dried TPO, MA (6 wt%) and DCP (0.5 wt%) were introduced into the compounder through the hopper, with the rotor speed set at 100 rpm at 180 °C. After 10 min, a small portion of the MA-g-TPO (i.e., MA-g-PP, MA-g-PPb and MA-g-PEp) was extruded for ex situ analysis (such as FT-IR and titration). Subsequently, varied amounts of di-epoxy crosslinker (DGEBA, see Fig. 2) and the transesterification catalyst, Zn(acac)2 (1 wt%) were introduced into the compounder with the screw speed maintained at 100 rpm and the temperature at 180 °C. The total residence time for the epoxy–anhydride curing was 10 min. The axial force and the pressure were followed to qualitatively understand the evolution of effective viscosity during the reactive extrusion.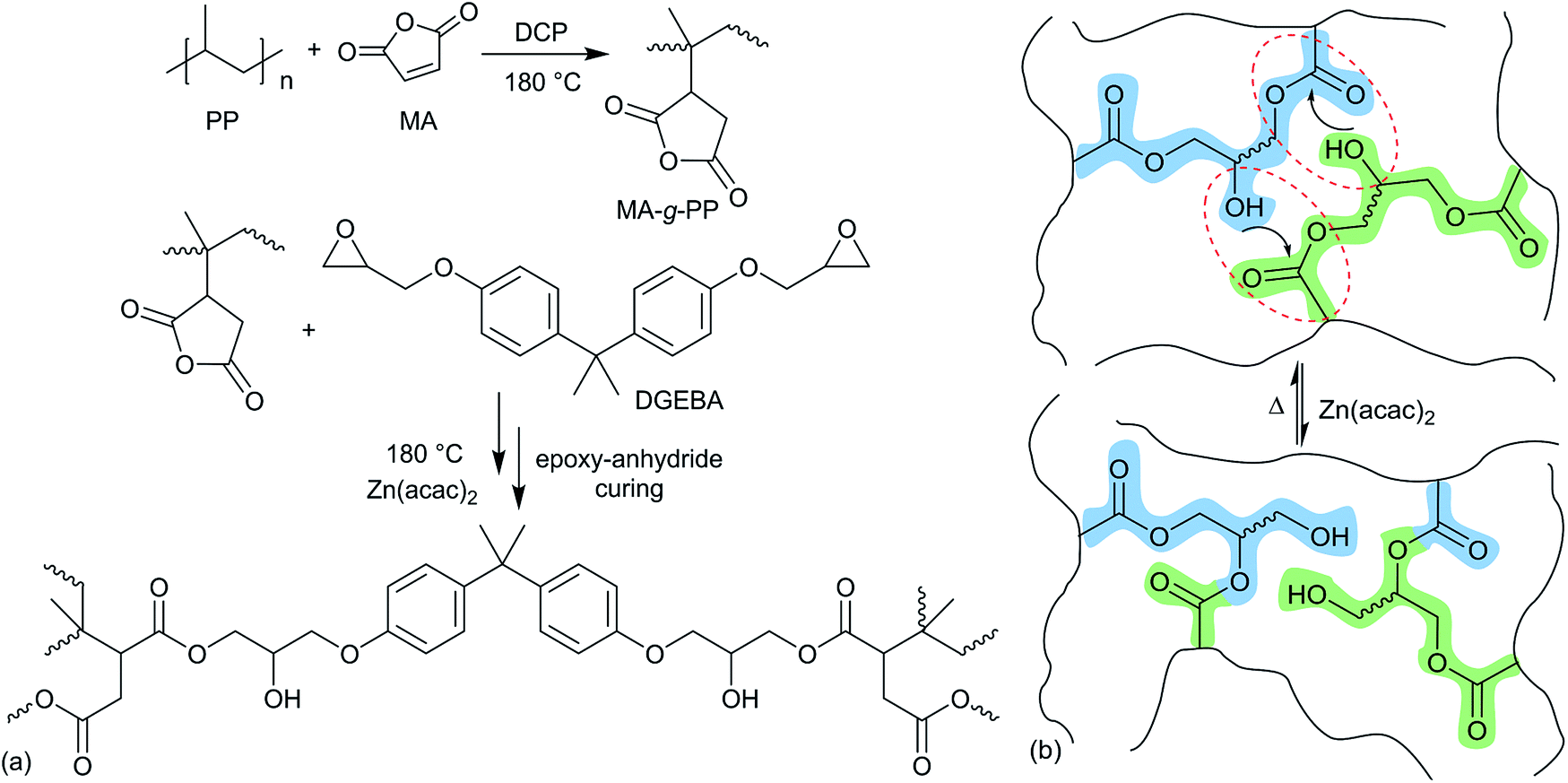 | ||
| Fig. 2 Synthesis of the polyolefin vitrimer through reactive extrusion. (a) In the first step, the graft copolymer (MA-g-PP) is prepared using DCP as a free radical initiator. In the subsequent step, crosslinking of MA-g-PP is performed with a di-functional epoxy (DGEBA) in the presence of catalyst Zn(acac)2. (b) Thermo-reversible associative bond exchange in the PP-vitrimer through transesterification (representative bond exchange out of several possible exchanges among segments in the crosslinked network). | ||
FT-IR
Infrared spectroscopy of the polymers was carried out with a PerkinElmer Spectrum 100 FTIR spectrometer in ATR mode in the range of 3500–500 cm−1.Gel fraction
The gel content in the crosslinked network was measured by a solvent extraction method. A single specimen weighing 200–500 mg was immersed in hot xylene (at 120 °C) for 24 hours while frequently (6 h intervals) replacing it with fresh solvent. Subsequently, the insoluble part was dried to constant weight. Gel content was measured as follows: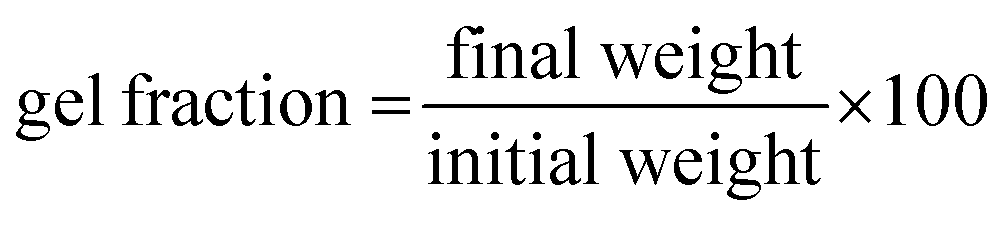
Tensile test
Uniaxial tensile tests of PP and crosslinked-PP (vitrimer) were performed on dumbbell-shaped specimens at room temperature using a Tinius Olsen 1ST universal testing machine mounted with a 2kN cell with a fixed crosshead speed of 10 mm min−1.Differential scanning calorimetry (DSC)
Melting (Tm) and crystallization transitions (Tc) of the neat thermoplastic and the crosslinked polymers were measured in a DSC4000 from PerkinElmer. Typically, 5–10 mg of the samples were vacuum dried at 80 °C for 6 h before running the temperature scan in the DSC. Similar temperature profiles were followed for all the samples. In the first cycle of the temperature scan the sample was heated to 210 °C and kept isothermally for 5 min to erase the thermal history. The crystallization temperature was determined from the exothermic peak while cooling the sample to 30 °C. A second heating cycle was performed to determine melting temperature from the endothermic peak. In all the temperature scanning steps 10 °C min−1 heating/cooling was used under a nitrogen flow of 50 ml min−1. The crystallinity fraction, χc, was calculated as31where ΔHm is the heat of fusion per g calculated from the area under the melting endotherm, and ΔH0m is the standard heat of fusion of the 100% crystalline polymer. χc was estimated taking the theoretical value of the complete crystalline polymer – ΔH0m is 209 J g−1 for PP,32 and ΔH0m is 294 J g−1 for HDPE.33
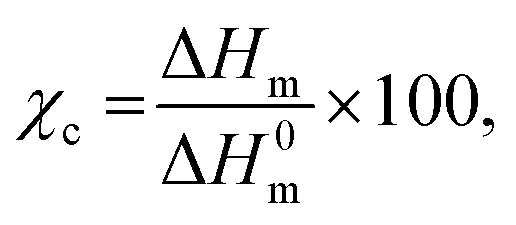
Constant-force (“iso-stress”) measurement
Iso-stress tests of the crosslinked networks and the corresponding precursor TPOs were conducted using a DMA 850 (TA Instruments) in tensile mode. Rectangular specimens with dimensions ≈ 15 mm × 5 mm × 0.9 mm were initially equilibrated at the starting temperature of 50 °C after which 10 kPa stress was applied. The extensional strain of different specimens was observed under the constant tensile stress while they were subject to a temperature ramp of 2 °C min−1.Dynamic-mechanical thermal analysis (DMTA)
A DMA 850 was employed for the measurement of thermomechanical properties. A temperature ramp was conducted in tensile mode on rectangular specimens with dimensions ≈ 15 mm × 5 mm × 0.9 mm at 2 °C min−1 and frequency = 1 Hz with 0.01% strain in the temperature ranges of 40–300 °C for PP and 50–220 °C for PPb and PEp. Prior to starting the temperature scan the DMA was equilibrated at 40 °C for 5 min.Stress relaxation (“iso-strain”) measurement
Stress relaxation was performed on the DMA 850 with fixed strain in the strain mode. Specimens with dimensions ≈ 15 mm × 5 mm × 0.9 mm were used. All the specimens were tested under uniaxial strain which was held constant isothermally for 300 min at probe temperatures of 210, 220, 230 and 240 °C. Prior to imposing the strain the DMA was equilibrated to the probe temperature for 5 min. Time dependent stress σ(t) was normalized by the initial stress σmax and the relaxation time was determined by the fitting of the exponential relaxation function, which in this case is equivalent to the time taken to reduce the stress to 1/e (ca. 37%) at various temperatures.Shape memory
A physical demonstration of shape memory was performed with a rectangular sample of crosslinked network which was deformed at 210 °C into a temporarily twisted shape. With the maintained stress the sample was cooled to ambient temperature (50 °C, which is below Tc) that freezes the sample in its temporary shape. The stress was released, and a subsequent reheating to 210 °C leads to the recovery to the initial reference shape. The DMA 850 was used to quantitatively assess the shape memory behaviour. A rectangular specimen with dimensions ≈ 15 mm × 5 mm × 0.9 mm was placed between DMA clamps in tensile mode. Shape memory behaviour was characterized using a four-step program starting from a pre-equilibrated temperature (210 °C) higher than the corresponding melting temperature (T > Tm) of the sample: (1) deformation: the rectangular strip was elongated by applying load from 0 to 20 kPa with a constant stress ramp of 20 kPa min−1 at 210 °C. (2) Cooling/fixing: The sample was cooled (at 10 °C min−1) to a sufficiently lower temperature (T < Tc) under the applied load (20 kPa). Below the crystallization temperature, the sample was observed to almost completely hold the temporal shape. (3) Unloading: The load was completely removed at a constant stress ramp of 20 kPa min−1. This step reveals the extent of shape fixing (fixity, Rf) of the crosslinked network. (4) Recovery: The specimen was heated (at 10 °C min−1) to the initial temperature (T > Tm). The extent of heat-induced recovery (Rr) of the ‘memorized’ shape from the temporal shape is estimated after equilibrating the specimen at this elevated temperature (210 °C). This thermomechanical protocol consisting of the four steps was repeated over several cycles to observe the quality of shape memory behaviour.Welding
