
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithiation of V2O3(SO4)2 – a flexible insertion host†
Stephanie F.
Linnell
 a,
Julia L.
Payne
a,
David M.
Pickup
b,
Alan V.
Chadwick
b,
A. Robert
Armstong
a and
John T. S.
Irvine
*a
a,
Julia L.
Payne
a,
David M.
Pickup
b,
Alan V.
Chadwick
b,
A. Robert
Armstong
a and
John T. S.
Irvine
*a
aSchool of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: jtsi@st-andrews.ac.uk
bSchool of Physical Science, University of Kent, Canterbury, CT2 7NH, UK
First published on 10th September 2020
Abstract
Materials that display strong capabilities for lithium insertion without significant change in unit cell size on cycling are of considerable importance for electrochemical applications. Here, we present V2O3(SO4)2 as a host for lithium-ion batteries. Electrochemically, 2.0 Li+ ions can be inserted, giving Li2V2O3(SO4)2 with an oxidation state of V4+, as determined by X-ray absorption spectroscopy. The capacity of V2O3(SO4)2 can be increased from 157 mA h g−1 to 313 mA h g−1 with the insertion of two additional Li+ ions which would drastically improve the energy density of this material, but this would be over a wider potential range. Chemical lithiation using n-butyllithium was performed and characterisation using a range of techniques showed that a composition of Li4V2O3(SO4)2 can be obtained with an oxidation state of V3+. Structural studies of the lithiated materials by X-ray diffraction showed that up to 4.0 Li+ ions can be inserted into V2O3(SO4)2 whilst maintaining its framework structure.
Introduction
Materials that can reversibly incorporate large numbers of ions balanced by redox changes in the host lattice are a subject of much interest, with important applications such as electrochromics and battery electrodes. One such application is in the search for less expensive positive electrode materials for high energy density lithium-ion batteries (LIBs). Consequently, great efforts have been devoted to the search for crystal structures that allow a large amount of lithium storage at high redox potentials but remain structurally stable on cycling.1 Polyanion compounds (phosphates, sulfates) are promising candidates due to their structural stability and higher operating potentials compared to oxides.2,3 Among them LiFePO4 has been extensively studied and commercialised, but its energy density (∼580 W h kg−1) is limited due to its relatively low operating voltage of 3.45 V vs. Li+/Li0.4,5 Tavorite-structured compounds including LiFeSO4F,6,7 LiVPO4F,8–12 and LiVPO4O,13–15 operate at higher voltages, are structurally stable and are considered attractive alternatives. In particular, the vanadium based materials have attracted considerable interest, due to the ability to adopt a range of oxidation states, 2+, 3+, 4+ and 5+, enabling them to insert/extract reversibly-more than one Li+ ion per transition metal ion. The Li+ extraction/insertion in LiVPO4F is associated with two redox processes and when the two reactions are utilised this material can achieve a specific capacity of 312 mA h g−1. The insertion of Li+ occurs at 1.80 V vs. Li+/Li0 and is associated with the V3+/V2+ redox couple and extraction of Li+ occurs via two plateaux at 4.24 V and 4.28 V vs. Li+/Li0 associated with the V4+/V3+ redox couple.12 Structural studies have revealed shifts from the triclinic LiVPO4F pristine phase to monoclinic Li2VPO4F and VPO4F phases along with unit cell volume expansions/contractions but with retention of the VPO4F framework on cycling.12 As for LiVPO4O, this material can also exploit two redox couples and thereby deliver a specific capacity of 318 mA h g−1.12 The insertion of Li+ is associated with the V3+/V4+ redox couple, with three short plateaux at 2.45 V, 2.21 V and 2.04 V attributed to the formation of Li1.5VPO4O, Li1.75VPO4O and Li2VPO4O, respectively. Over the higher voltage range, Li+ is extracted with one plateau observed at 3.95 V associated with the V4+/V5+ redox couple and the formation of ε-VPO4O.13 The LixVPO4O (0 < x < 2) phases can be indexed to different space groups, showing unit cell volume expansions/contractions and the VPO4O framework is maintained on cycling vs. Li+/Li0 with changes in the V–O bond distances for the different phases, due to the formation of the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond within the VO6 octahedra for the V4+ and V5+ phases, which is absent for the V3+ phase.13
O bond within the VO6 octahedra for the V4+ and V5+ phases, which is absent for the V3+ phase.13
Other materials of interest include the vanadyl phosphate, Li4VO(PO4)2. This material operates at a relatively high voltage of 4.1 V vs. Li+/Li0 and exhibits a reversible capacity of 70 mA h g−1 which is associated with the V4+/V5+ redox couple.16,17 The PO43− polyanion in Li4VO(PO4)2 has been replaced with the more electronegative SO42− polyanion to give Li2VO(SO4)2.18 Li2VO(SO4)2 shows a plateau at 4.7 V vs. Li+/Li0 associated with the V5+/V4+ redox couple and delivers a reversible capacity of 50 mA h g−1.18 Despite reaching a higher potential, vanadium-based materials that utilise the SO42− polyanion remain underexplored.18–21 Vanadium sulfates are attractive candidate compounds and while the search for new crystal structures and compositions is ongoing, there are existing vanadium sulfates that can be explored.18,22
V2O3(SO4)2 can accept four Li+ ions, delivering a theoretical capacity of 313 mA h g−1. Here, we study the Li+ insertion into V2O3(SO4)2 using two methods (1) electrochemical lithiation and (2) chemical lithiation using n-butyllithium. V2O3(SO4)2 positive electrodes were cycled in Swagelok-type cells between 4.20 V and 1.95 V vs. Li+/Li0 and examined ex situ by means of X-ray absorption spectroscopy, infrared spectroscopy and powder Xray diffraction studies to reveal the phase evolution and structural changes which occur at different stages during the lithiation and delithiation process. V2O3(SO4)2 was also chemically lithiated to insert two additional Li+ ions. X-ray diffraction studies, inductively coupled plasma optical emission spectroscopy analysis and X-ray absorption spectroscopy measurements were carried out to understand the structural evolution accompanying the chemical insertion of Li+ ions.
Experimental
Synthesis of V2O3(SO4)2
V2O3(SO4)2 was synthesised by modifying a method reported by Richter et al.23 V2O5 (Sigma Aldrich, ≥99.6%, 3.64 g) was stirred with an excess of concentrated sulfuric acid (Fischer, >95%, 30 mL) at 140 °C for 48 h according to eqn (1).| V2O5 + 2H2SO4 → V2O3(SO4)2 + 2H2O | (1) |
Once cooled, the product was filtered under vacuum and washed with cold concentrated H2SO4, followed by a further washing with CF3COOH (Alfa Aesar, 99%) and finally dried at 200 °C overnight. The product was stored and handled in an argon filled glovebox due to its hygroscopicity.
Electrochemical lithiation
Electrodes were prepared by ball-milling 80 wt% active material with 20 wt% carbon super C65 (Imerys) at 300 rpm under argon for 30 min. A Fritsch Pulverisette planetary ball mill was used with 10 mm diameter zirconia milling balls. Electrochemical tests of the V2O3(SO4)2 positive electrodes were performed in Swagelok type cells which were assembled in an argon filled glovebox. Positive electrodes were separated from the Li metal disc negative electrode by a Whatman GF/D borosilicate glass fibre sheet saturated with 1 M LiPF6 in ethylene carbonate/dimethyl carbonate [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w)] (LP30, BASF) as the electrolyte. In order to evaluate the electrochemical lithiation of V2O3(SO4)2 as a positive electrode, cells were cycled in a temperature-controlled environment at 30 °C using a Mac Pile Bio Logic II system in galvanostatic mode. To determine the cycling performance a cell was cycled over different rates (C/20, C/10, C/5, C/2 and C) at 30 °C using a Maccor Series 4200 system.
:1 (w/w)] (LP30, BASF) as the electrolyte. In order to evaluate the electrochemical lithiation of V2O3(SO4)2 as a positive electrode, cells were cycled in a temperature-controlled environment at 30 °C using a Mac Pile Bio Logic II system in galvanostatic mode. To determine the cycling performance a cell was cycled over different rates (C/20, C/10, C/5, C/2 and C) at 30 °C using a Maccor Series 4200 system.