Welding of PP- and PE-vitrimers was performed at 210 °C. Rectangular specimens of PP-vitrimer and PE-vitrimer with an overlap joint area of 1 cm2 were placed between polytetrafluoroethylene (PTFE) plates with a weight (500 g, giving a pressure of 49 kPa) placed atop. Two contact times were tested: 40 min and 60 min. The weight was removed from the overlapping joints, and the plastic slowly cooled to room temperature before testing. Lap-shear tests were performed at room temperature with a fixed crosshead speed of 0.5 mm min−1 using a Tinius Olsen 1ST universal testing machine mounted with a 2kN cell.Results and discussion
We synthesized dynamically crosslinked TPOs using epoxy–anhydride curing in the presence of transesterification catalyst Zn(acac)2. To demonstrate the broad applicability of our method, we used three different TPOs, abbreviated as PP, PPb and PEp, which are PP from a standard laboratory supplier, a PP bottle recovered from recycling (Fig. S1a†), and HDPE packaging collected from a waste bin (Fig. S1b†). Reactive extrusion was performed with the TPOs using a melt compounder, which has been shown to be highly effective in reacting with and processing thermoplastics.34 To graft the required functional moiety on the inert C–C backbone of the TPOs, we used the reaction with maleic anhydride (MA),35 carried out with the aid of free-radical initiator dicumyl peroxide (DCP) in a twin-screw melt compounder at high temperature (180 °C) and shear. The grafted MA-g-PP polymer (or MA-g-PPb and MA-g-PEp for the case of our other plastics) was then crosslinked with di-functional epoxy bisphenol A diglycidyl ether (DGEBA, see Fig. 2). The Zn(acac)2 catalyst was added at the crosslinking stage to aid the anhydride–epoxy curing.36 For the sake of conceptualizing the intricate crosslinked network, in Fig. 2(a) we present a straightforward case where two carboxylate moieties generated on different MA-g-PP chains react with a di-functional DGEBA unit. However, the epoxy and cyclic-anhydride curing is complex, with the overall mechanism generally regarded as classical anionic ring-opening copolymerization between an epoxy and anhydride.36,37 We envisage that the curing process between MA-g-PP (also for other functionalized polyolefins) and DGEBA is initiated by the Zn(acac)2 catalyst with generation of alkoxide intermediates (see Fig. S2† for details). Alkoxides could terminate immediately, or attack subsequent anhydride moieties producing carboxylate intermediates. Carboxylates terminate (through the β-elimination mechanism assisted by the Zn salt),36,38 or again enchain the epoxy groups forming alkoxides, and so on. The curing process yields abundant ester and hydroxyl functional groups in the crosslinked network. It is important that the same Zn(acac)2 catalyst also promotes the subsequent transesterification, see Fig. 2(b), of the ester–hydroxyl groups of the established crosslinkers.39 The dynamically crosslinked polymer network (vitrimer) was extruded from the melt compounder and hot-pressed into the required shapes for thermomechanical analysis. Despite the permanently crosslinked nature, this ability of extruding or moulding into desired dimensions gives tremendous flexibility to design materials for specific application.MA functionalization of PP through free radical initiation is a relatively fast process, and has been shown to be efficient in a melt compounder.34,40 We have carried out the MA grafting and the subsequent crosslinking reaction of MA-g-PP with DGEBA in the presence of Zn(acac)2 at 180 °C with a total residence time of 20–22 min. To understand the processing conditions, including the optimal total residence time of the reactive mixing, the process torque and pressure profiles (see Fig. S3†) were monitored in the twin-screw extruder. As expected, the small molecule functionalization does not alter the overall viscosity: the torque remained constant during this stage of the process. At the next stage, epoxy and Zn(acac)2 were introduced for the preparation of the crosslinked vitrimer. The initial sharp rise in torque and pressure indicates the rise in viscosity from the crosslinked PP matrix. However, the torque and pressure reach a plateau within about 5 minutes after the addition of the DGEBA and Zn(acac)2 mixture, indicating the near completion of epoxy–anhydride curing (which is what dictated our choice of optimal residence time). The level of the torque plateau is determined by the rate of transesterification exchange at this temperature (180 °C), which determines the plastic flow under shear stress, and could be inferred from the effective viscosity of the matrix.29
Covalent functionalization of MA onto PP through reactive extrusion was verified by infrared spectroscopy (FTIR). The absorbance band centred at 1785 cm−1 is attributed to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) symmetric stretching arising from MA moieties (see Fig. S4†). To demonstrate the difference between the MA bonded onto PP chains, and the non-bonded (free) MA mixed into the PP melt, we carried out the identical reaction without adding the DCP initiator. Without DCP, MA would not react with PP, giving us a reference of a melt-mixture of PP and MA (and a similar FTIR signal). In both cases, the extruded PP–MA was dissolved in hot xylene, precipitated in acetone, and subsequently dried further for FTIR experiments. Since MA readily dissolves in acetone, it was separated from PP during the precipitation in case of unreacted PP–MA. This is associated with the disappearance of the peak at 1785 cm−1 after the precipitation in acetone. In contrast, the absorbance band corresponding to the CO symmetrical stretching is retained after the precipitation in acetone for the sample with DCP, confirming the covalent functionalization of MA randomly onto PP chains. The ESI† also gives details of a titration method to quantitatively estimate the efficiency of MA grafting, and the result we obtained is 2.4 wt% of grafted MA moieties (for the 6 wt% of MA initially added into the reaction mixture).
O) symmetric stretching arising from MA moieties (see Fig. S4†). To demonstrate the difference between the MA bonded onto PP chains, and the non-bonded (free) MA mixed into the PP melt, we carried out the identical reaction without adding the DCP initiator. Without DCP, MA would not react with PP, giving us a reference of a melt-mixture of PP and MA (and a similar FTIR signal). In both cases, the extruded PP–MA was dissolved in hot xylene, precipitated in acetone, and subsequently dried further for FTIR experiments. Since MA readily dissolves in acetone, it was separated from PP during the precipitation in case of unreacted PP–MA. This is associated with the disappearance of the peak at 1785 cm−1 after the precipitation in acetone. In contrast, the absorbance band corresponding to the CO symmetrical stretching is retained after the precipitation in acetone for the sample with DCP, confirming the covalent functionalization of MA randomly onto PP chains. The ESI† also gives details of a titration method to quantitatively estimate the efficiency of MA grafting, and the result we obtained is 2.4 wt% of grafted MA moieties (for the 6 wt% of MA initially added into the reaction mixture).
The extent of crosslinking depends on two main parameters. The first is the abundance of anhydride reactive sites along the C–C backbone. The other parameter is the epoxy content. In this work, we have fixed the fraction of MA to a moderate concentration (6 wt%), since a more extensive functionalization of the thermoplastic with MA results in deteriorated mechanical performance of the solid plastic.34 We varied the fraction of added epoxy (DGEBA), while keeping the catalyst (Zn(acac)2) content fixed at 1 wt%. Under these conditions, we expect the gel fraction to initially increase with the increase of DGEBA mole fraction, and then saturate when most of the MA-functionalized groups are used up, and the excess epoxy remains as a sol fraction in the network. The extent of crosslinking was estimated by measuring the gel fraction of the resulting product. The insoluble fraction from the crosslinked network was obtained after prolonged immersion in hot xylene (see Fig. S5†). It is worth noting that polypropylene and polyethylene dissolve in hot xylene at about 120 °C. The results of the gel fraction test (Fig. S6†) clearly show the optimal ratio between the epoxy and the MA content. At this optimal ratio (DGEBA/MA = 1.7), the gel fraction is 58% for PP and 66% for PEp, which is much higher than those previously reported in studies of crosslinked polyethylene.19 At a higher DGEBA content, we find an increasing sol fraction of non-bonded epoxy, which would eventually degrade the mechanical properties of the vitrimer. In addition, we observe that there is no formation of a crosslinked network in the absence of catalyst Zn(acac)2 during the reactive extrusion of the MA-g-PP and DGEBA, suggesting that epoxy–anhydride curing is indeed assisted by this catalyst. The extruded strand without Zn(acac)2 dissolves in hot xylene, in contrast to the PP-vitrimer (Fig. S7†). Mechanical properties at room temperature were examined through a uniaxial tensile test. We observed that crosslinked-PP showed improved tensile strength and maintained a comparable elongation at break to the precursor thermoplastic PP (see Fig. S8†).
One of the intriguing aspects of crosslinking semi-crystalline polymers like PP and PE is that the side chain functionalization mostly occurs in the amorphous region while the distribution of crystalline domains remains largely unaffected,39 implying that the bulk of the crosslinked PP (also PPb and PEp) is expected to show the usual melting endotherm and crystallization exotherm of its thermoplastic precursor. The crosslinked network can be schematically visualized as in Fig. S9(a).† To investigate the degree of crystallization of the crosslinked polyolefins, we carried out calorimetry studies to probe their thermal characteristics such as melting temperature (Tm), crystallization temperature (Tc) and the fraction of crystallinity (χc). In the first heating run the sample under investigation was annealed at a sufficiently higher temperature (210 °C), so that the internal relaxation would erase the thermal history of sample preparation. Tc was identified from the subsequent cooling thermogram, and Tm from the second heating run. The parameters Tm, Tc and χc for the crosslinked vitrimers, and their thermoplastic precursors are listed in Table 1, and the calorimetric thermograms are shown in Fig. S9(b), and S10(a–e).† It is interesting to point out that the melting point of PP in the recycled bottle is about 16 °C lower than in the pure PP (presumably due to additives in PPb), but the degree of crystallinity is almost the same (since the impurity concentrates in the amorphous regions).