To understand the Li+ insertion/extraction in V2O3(SO4)2, multiple cells were discharged at a rate of C/20 to various states of discharge and charge. The positive electrodes were recovered from the V2O3(SO4)2‖Li cells after cycling, washed with dimethyl carbonate (DMC, Sigma Aldrich, ≥99%) three times in an argon filled glovebox, and dried under vacuum overnight.
Chemical lithiation
Four chemical lithiation reactions were performed with n-butyllithium (1.6 M in hexanes, Sigma Aldrich) in an argon filled glovebox at room temperature. V2O3(SO4)2 (1.46 mmol) was dispersed in n-hexanes (Sigma Aldrich, 95%, ≤20 mL) and four different amounts of n-butyllithium were added, as detailed in Table 1. The dispersions were stirred continuously for two weeks. The products were filtered, washed with n-hexanes, dried under vacuum and handled in an argon filled glovebox.| V2O3(SO4)2/mmol | n-Hexane/mL | n-Butyllithium | |
|---|---|---|---|
| Moles/mmol | Volume/mL | ||
| 1.46 | 10 | 1.46 | 0.91 |
| 1.46 | 10 | 2.92 | 1.83 |
| 1.46 | 20 | 5.85 | 3.65 |
| 1.46 | 20 | 11.70 | 7.30 |
Materials characterisation
The pristine phase, V2O3(SO4)2, and lithiated samples were examined using inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis, powder X-ray diffraction (PXRD), neutron powder diffraction (NPD), X-ray absorption spectroscopy (XAS) and infrared (IR) spectroscopy, optical microscopy and scanning electron microscopy (SEM).ICP-OES analysis was performed on the chemically lithiated samples using a Thermo Fisher Scientific ICP-OES iCAP 6000. Standards were prepared for lithium by diluting concentrated commercial ICP-OES standards to four different concentrations. The chemically lithiated samples were dissolved using aqua regia (solution of nitric acid and hydrochloric acid at a 1:3 ratio by volume).
Laboratory PXRD patterns were collected on a PANanalytical Empyrean diffractometer, using Mo-Kα1,2 radiation (λ = 0.7107 Å) with a Zr β-filter and X'celerator detector. Data were collected in the 2θ range, 3.5–35.0° over 16 h per scan with a step size of 0.0167°, in capillaries at room temperature. Samples were loaded into glass capillaries (0.7 mm diameter) in an argon filled glovebox. An NPD pattern was obtained on the GEM diffractometer at the ISIS Neutron and Muon Source, UK. The powdered sample was loaded into a 8 mm vanadium can under argon and sealed using an indium wire and the measurement was made under ambient conditions over 1 h. Diffraction data were analysed using the Rietveld method using the general structural analysis system (GSAS) and its associated graphical user interface program (EXPGUI).24,25
Vanadium K-edge (5.4651 keV) X-ray absorption spectra were acquired in transmission on B18 beamline at Diamond Light Source, UK. The powdered samples (8 mg of active material) were ground for 30 min with cellulose (150 mg) and pressed into 13 mm diameter pellets, and then sealed into aluminium bags in an argon filled glovebox. XAS spectra of the vanadium metal foil were acquired simultaneously with the experimental samples. All spectra were acquired in triplicate for each sample. XAS spectra were aligned, merged and normalised using the software program Athena and EXAFS spectra were fitted using the program Artemis.26,27
An IRAffinity-1 Shimadzu Fourier transform IR spectrometer was used to obtain IR spectra. The powdered samples were placed directly onto the diamond crystal and IR spectra collected at room temperature between 4000 and 400 cm−1. Optical microscopy images were collected using a Meiji Techno microscope fitted with a Canon PowerShot G6 digital camera. A JEOL JSM-5600 SEM fitted with a tungsten filament electron source and a secondary electron detector were used to examine the morphology of the samples.
Results and discussion
Characterisation of V2O3(SO4)2
V2O3(SO4)2 was successfully prepared using a lower temperature of 140 °C, compared to the previously reported synthesis temperature of 180 °C.23 The SEM micrograph of V2O3(SO4)2, shown in Fig. 1(a) reveals a homogeneous morphology consisting of acicular crystallites.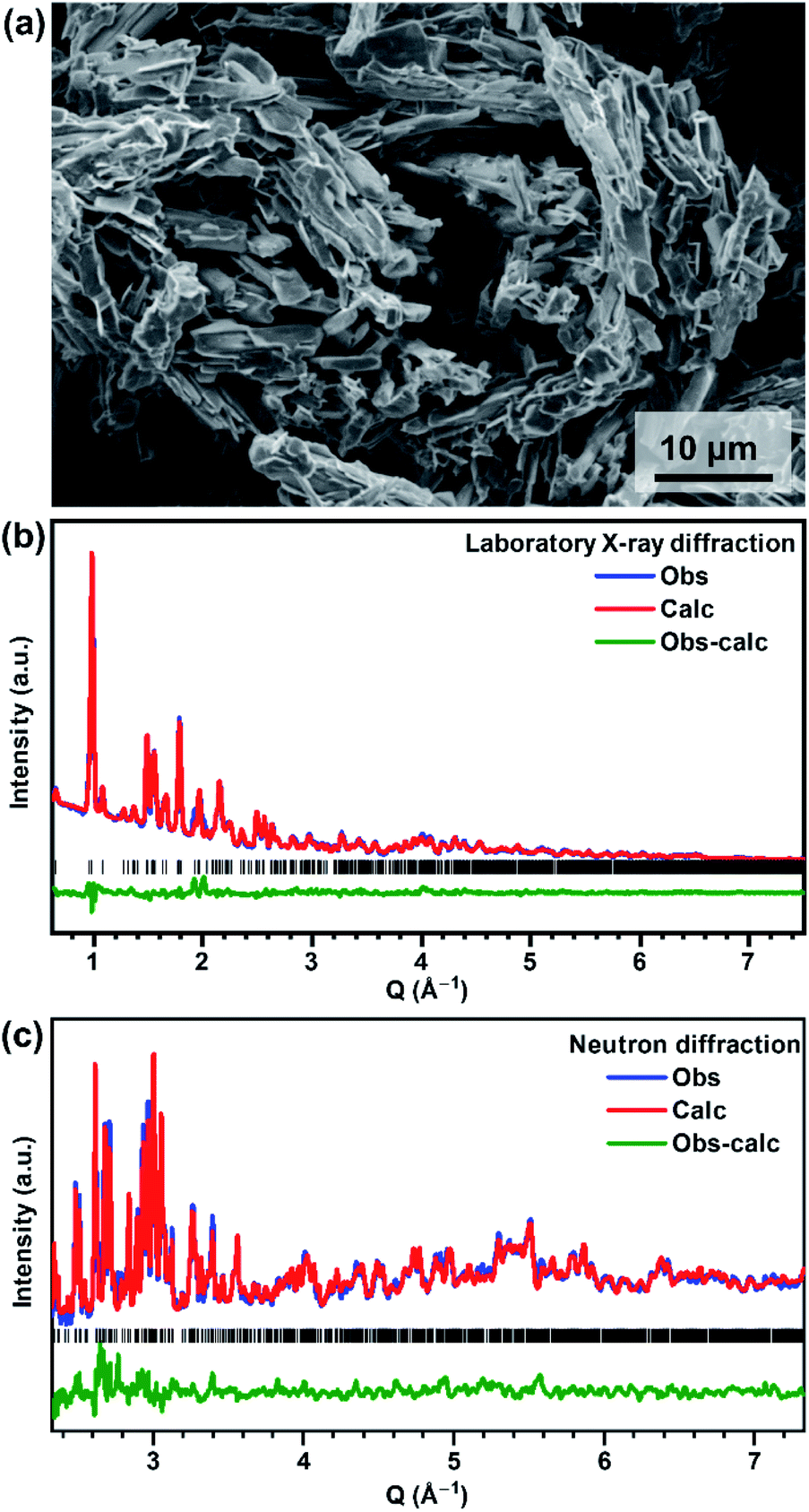 | ||