| Polyolefin source | Content | T m (°C) | T c (°C) | χ c (%) | Gel fraction (%) | Dynamically crosslinked polyolefin | |
|---|---|---|---|---|---|---|---|
| MA (wt%) |
|
||||||
| PP commercial source (PP) | 0 | 0 | 166 | 118 | 40 | 0 | PP-vitrimer |
| 6 | 1.2 | 164 | 128 | 37.5 | 42 | ||
| 6 | 1.5 | 162 | 128 | 36.8 | 48 | ||
| 6 | 1.7 | 162 | 124 | 36 | 58 | ||
| 6 | 1.8 | 163 | 126 | 35 | 52 | ||
| 6 | 1.9 | 162 | 124 | 32 | 46 | ||
| PP recycled bottle (PPb) | 0 | 0 | 149 | 103 | 36.4 | 0 | |
| 6 | 1.7 | 150 | 112 | 32.2 | 54 | PPb-vitrimer | |
| HDPE packaging (PEp) | 0 | 0 | 133 | 108 | 53.4 | 0 | |
| 6 | 1.7 | 129 | 112 | 24.8 | 66 | PEp-vitrimer | |
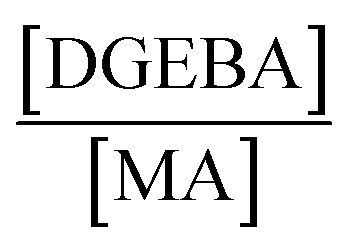
The onset of crystallization generally increased upon crosslinking (see Fig. S9b†), suggesting the presence of heterogeneous microdomains that act as nucleation sites for crystallization. For crosslinked PP with a gel fraction of 42%, the crystallization point Tc is increased by 10 °C compared to that of the neat PP (Tc = 118 °C). With further addition of DGEBA, Tc does not increase, suggesting that the non-bonded epoxy is aggregated in the amorphous regions of the material. In contrast, the crystallinity fraction decreases monotonically with the increase of DGEBA concentration. The segmental alignment in the polyolefin chain would be significantly disrupted by the crosslinking constraints, thereby decreasing the fraction of crystallinity. Two opposing factors are engaged simultaneously with the increase of DGEBA loading: although offering heteronucleation sites for crystallization, the crosslinked microdomain places a physical barrier to be surmounted for chain packing that reduces the ability to crystallize. At lower DGEBA/MA molar ratios, these two factors balance each other, and with a further increase of DGEBA content, nucleation does not increase significantly, but the crosslinking becomes a dominant factor in crystallization. As a result, with DGEBA/MA ratio = 1.9, the fraction χc reduces compared to the system with DGEBA/MA = 1.2. The interplay between heteronucleation and the ability to crystallize is expected to be dependent on the nature of the thermoplastic precursor, since its mobility in the dense matrix under shear is the key factor in being able to find the bonding sites. The reduction of χc for the crosslinked PEp is observed to be significantly higher than that of crosslinked PP with identical DGEBA content (DGEBA/MA = 1.7). For further study, we selected the network with highest crosslinking (DGEBA/MA = 1.7) for different polyolefins: PP-vit, PPb-vit and PEp-vit.
Different thermomechanical profiles are expected from the crosslinked polymers and their precursor TPOs. The elastic–plastic transition of the vitrimers was assessed through the iso-stress experiment. Fig. 3(a) shows the results of iso-stress testing on a temperature ramp (which is frequently called ‘dilatometry’ in the literature: such a term is misleading since the experiment tests elastic or plastic elongation at nearly constant volume). As the elastic modulus gradually decreases on heating, the strain increases – until in the precursor TPO the modulus drops to zero at the melting point Tm (different for PP, PPb and PEp, see Table 1) and the sample flows, the tensile strain diverging. The crosslinked vitrimers (PP-vit, PPb-vit and PEp-vit) show a different response. The materials remain rubber-elastic above the melting point Tm, which is seen by a region of rubber-elastic plateau modulus. However, as the temperature increases and the bond-exchange reaction accelerates, the elastic–plastic transition takes place and we see a further increase in strain. This could be interpreted as plastic creep, although ‘creep’ is usually understood as a time progression of strain. The elastic–plastic transition is affected by the applied stress, and by the rate of heating, so this test is only indicative (both here and in the literature). In fact, at a higher stress, we see no rubber plateau: only the gradual plastic creep in all samples at high temperature. Only at a low stress of 10 kPa presented in Fig. 3(a) do we see an indication of the ‘rubber plateau’ one might expect in an ordinary crosslinked network.
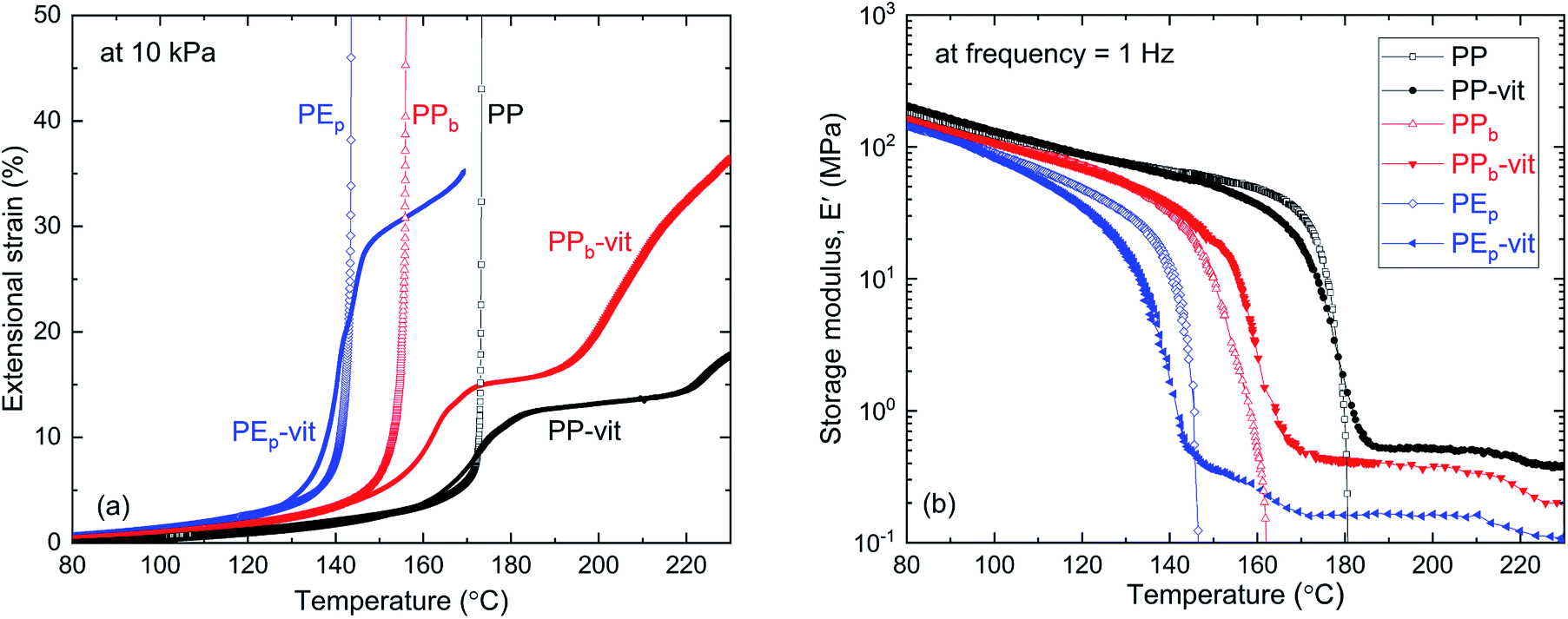 | ||
| Fig. 3 Thermomechanical properties of the vitrimers and the corresponding TPOs. (a) The iso-stress experiment: all samples were subjected to a constant engineering stress (10 kPa), with a temperature ramp of 2 °C min−1. (b) DMTA profiles of the linear storage modulus E′ for PP and PP-vit, PPb and PPb-vit, and PEp and PEp-vit, measured at a frequency of 1 Hz, with a temperature ramp of 2 °C min−1. Vitrimers display a clear rubber plateau above the melting point, while retaining the solid plastic state below Tm. | ||
To further investigate the rubber-elastic regime of these vitrimers, we studied the temperature dependence of the dynamic modulus, which is critical to assess the integrity of the polymer network. Dynamic-mechanical thermal analysis (DMTA) was employed at a low constant frequency of 1 Hz, and the results are presented in Fig. 3(b). A slow heating ramp reveals the familiar trend in the storage modulus E′, found in semi-crystalline polymers. Each of the precursor TPOs and their corresponding vitrimers show the same monotonic decrease of E′ until the temperature approaches the melting point (different for the three materials). A well-defined drop in E′ occurs at the melting point. However, with heating continued, a stable rubber modulus emerges for all vitrimers, a hallmark that distinguishes the permanently crosslinked network from its thermoplastic precursor melt. This result corroborates our swelling experiment discussed earlier, as the insoluble fraction (see Fig. S7† for PP-vit as an example) represents the permanently crosslinked network, which is rubbery above Tm. There is a correlation between the dynamic rubber modulus seen in Fig. 3(b) and the ‘rubber plateau’ in strain under constant stress in Fig. 3(a), although there is a difference from the static rubber modulus relevant in the latter. The strength of the crosslinked network can be correlated with the rubber plateau modulus, which has been the focus of recent literature.30 In our study, we find a dynamic rubber modulus of 0.44 MPa for PP-vit, 0.30 MPa for PPb-vit, and 0.18 MPa for PEp-vit, respectively. Our ongoing investigation aims to understand the interplay among several factors such as MA content, molecular architecture of epoxy and choice of catalyst to tune the rubber plateau modulus.
Similar to the high-temperature end in Fig. 3(a), we see that the rubber plateau starts to drop at temperatures above 200 °C, which is another indication of the plastic flow onset (in contrast with the classical rubber modulus that must increase with temperature). The DMTA data do not reveal the clear elastic–plastic transition that is so prominent in the iso-stress data in Fig. 3(a). This is because the 1 Hz cycle is too fast for any significant plastic creep to occur even at a very high temperature of 210 °C. The stress relaxation results in Fig. 4 reveal that the relevant time scales are much longer (e.g. 3000 s at 210 °C), and so in dynamic measurement the vitrimers above Tm behave as ordinary rubbers. It is worth noting that the formation of a crosslinked network is precluded in the absence of the catalyst, as shown in Fig. S11.† Without Zn(acac)2 no evidence was found for the signature rubbery plateau in the DMTA either.
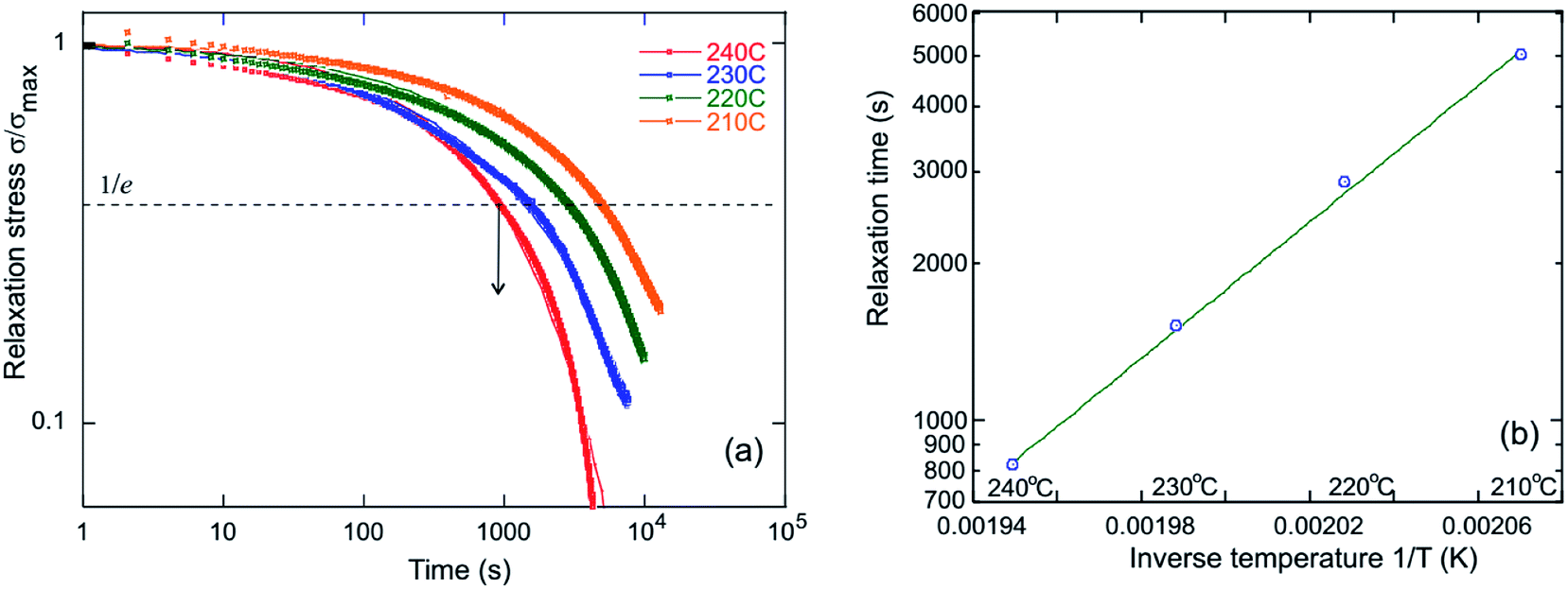 | ||
| Fig. 4 Dissipation of internal stress through bond exchange in a dynamic network. (a) Scaled stress relaxation σ(t)/σmax for PP-vit probed at different temperatures. The guiding line corresponds to the fit with the exponential relaxation function from where relaxation time, τ, is derived (indicated as arrow). (b) The Arrhenius plot for the relaxation time; the slope of the linear fit gives single value activation energy, Ea ≈ 124 kJ mol−1 for PP-vit. | ||
Stress relaxation (iso-strain) experiments were conducted to assess the degree of plasticity in the crosslinked network, and to estimate the relaxation times and the activation energy of bond exchange. Iso-strain steady stress relaxation is the only method that probes the rate of bond exchange directly, if the relaxation follows the exponential law.16 Internal stress is dissipated through thermo-reversible bond exchange. Stress relaxation exhibited by PP-vit is presented in Fig. 4(a), as an illustration, when an instantaneous fixed-strain was imposed at a predetermined equilibrated temperature (which was maintained throughout the test). Stress relaxation of the dynamically bonded network is often sufficiently well described by the simple exponential function
σ(t) = σmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ), exp(−t/τ), |
where τ0 is the pre-exponential factor representing the inverse rate of attempts of the reaction, and Ea is the activation energy which is calculated from the slope of the fit lines, as in Fig. 4(b). We obtained the activation energy for transesterification: Ea ≈ 124 kJ mol−1 for PP-vit. This is much higher than the quoted value for dioxaborolane metathesis, Ea ≈ 16 kJ mol−1,19 and also higher than Ea ≈ 108 kJ mol−1 for the exchange reaction of vinylogous urethanes,21 suggesting our PP-vit constitutes a more robust dynamic network.