| Fig. 1 (a) SEM micrograph of V2O3(SO4)2, (b) laboratory X-ray diffraction Rietveld fit and (c) neutron diffraction Rietveld fit for V2O3(SO4)2 using the structural model published by Richter et al. as the starting model.23 | ||
V2O3(SO4)2 can be indexed to a monoclinic unit cell with the space group P21/a and unit cell parameters a = 9.464(12) Å, b = 8.908(13) Å, c = 9.939(12) Å, β = 104.732(4) °. All peaks were indexed and the unit cell parameters correspond well with those reported by Richter et al.23 A Rietveld refinement was performed using the structural model published by Richter et al.23 The Rietveld fits of the PXRD and NPD data are shown in Fig. 1(b) and (c), respectively, and the atomic coordinates and isotropic thermal parameters are given in the ESI, Table S1.†
The structure of V2O3(SO4)2 is illustrated in Fig. 2. V2O3(SO4)2 is made up of pairs of vanadium octahedra linked by a bridging oxygen atom and two corner-sharing SO4 tetrahedra, forming the [V2O3]4+ subunit which consists of the two species, [V(1)O2]+ and [V(2)O]3+, as shown in Fig. 2(a). The corner sharing VO6 distorted octahedra and SO4 tetrahedra create a three-dimensional network with open channels running down the c axis as illustrated in Fig. 2(b). The VO bond lengths in V2O3(SO4)2 are summarised in Table S2.†
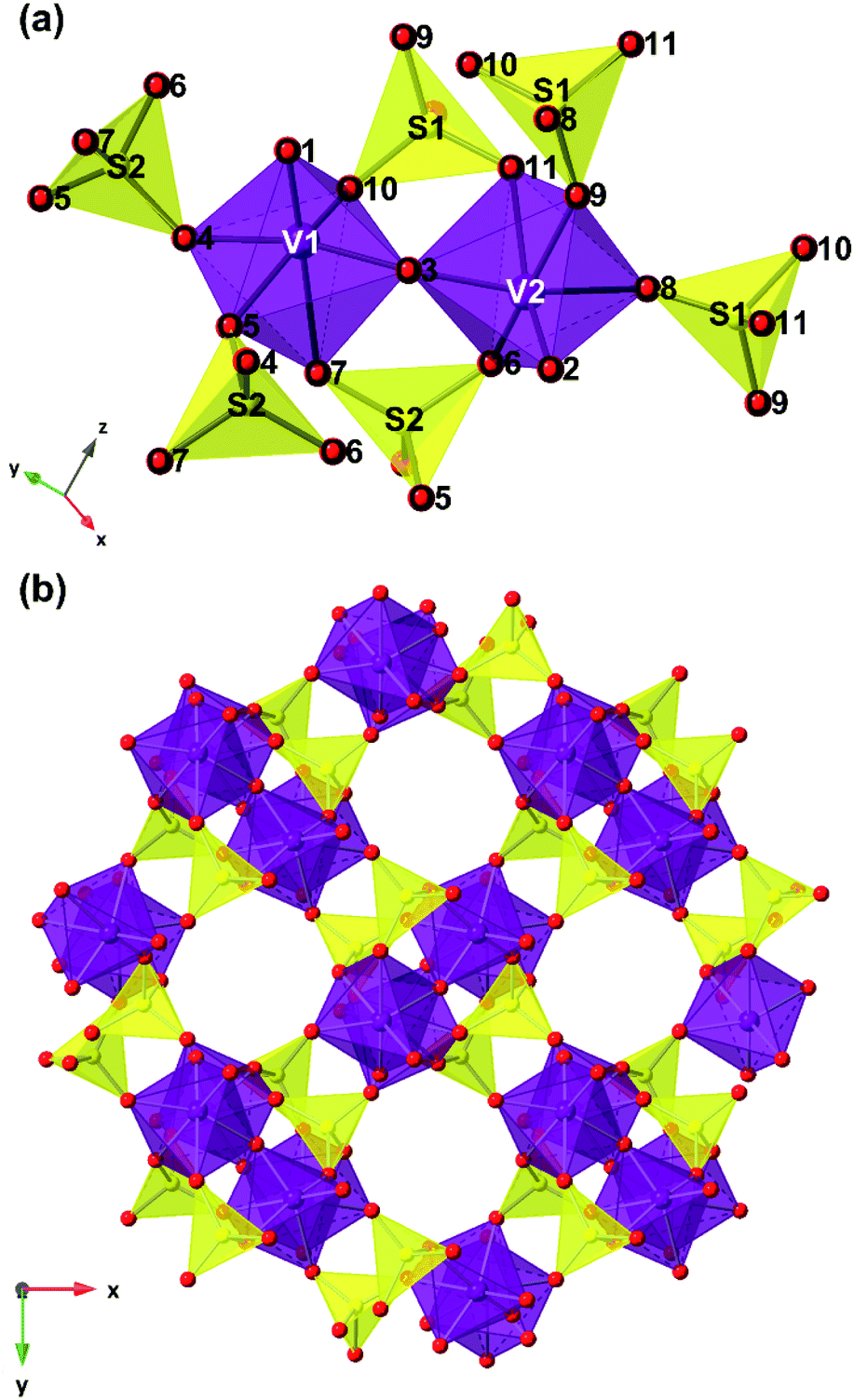 | ||
| Fig. 2 Structural model of V2O3(SO4)2 showing (a) the [V2O3]4+ subunit made up of pairs of vanadium octahedra (purple polyhedra) linked to each other by a bridging oxygen atom (red spheres) and two bridging SO4 tetrahedra (yellow polyhedra) and (b) the open channels down the c-axis. | ||
Electrochemical lithiation
The electrochemical Li+ insertion/extraction process in V2O3(SO4)2 was explored. Cells were cycled in galvanostatic mode between 4.20 V and 1.95 V at a rate of C/20, starting on discharge. The galvanostatic discharge–charge curves for the first, second, third and fifth cycles and the derivative dx/dV curve of the first cycle are shown in Fig. 3(a) and (b), respectively. The galvanostatic curve (Fig. 3(a)) reveals four sloping plateaux on discharge and charge, and approximately 2.0 Li+ (160 mA h g−1) were inserted on the first discharge, hence the average vanadium oxidation state reached +4.00. The dx/dV curve (Fig. 3(b)) shows four redox peaks centred about 4.09 V, 3.25 V, 2.83 V and 2.23 V vs. Li+/Li0. The intensities of the reduction peaks are greater than those of the oxidation peaks, suggesting that not all the Li+ that was inserted could be extracted. As for the redox processes centred about 3.25 V, the dx/dV curve shows two reduction peaks at 3.23 V and 3.14 V and two oxidation peaks at 3.15 V and 3.11 V. These peaks suggest charge-ordering of the vanadium atoms or vacancy-ordering of Li+ which has been reported for LixMn2O4 among other systems.28–30 Upon subsequent cycling, the electrode can reversibly insert approximately 1.5 Li+, leading to a reversible capacity of approximately 120 mA h g−1, which progressively decays upon cycling. Additionally, a cell was cycled in potentiostatic mode at a slow rate of 5.6 μV s−1 between 4.20 V and 1.95 V. The voltammogram is shown in Fig. S1† and corresponds well with the dx/dV curve obtained from galvanostatic cycling (Fig. 3(b)), showing the same four redox processes occurring at 4.09 V, 3.25 V, 2.83 V and 2.23 V vs. Li+/Li0.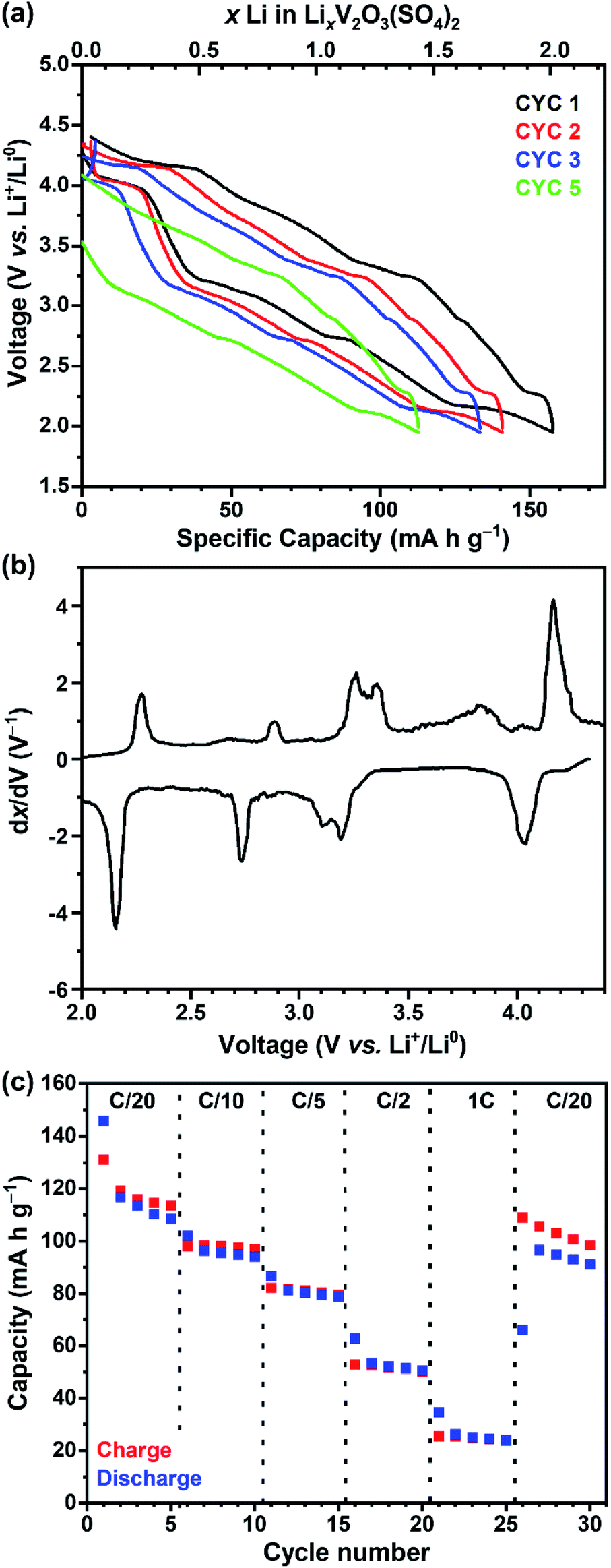 | ||
| Fig. 3 (a) V2O3(SO4)2‖Li cell cycled at a rate of C/20 between 4.20 V and 1.95 V showing the voltage–capacity curve for the first (black), second (red), third (blue) and fifth cycles (green), (b) the derivative dx/dV curve of the first cycle and (c) the specific capacity versus different C-rates (C/20, C/10, C/5, C/2, 1C) of V2O3(SO4)2 cycled between 4.20 V and 1.95 V vs. Li+/Li0. | ||