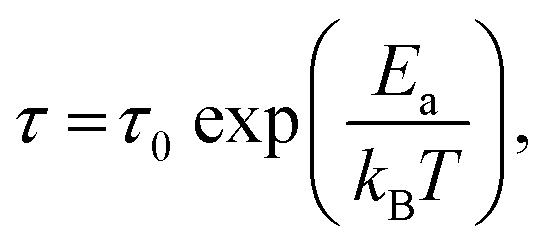
As the bond exchange is triggered by a thermal stimulus, we assessed the shape memory characteristics in a dynamically crosslinked network. The shape memory effect is the ability to recover to the original shape from a programmed temporary shape in the presence of a stimulus, here heat.41 These stimuli responsive materials have shown great potential in sensors, actuators, morphing structures in aerospace vehicles and bio-medical devices.14,42,43 The classical shape-memory effect41 relies on the combination of a mechanical reference state of the crosslinked network, and the mechanical freezing of shape in a semicrystalline state. Although in our case the reference state could be altered by plastic deformation of the dynamically crosslinked vitrimer network, if the deformation is produced by a sufficiently low stress and sufficiently fast, as indicated in Fig. 3(a and b), no plastic creep would occur and one would expect a good shape recovery. Fig. 5(a) illustrates this effect on a PP-vit sample. At ambient temperature, the material is a solid plastic (with many structural applications owing to its high mechanical strength). This is its reference shape A, in this case formed by pressure moulding of the sample. On heating it just above its melting point Tm, we deform the rubbery network into shape B, and then cool it below Tc, which freezes shape B in the solid plastic. Then on re-heating the sample above Tm, with no stress, we have it returning to shape A (Movie S1, ESI†). A control experiment with thermoplastic PP showed no shape recovery from the programmed temporal shape (Fig. S12†). This striking difference opens several unique application possibilities for dynamically crosslinked structural TPOs.
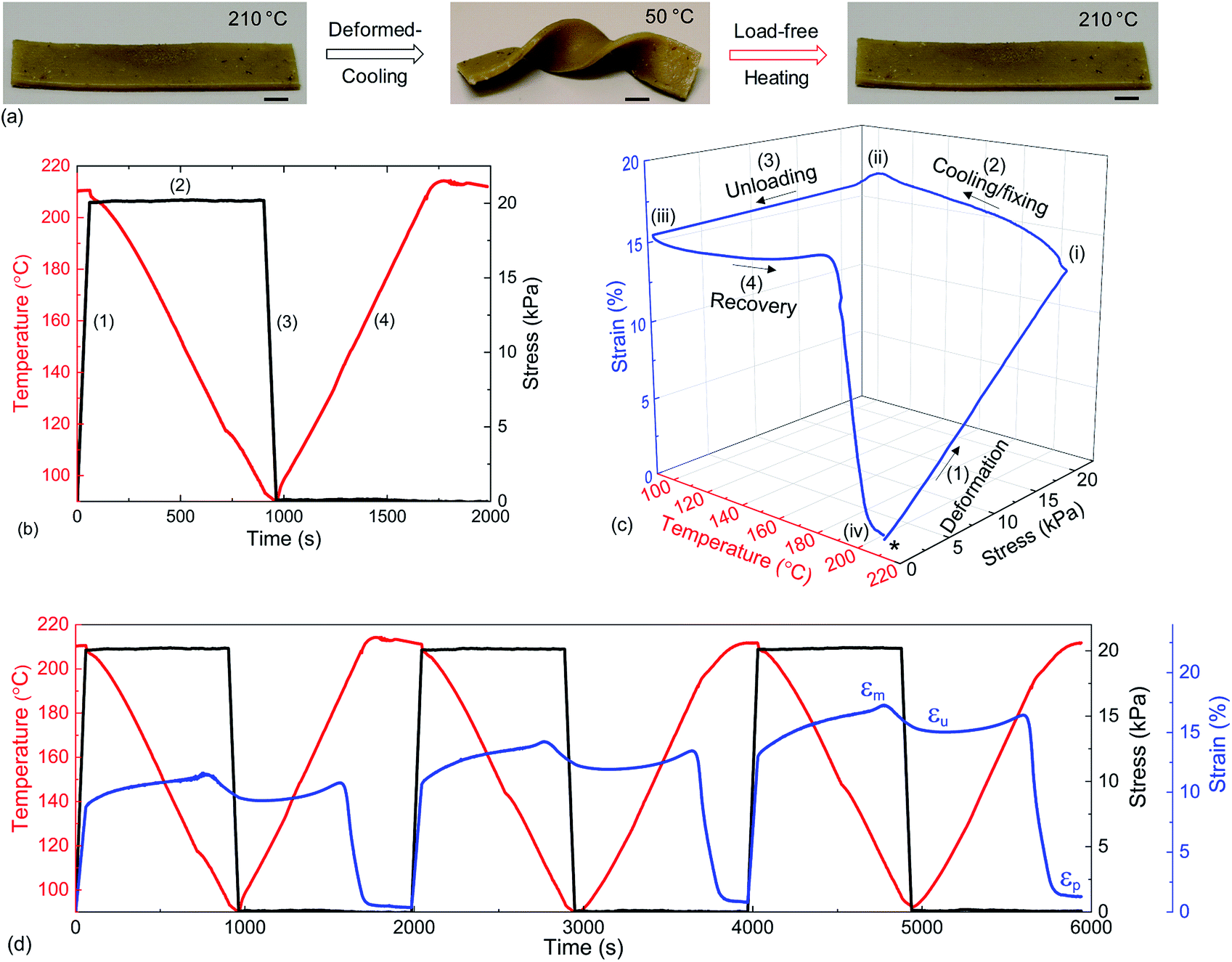 | ||
| Fig. 5 Illustration of the shape-memory effect. (a) PP-vit originally in its reference shape A was deformed at high temperature to fix and freeze to a temporary shape B. Subsequent heating leads to recovery to the initial reference shape A (scale bar, 5.0 mm). (b) Thermomechanical protocol in DMA applied on a rectangular specimen of PP-vit (see text). (c) Evolution of extensional strain on PP-vit: DMA was equilibrated at 210 °C prior to starting the experiment. A heated, unloaded PP-vit specimen (asterisk) was deformed to state (i), loaded and cooled to state (ii), and then unloaded to state (iii) and reheated (210 °C) to state (iv), leading to recovery. (d) Three thermomechanical cycles demonstrating the repeatability of fixity and recovery. | ||
We quantitatively estimated the shape fixing (Rf, fixity) and the recovery of original shape (Rr, recovery) through DMA in tensile mode. A thermomechanical protocol in Fig. 5(b), consisting of four steps, was applied on PP-vit, and the resulting evolution of strain is presented in Fig. 5(c). PP-vit was deformed at 210 °C (above Tm) while it is in a rubbery state. With the maintained stress, a subsequent cooling leads to the solid semi-crystalline state that immobilizes the crosslinked network in a temporal shape. The shape fixity can be calculated as42
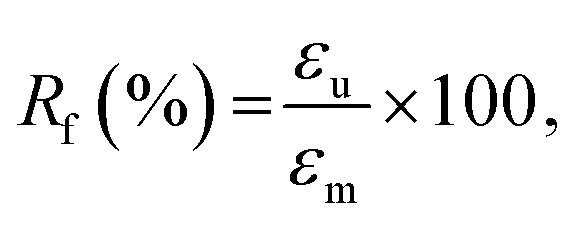