Fig. 3(c) shows the specific capacities over different C-rates (C/20, C/10, C/5, C/2 and C). The first cycle at C/20 shows some irreversible capacity (coulombic efficiency of 90%) but subsequent cycles at C/20 show a lower capacity but greater coulombic efficiency of 97%. The discharge and charge capacities decreased to 25 mA h g−1 at 1C. After cycling at the faster C-rates a reasonable capacity (110 mA h g−1, coulombic efficiency of 92%) was reached by the 27th cycle at C/20, suggesting that V2O3(SO4)2 did not degrade after cycling at faster rates. The first cycle of each C-rate shows greater irreversible capacity than the subsequent cycles, indicating that the redox processes are rate dependent.
To understand the electrochemical Li+ insertion/extraction in V2O3(SO4)2 multiple cells of V2O3(SO4)2‖Li were discharged to the potentials 3.50 V, 2.95 V, 2.40 V and 1.95 V and charged to 4.00 V. These potentials were selected because they correspond to the inflection points observed between the plateaux in the voltage profile and dx/dV curves (Figure S2†).
Chemical lithiation
ICP-OES data were collected to determine the amount of lithium in the chemically lithiated samples and their compositions are given in Table 2. The oxidation states of vanadium are also given in Table 2. These were calculated based on charge balance considerations, assuming the oxidation states of lithium and oxygen are +1 and −2, respectively, and that all the lithium present in the sample was incorporated into V2O3(SO4)2.| Moles of n-butyllithium/mol | x Li from ICP-OES analysis | Experimental composition | Vanadium oxidation state based on composition |
|---|---|---|---|
| 0.00 | 0.00(0) | V2O3(SO4)2 | +5.00(0) |
| 1.46 | 0.97(2) | Li0.97V2O3(SO4)2 | +4.52(1) |
| 2.92 | 1.64(16) | Li1.64V2O3(SO4)2 | +4.18(8) |
| 5.85 | 2.47(3) | Li2.47V2O3(SO4)2 | +3.77(2) |
| 11.70 | 3.97(5) | Li3.97V2O3(SO4)2 | +3.02(3) |
Chemical lithiation of V2O3(SO4)2 resulted in colour changes as shown in Fig. 4, from the yellow pristine phase, V2O3(SO4)2, to green Li0.97V2O3(SO4)2, darker green Li1.64V2O3(SO4)2 and Li2.47V2O3(SO4)2, to dark blue Li3.97V2O3(SO4)2. Optical images, also presented in Fig. 4, show the samples getting darker in colour with increasing Li+ content. These colour changes are consistent with changes in the vanadium oxidation state as discussed below. SEM micrographs of the chemically lithiated samples, shown in Fig. 4, show some needle-shaped crystallites which are consistent with V2O3(SO4)2 (Fig. 1(a)), suggesting that Li+ insertion did not cause the morphology of V2O3(SO4)2 to break down completely.
 | ||
| Fig. 4 Photographs (top), optical microscopy images (middle) and SEM micrographs (bottom) showing the colours and morphologies of (a) V2O3(SO4)2, (b) Li0.97V2O3(SO4)2, (c) Li1.64V2O3(SO4)2, (d) Li2.47V2O3(SO4)2 and (e) Li3.97V2O3(SO4)2. | ||
Vanadium oxidation state of LixV2O3(SO4)2
To confirm that the Li+ insertion was accompanied by a change in the vanadium oxidation state, vanadium K-edge XAS measurements were performed. Data were collected on V2O3(SO4)2 electrodes at various states of discharge and charge, the chemically lithiated phases, as well as vanadium foil, V2O3, VOSO4·3H2O and V2O5 reference compounds with vanadium formal oxidation states of 0, +3, +4, and +5, respectively. The XANES spectra for V2O5, VOSO4·3H2O and V2O3 reference compounds, the pristine phase, V2O3(SO4)2 and the electrochemically lithiated samples are compared in Fig. 5(a) and the chemically lithiated samples are shown in Fig. 5(b).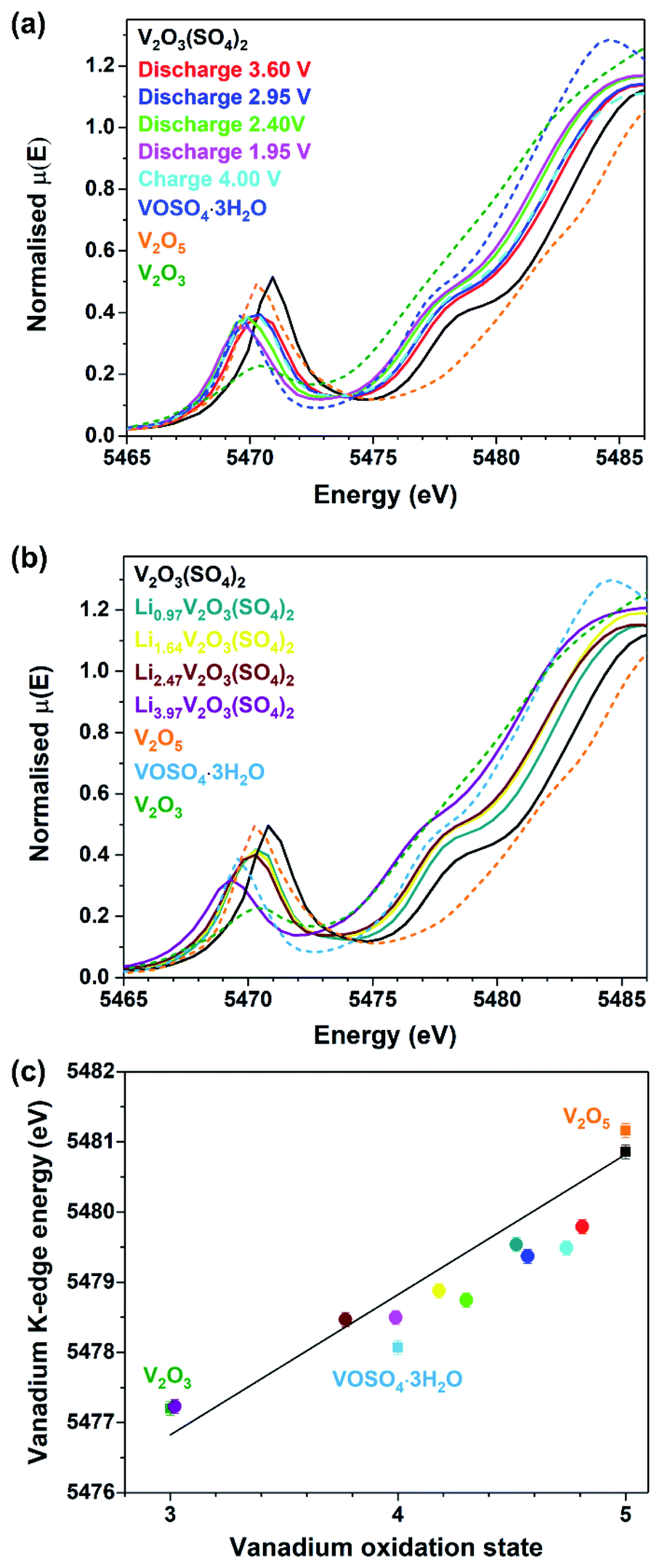 | ||
| Fig. 5 Vanadium K-edge XANES data showing (a) normalised spectra for the V2O3(SO4)2 electrodes at various states of discharge and charge compared to the reference materials, V2O5, VOSO4·3H2O and V2O3, (b) normalised spectra for the chemically lithiated V2O3(SO4)2 samples compared to the reference materials and (c) vanadium K-edge energy (at half-height) as a function of oxidation state. | ||
Fig. 5(a) and (b) show the intensity of the pre-edge peak decreases with increasing Li+ content. The intensity of the pre-edge peak is correlated to the size of the VOx coordination sphere which is 55% of the total intensity.31 This suggests that the shorter, vanadyl VO bonds increase in length and thereby the VO6 octahedra become more symmetrical.
This is consistent with a V5+ to V4+ transition for the electrode discharged to 1.95 V and a transition to V3+ for Li3.97V2O3(SO4)2.31,32
Furthermore, Fig. 5(a) and (b) reveal a small difference in the edge energies for V2O5 and V2O3(SO4)2, despite these materials having the same oxidation state because the edge energy of a material is also influenced by differences in coordination number and electronegativity of the ligands.31 The edge positions of the lithiated phases shift progressively to lower energies with increasing Li+ content relative to V2O3(SO4)2. The edge energy of the V2O3(SO4)2 electrode discharged to 1.95 V is similar to the reference material, VOSO4·3H2O, suggesting that approximately 2.0 mol Li per mol V2O3(SO4)2 were inserted electrochemically, which is in agreement with the electrochemical data (Fig. 3). The edge energy of the V2O3(SO4)2 electrode charged to 4.00 V was shifted to a higher energy relative to the electrode discharged to 1.95 V, suggesting an increase in the vanadium oxidation state upon Li+ extraction, as expected.33 However, the electrode charged to 4.00 V shows a similar edge energy to the electrode discharged to 3.60 V, implying that the electrochemical process was partially irreversible.
The edge energies of the chemically lithiated materials also shift to lower energies with increasing Li+ content, suggesting Li+ was incorporated into the structure of V2O3(SO4)2 in agreement with the colour changes observed (Fig. 4). Li3.97V2O3(SO4)2 has a very similar edge energy to V2O3, implying approximately 4.0 Li+ per mol was incorporated which agrees with the ICP-OES analysis for this phase. The absorption edge energies for the reference compounds V2O3, VOSO4·3H2O, V2O5, V2O3(SO4)2 are plotted in Fig. 5(c); these edge energies exhibit an essentially linear relationship with formal oxidation state. The edge energies of the reference materials are similar to those previously reported, thus a comparable linear relationship was obtained.31,33,34 To determine if the energy shifts of the absorption energies are consistent with the Li+ content, the energies of the absorption edges taken at half-height of the normalised spectra, were plotted against vanadium oxidation state. The Li+ content was derived from the electrochemical data for the electrochemically lithiated samples and ICP-OES analysis for the chemically lithiated samples. The average oxidation state of vanadium was calculated from the composition by assuming that the oxidation states of lithium and oxygen were +1 and −2, respectively. The edge energies for the electrochemically lithiated samples suggest that the vanadium oxidation states are comparable to those estimated based on the electrochemical data. Similar edge shifts have also been reported for other vanadium-based electrodes, including for example LixV2O5, LiVOPO4 and NaxVPO4.8F0.7.14,32,33,35,36 As for Li3.97V2O3(SO4)2, its edge energy shows 4.0 Li+ per mol of V2O3(SO4)2 were inserted, thereby demonstrating that two additional mol of Li per mol of V2O3(SO4)2 can be inserted chemically.
The experimental compositions and vanadium oxidation states for the lithiated samples are given in Table 3. The experimental compositions for the V2O3(SO4)2 electrodes were estimated based on the electrochemical data (Fig. 3(a)), assuming the oxidation states of lithium and oxygen were +1 and −2, respectively. The compositions of the chemically lithiated samples were obtained from ICP-OES analysis. The vanadium oxidation states of the lithiated samples are also given based on the compositions and the absorption edge energy, at half-height of the normalised XANES spectra.
| Sample | Composition based on experimental data | Vanadium oxidation state based on composition | Edge energy/eV | Vanadium oxidation state based on edge energy |
|---|---|---|---|---|
| V2O3(SO4)2 | V2O3(SO4)2 | +5.00 | 5480.9(1) | +5.04(5) |
| Discharge 3.60 V | Li0.39V2O3(SO4)2 | +4.81 | 5479.8(1) | +4.48(5) |
| Discharge 2.95 V | Li0.89V2O3(SO4)2 | +4.57 | 5479.4(1) | +4.27(5) |
| Discharge 2.40 V | Li1.40V2O3(SO4)2 | +4.30 | 5478.8(1) | +3.87(5) |
| Discharge 1.95 V | Li2.01V2O3(SO4)2 | +3.99 | 5478.5(1) | +3.84(5) |
| Charge 4.00 V | Li0.53V2O3(SO4)2 | +4.74 | 5479.5(1) | +4.33(5) |
| 1.46 mmol nBuLi | Li0.97V2O3(SO4)2 | +4.52(1) | 5479.5(1) | +4.34(5) |
| 2.92 mmol nBuLi | Li1.64V2O3(SO4)2 | +4.18(8) | 5478.9(1) | +4.03(5) |
| 5.85 mmol nBuLi | Li2.47V2O3(SO4)2 | +3.77(2) | 5478.5(1) | +3.82(5) |
| 11.70 mmol nBuLi | Li3.97V2O3(SO4)2 | +3.02(3) | 5477.2(1) | +3.20(5) |
In addition to using XANES analysis to confirm the change in vanadium oxidation state, IR spectra were collected. The IR spectra of V2O3(SO4)2, the V2O3(SO4)2 electrodes and the chemically lithiated phases are presented in Fig. 6(a) and (b), respectively. The IR spectrum of V2O3(SO4)2 exhibits absorption bands previously reported for V2O3(SO4)2, including the short vanadyl bond (993 cm−1), intermediate V–O bond (948 cm−1) and V–O–V bond (770 cm−1).23,37 It was expected that the vanadium oxidation state should decrease with increasing Li+ content which is in agreement with the XANES analysis. As a result, one of the short VO bonds of the [VO2]+ species and the VO bond of the [VO]3+unit should increase in length on insertion of 2.0 Li+ which would form two [VO]2+ species. On insertion of 4.0Li+, the Li3.97V2O3(SO4)2 phase should not contain any short VO bonds, as these are absent in V3+ phases.38 The absorption band (993 cm−1) corresponding to the short VO bond shifts with varying Li+ content.23Fig. 6(a) shows the VO absorption band of V2O3(SO4)2 discharged to 3.50 V, 2.95 V, 2.40 V and 1.95 V shifts to lower frequencies of 983 cm−1, 985 cm−1, 975 cm−1 and 975 cm−1, respectively, relative to V2O3(SO4)2 (993 cm−1). While the VO absorption band of the V2O3(SO4)2 electrode charged to 4.00 V shifted to a higher frequency 989 cm−1, relative to the electrode discharged to 1.95 V (975 cm−1). This shift suggests the VO bond length increases with increasing Li+ content and contracts on extraction of Li+ which coincides with the XANES data.