where εp is the strain achieved after the recovery (4th) step, and εu and εm are the same as earlier. The evolution of strain in PP-vit was observed over several thermomechanical cycles, Fig. 5(d), to assess the quality of shape memory. We achieved a reproducible measurement for PP-vit with the average Rf = 91% and Rr = 90%.
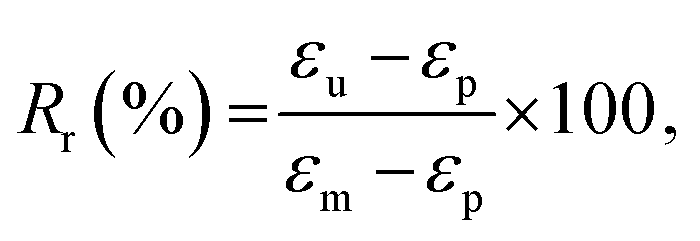
Modern technologies including electronic devices and robotics benefit from assembling several polymer structures which is rather difficult to obtain with incompatible polymer interfaces. We show that the incompatible PP/PE interface can be assembled in their vitrimer form (Fig. S13a†) by a simple welding process at 210 °C within 60 min of heat exposure (Fig. 6(a)). The welding is achieved through the formation of covalent bonds that create bridges across the interface through dynamic bond exchange (Fig. 6(b)). Our primary investigation shows that the welding process produces stable joints (Fig. S13(b and c)†). A detailed study would be required to tailor the interfacial bonding by optimizing the welding parameters such as the applied load atop the overlapping joints, catalyst content, welding temperature and the duration of welding. However, in the present case, within a short welding time span, significant adhesion was achieved between PP-vit and PEp-vit. From the lap shear test in Fig. 6(c), the interfacial failure force was estimated to be 65 N for 40 min and 82 N for 60 min of welding time (lap joint area = 1 cm2 giving a lap shear stress of 0.65 and 0.82 MPa, respectively). This shows a way toward assembling complex objects with incompatible interfaces without the use of any adhesive or mechanical fasteners.
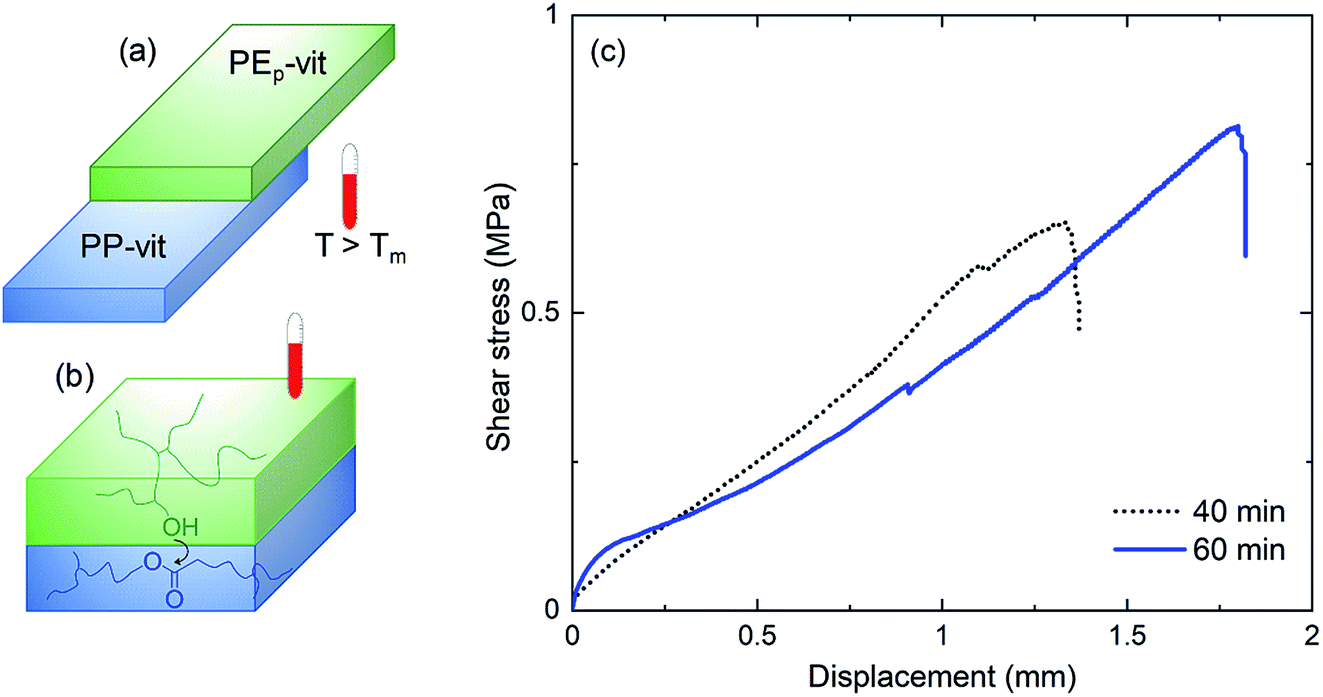 | ||
| Fig. 6 Welding of incompatible PP/PE interfaces. (a) Model of flat surfaces of PP-vit and PEp-vit pressed at high temperature (210 °C) for 40 min and 60 min. Subsequent cooling to ambient temperature leads to a single welded object. (b) Bonding across interfaces by the formation of covalent bonds through dynamic exchange. (c) Lap shear test of welded plates with an overlap joint area of 1 cm2 performed with a fixed crosshead speed of 0.5 mm min−1. All the lap joints broke due to interfacial detachment (adhesive failure). | ||
The key advantage of a vitrimer over a thermoset is the ease of processing and remoulding above its elastic–plastic transition. Thermally triggered bond exchange reaction allows the material to flow plastically without risking the bond cleavage (in simple terms: fracturing of the network), thereby altering the internal network topology on observable timescales. Small pieces of the PP-vit were fused to make a single sample of rectangular shape with the aid of a hydraulic press at 210 °C and the thermomechanical properties were subsequently measured to verify how the re-moulding cycles affect the material's properties. There is only a slight and non-systematic change in the storage modulus profile against temperature (see Fig. S14†), which suggests that the material retains its mechanical properties after multiple cycles of processing. We observed that the ease of reprocessing is higher in the PP-vitrimer with lower DGEBA content. Despite having permanent crosslinks, this remoulding ability of a vitrimer is in high demand since reprocessing is increasingly becoming a prerequisite for the development of sustainable and affordable technologies.