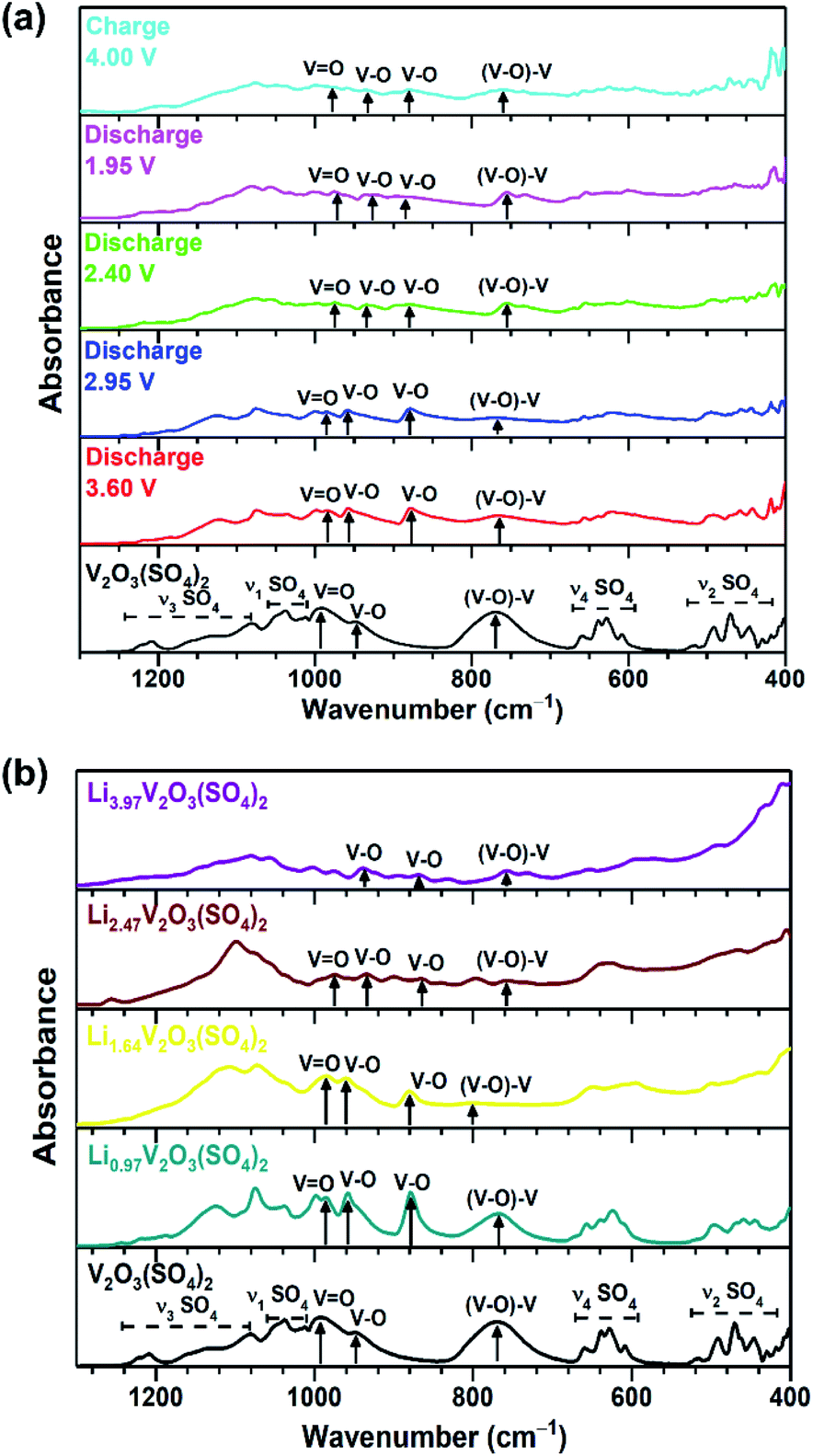 | ||
| Fig. 6 IR spectra of (a) V2O3(SO4)2 (black) and the V2O3(SO4)2 electrode discharged to 3.60 V (red), discharged to 2.95 V (blue), discharged to 2.40 V (green), discharged to 1.95 V (pink), charged to 4.00 V (cyan) and (b) Li0.97V2O3(SO4)2 (teal), Li1.64V2O3(SO4)2 (yellow), Li2.47V2O3(SO4)2 (maroon), Li3.97V2O3(SO4)2 (purple). | ||
Fig. 6(b) shows that VO absorption band also shifts to lower frequencies of 985 cm−1, 984 cm−1 and 975 cm−1 for Li0.97V2O3(SO4)2, Li1.64V2O3(SO4)2 and Li2.47V2O3(SO4)2, respectively, demonstrating that the VO bond length increases. In the case of Li3.97V2O3(SO4)2, the intensity of the VO bond almost disappears, suggesting that Li3.97V2O3(SO4)2 does not contain the short vanadyl bond – consistent with a V3+ phase.14,38
Interestingly, the lithiated phases exhibit an additional absorption band at 880 cm−1, not observed for V2O3(SO4)2. This band (880 cm−1) is consistent with a longer V–O bond.23,39,40 Its frequency suggests that it is longer than the other V–O bonds which exhibit absorption bands in the frequency range 934–960 cm−1. This implies a change in the local structure around vanadium which will be discussed further in the subsequent section.
Structural evolution
To reveal the structural changes with Li+ content, EXAFS analysis and PXRD studies were performed. Information on the changes in the local structure of the vanadium atoms was obtained by fitting the EXAFS data. A model based on the structure of the pristine phase, V2O3(SO4)2, was used to fit the EXAFS spectra. The local structure of vanadium in V2O3(SO4)2 is complex and the local environment of the V(1) and V(2) atoms is shown in Fig. 7.24 Each vanadium atom is coordinated via six V–O bonds (Table S2†). The vanadium atoms are both coordinated to a short terminal oxygen atom, V(1)O(1) at 1.598(8) Å and V(2)O(2) at 1.590(3) Å, and interconnected by a bridging oxygen atom, O(3), with V(1)O(3) at 1.686(7) Å and V(2)–O(3) at an intermediate length of 1.781(4) Å, forming [V(1)O2]+ and [V(2)O]3+. Each vanadium atom also shows three intermediate bonds, V–Oeq (∼1.95(5) Å), and one long bond, V⋯O (∼2.32(2) Å). Since the environments of V(1) and V(2) are similar, this structure was simplified into a five-path model consisting of a path for the short vanadyl bond (VO), one for the short intermediate bond (V–O), one for the three equatorial bonds (V–Oeq), one for the long bond (V⋯O) and one for the V–O–S coordination, as illustrated in Fig. 7. During the fitting process, the atomic separations were refined, the coordination number (N) was held constant and one photoelectron energy shift (Eo) was fitted for the five paths. An amplitude reduction factor (So2) of 1.0 was obtained by fitting the data from the pristine phase and used in the fitting of the data from lithiated materials. A different disorder factor (σ2) for each path was used and fitting ranges of 4.0 < k < 13.0 Å−1 and 1.0 < R < 4.0 Å were employed. The fit values for Eo remained within ±10 eV. The experimental k3-weighted EXAFS data fitted with the five-path model and the k3-weighted Fourier Transform of the EXAFS for the electrochemically lithiated samples are shown in Fig. 8(a) and (b), respectively. The experimental k3-weighted EXAFS data fitted with the five-path model and the k3-weighted Fourier transform of the EXAFS for the chemically lithiated samples are shown in Fig. 8(c) and (d), respectively. Note that the peaks of the k3-weighted Fourier Transform have not been phase-shift corrected and appear at shorter distances than the atomic separations they represent.
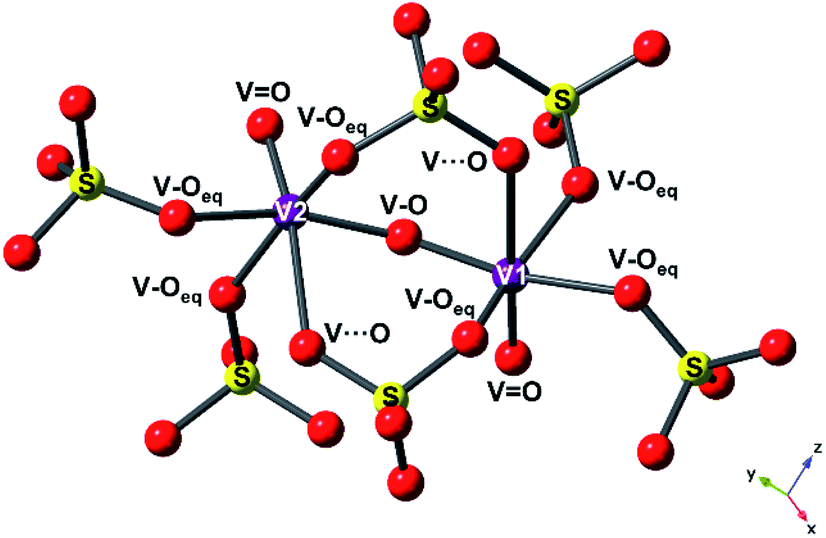 | ||
| Fig. 7 Local environment of V(1) and V(2) atoms in V2O3(SO4)2, showing the different paths used in the EXAFS fitting model ((VO) + (V–O) + (V–Oeq) + (V⋯O) + (V–O–S)), with the vanadium atoms shown as purple spheres, oxygen atoms as red spheres and sulfur atoms as yellow spheres. | ||
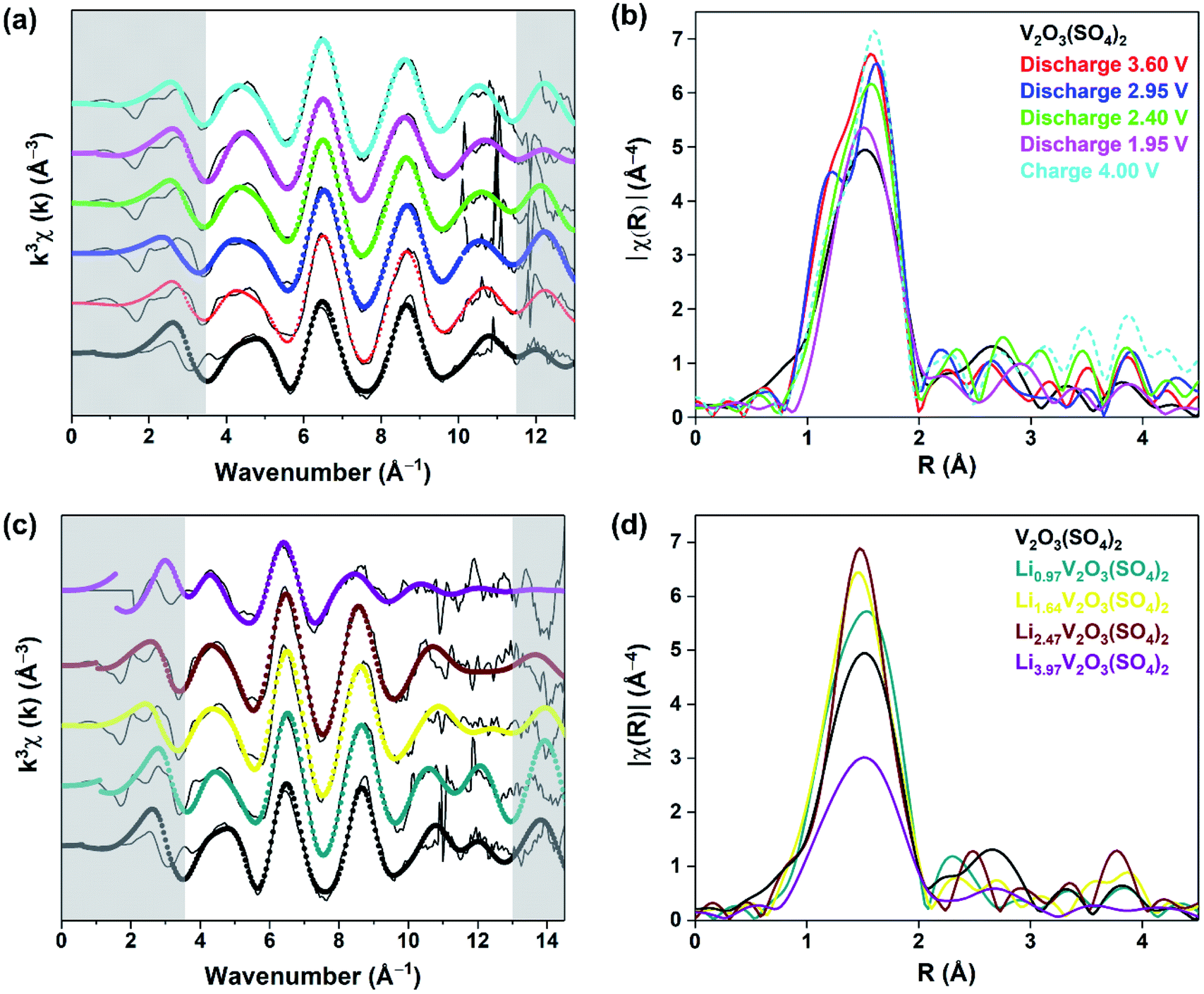 | ||
| Fig. 8 (a) Experimental k3-weighted EXAFS data (black lines) fitted with the five-path model (filled circles) and (b) their corresponding vanadium K-edge EXAFS spectra Fourier Transform in the R-space for the V2O3(SO4)2 electrode at various states of discharge and charge, (c) experimental k3-weighted EXAFS data (black lines) fitted with the five-path model (filled circles) and (d) their corresponding vanadium K-edge EXAFS spectra Fourier Transform in the R-space for the chemically lithiated phases. | ||
Fig. 8(b) and (d) each show one distinct peak positioned in the 1–2 Å range, associated with the V–O local environment of the first coordination sphere with the majority of this peak originating from the three V–Oeq bonds.32,33 The following peaks extending to 3.5 Å are dominated by contributions from the long V⋯O bond and the V–S single scattering paths. They show variations in the amplitude of the V–O contribution with Li+ content, suggesting changes in the local arrangement of the first coordination sphere surrounding the vanadium atoms. Fig. 8(b) shows that the amplitude of the first peak increased after discharging to 3.60 V due to Li+ insertion. The amplitude of this peak decreases on insertion of more Li+ and increases again after extraction of Li+.
The results from the fits of the EXAFS for the electrochemically lithiated samples are presented in Table S4.† The bond lengths for V2O3(SO4)2 are comparable to the bond lengths obtained from PXRD analysis (Table S2†). On cycling V2O3(SO4)2vs. Li+/Li0, the VO bond length fluctuated but remained within the range of a vanadyl bond (1.56(6)–1.60(4) Å).38 This result was expected since the electrochemical data and XANES spectra both suggested that V5+ was reduced to V4+ which also contains a VO bond.38 The V–O bond, which models the bridging oxygen atom between to the two vanadium atoms increased in length to 1.83(5) Å after discharge to 1.95 V. This is consistent with a transition from V5+ to V4+ (i.e., [VO2]+ to [VO]2+) on insertion of 2.0 Li+ per mol of V2O3(SO4)2. On extraction of Li+, the V–O bond contracted again to 1.78(1) Å consistent with a [VO]2+ to [VO2]+ transition, in agreement with the XANES data. Fig. 8(d) shows an increase in the amplitude of the first peak with increasing Li+ content which can be attributed to a higher degree of symmetry surrounding the vanadium atoms with increasing Li+ content.32,33
However, in the case of the Li3.97V2O3(SO4)2 phase, the intensity of the V–O contribution decreased significantly. Since the symmetry surrounding the vanadium atoms continued to increase with increasing Li+, this effect was probably outweighed by an increase in the disorder as the V–O bonds became longer and weaker. Fig. 8(c) shows the spectrum of Li3.97V2O3(SO4)2 is noisy, with no signal beyond 11 Å−1 so the k-range was restricted to 3.6 < k < 10.8 Å−1 and 1.0 < R < 4.0 Å. However, with this range the five-shell model gave nonsensical parameters, including either negative or extremely large disorder factors and Eo values greater than 10 eV. Consequently, other models were tested. A satisfactory fit was obtained with a four-path model consisting of a path for the two short vanadyl bonds (VO), one for the three equatorial bonds (V–Oeq), one for the long bond (V⋯O) and one for the V–O–S coordination. The fitting parameters show an increase in the V–O bond lengths compared to the other samples. The short VO bond distance (1.67(2) Å) increased in length by 0.07 Å compared to the pristine phase and is midway between the VO and V–O–V bond.