Conclusions
This study contributes to the global effort to obtain ‘value-added’ materials from plastic litter. We have overcome key challenges to produce vitrimer networks from thermoplastic polyolefins. Our scheme can be replicated for various polyolefins without having to sort them. It appears that the bond exchange reaction in vitrimers is compatible with the unknown additives or contaminants in plastic litter. The economical aspect of the upcycling process is entirely preserved since the TPO functionalization is accomplished in an industrially relevant setting, i.e. through melt extrusion. We identified an optimum feed ratio of epoxy/MA to achieve maximum crosslinking. The transformation of a thermoplastic to an elastomeric network is characterized by the rubbery plateau in DMTA scans. The key features of the vitrimers exhibiting the stress relaxation behaviour mediated by catalysed dynamic exchange follows Arrhenius dependence with activation energy ≈ 124 kJ mol−1. This is much higher than the activation of other vitrimer systems recently reported,19 making these materials much more robust. The dynamically crosslinked network behaves like a classical shape-memory polymer with 90% recovery from a temporary programmed shape to the original permanent shape, showing enormous potential in developing sensors, actuators and ‘smart’ textiles.42 In addition, unlike traditional thermosets, our vitrimer can be reprocessed. Dimensional reconfiguration has been performed in several cycles preserving the network integrity. This is an intriguing aspect to be explored, as complex objects can be built by locally heating and welding without requiring precise temperature control because the viscosity of this family of materials changes slowly, obeying the Arrhenius law.12,29 This robust crosslinked network enabled by thermo-reversible bond exchange will further dissipate internal stress at the adjoining sites to reach mechanical equilibrium.Thus, we find that disposed thermoplastics, which increasingly threaten our only habitable ecosystem towards a disastrous consequence, can be utilized as feedstock for ‘smart’ plastic materials. The presented results provide a foundation to obtain high-value plastic from litter on an industrial scale. Concurrent progress in 3D-printing44–46 with this class of polymers showing mechanical responsiveness to thermal stimulation and recyclability promises next-generation materials in new technological areas.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (AdG No. 786659).References
- The New Plastics Economy – Rethinking the Future of Plastics, World Economic Forum, Ellen MacArthur Foundation, and McKinsey & Company, 2016 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS.
- C. J. Rhodes, Sci. Prog., 2018, 101, 207–260 CrossRef.
- J. Brahney, M. Hallerud, E. Heim, M. Hahnenberger and S. Sukumaran, Science, 2020, 368, 1257–1260 CrossRef CAS.
- C. Wilcox, E. Van Sebille and B. D. Hardesty, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11899–11904 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS.
- C. Soutis, Mater. Sci. Eng., A, 2005, 412, 171–176 CrossRef.
- A. Toldy, B. Szolnoki and G. Marosi, Polym. Degrad. Stab., 2011, 96, 371–376 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS.
- F. Meng, M. O. Saed and E. M. Terentjev, Macromolecules, 2019, 52, 7423–7429 CrossRef CAS.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- B. A. Helms and T. P. Russell, Chem, 2016, 1, 816–818 CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- F. Caffy and R. Nicolaÿ, Polym. Chem., 2019, 10, 3107–3115 RSC.
- J. Tellers, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polym. Chem., 2019, 10, 5534–5542 RSC.
- M. O. Saed, A. Gablier and E. M. Terentejv, Adv. Funct. Mater., 2019, 1906458 Search PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- F. Meng, R. H. Pritchard and E. M. Terentjev, Macromolecules, 2016, 49, 2843–2852 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS.
- A. Demongeot, R. Groote, H. Goossens, T. Hoeks, F. Tournilhac and L. Leibler, Macromolecules, 2017, 50, 6117–6127 CrossRef CAS.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- Y. Zhou, R. Groote, J. G. Goossens, R. P. Sijbesma and J. P. Heuts, Polym. Chem., 2019, 10, 136–144 RSC.
- H. Schönherr, W. Wiyatno, J. Pople, C. W. Frank, G. G. Fuller, A. P. Gast and R. M. Waymouth, Macromolecules, 2002, 35, 2654–2666 CrossRef.
- C. Ruiz-Orta, J. P. Fernandez-Blazquez, A. M. Anderson-Wile, G. W. Coates and R. G. Alamo, Macromolecules, 2011, 44, 3436–3451 CrossRef CAS.
- L. Amoroso, E. L. Heeley, S. N. Ramadas and T. McNally, Polymer, 2020, 201, 122587 CrossRef CAS.
- G. Moad, Prog. Polym. Sci., 1999, 24, 81–142 CrossRef CAS.
- D. Shi, J. Yang, Z. Yao, Y. Wang, H. Huang, W. Jing, J. Yin and G. Costa, Polymer, 2001, 42, 5549–5557 CrossRef CAS.
- T. Vidil, F. Tournilhac, S. Musso, A. Robisson and L. Leibler, Prog. Polym. Sci., 2016, 62, 126–179 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- X. Fernàndez-Francos, X. Ramis and À. Serra, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 61–75 CrossRef.
- Y. Zhou, J. G. Goossens, R. P. Sijbesma and J. P. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- W. Yuan, M. Guo, Z. Miao and Y. Liu, Polym. J., 2010, 42, 745–751 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- P. T. Mather, X. Luo and I. A. Rousseau, Annu. Rev. Mater. Res., 2009, 39, 445–471 CrossRef CAS.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- R. T. Shafranek, S. C. Millik, P. T. Smith, C.-U. Lee, A. J. Boydston and A. Nelson, Prog. Polym. Sci., 2019, 93, 36–67 CrossRef CAS.
- Z. Fang, H. Song, Y. Zhang, B. Jin, J. Wu, Q. Zhao and T. Xie, Matter, 2020, 2, 1187–1197 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta07339c |
| This journal is © The Royal Society of Chemistry 2020 |