The equatorial bonds (V–Oeq) also increased in length (2.04(4) Å) and are consistent with V3+–O bond lengths.41 These results are broadly consistent with the reduction of V5+ to V3+, since longer and weaker bonds were obtained from this analysis. Additionally, there is more disorder associated with the V–O bonds of Li3.97V2O3(SO4)2 which is also consistent with V3+–O bonds. Moreover, there is a correlation between the intensity of the pre-edge peak of the XANES spectrum and the size of the VOx coordination sphere.31 The reduced intensity of the pre-edge peak of Li3.97V2O3(SO4)2 relative to the other phases is consistent with the disappearance of the VO bond. This observation is consistent with the IR spectrum for this phase, suggesting Li3.97V2O3(SO4)2 consists of more regular VO6 octahedra compared to the distorted VO6 octahedra of V2O3(SO4)2.
To follow the long-range structural changes, PXRD patterns of the lithiated phases were collected. Fig. 9(a) compares the PXRD patterns of the electrochemically lithiated samples and Fig. 9(b) compares the PXRD patterns of the chemically lithiated samples. The lithiated materials show no additional reflections compared to V2O3(SO4)2, implying no secondary phases were formed on Li+ insertion. Fig. 9 reveals a shift of the reflections for the lithiated samples relative to V2O3(SO4)2, suggesting the unit cell volume changed on Li+ insertion. It can be assumed that the lithiated materials retained the monoclinic symmetry of V2O3(SO4)2 since the PXRD patterns of V2O3(SO4)2 and the lithiated phases are similar.
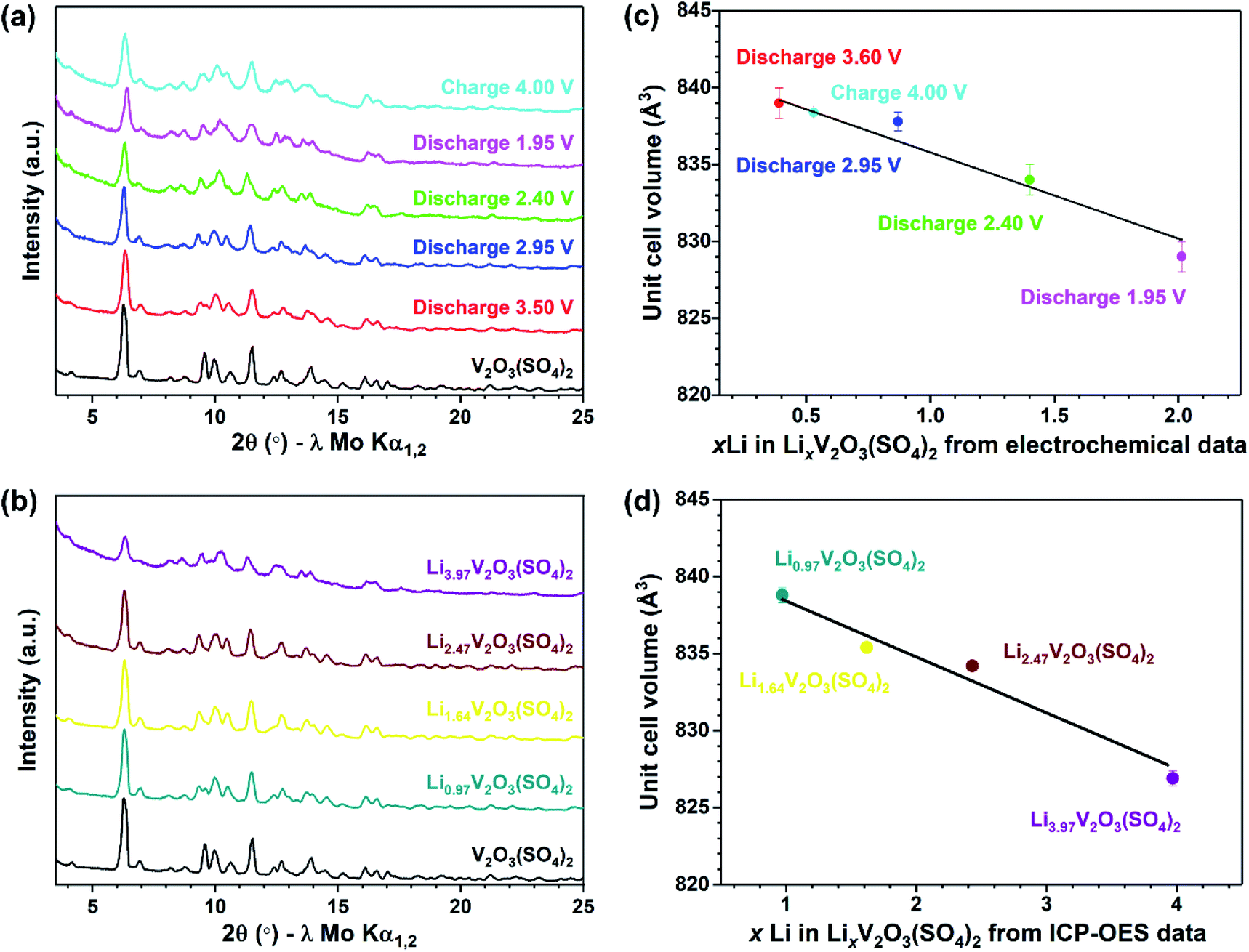 | ||
| Fig. 9 (a) PXRD patterns of V2O3(SO4)2 (black), V2O3(SO4)2 electrode discharged to 3.50 V (red), discharged to 2.95 V (blue), discharged to 2.40 V (green), discharged to 1.95 V (pink), charged to 4.00 V (cyan), (b) PXRD patterns of the chemically lithiated phases, Li0.97V2O3(SO4)2 (teal), Li1.64V2O3(SO4)2 (yellow), Li2.47V2O3(SO4)2 (maroon) and Li3.97V2O3(SO4)2 (purple), (c) the effect of xLi in LixV2O3(SO4)2 based on electrochemical data on the unit cell volume of electrochemically lithiated materials and (d) the effect of xLi in LixV2O3(SO4)2 as determined from ICP-OES analysis on the unit cell volume of chemically lithiated materials. | ||
To establish the changes in the unit cell parameters, a series of Rietveld refinements were performed using the structural model of V2O3(SO4)2 as the starting model.24 The same general procedure was followed for each refinement, the backgrounds were fitted using a Chebyshev polynomial with 36 terms, the unit cell parameters, zero-error and profile parameters were all refined. Good fits were obtained and the fits between 9–12° 2θ are presented in Fig. S3(a–j),† highlighting the shifts of the reflections relative to the pristine phase. The corresponding changes in the unit cell parameters are given in Table S3.† The relationship between the Li content (x Li in LixV2O3(SO4)2) and the unit cell volume for the electrochemically lithiated samples is illustrated in Fig. 9(c). This shows there was an expansion of the unit cell volume of roughly 3.5% on insertion of 0.39 mol Li+ after discharging to 3.60 V. This can be explained by the size-expansion of the VO6 octahedra due to the partial reduction of V5+ to V4+ and the insertion of Li+.42 Inserting more Li+ caused the unit cell volume to contract. This can be rationalised by the increased electrostatic attraction with increasing Li+ content which outweighed the size-expansion of the VO6 octahedra due to the reduction of V5+ to V4+. If 1.0 Li+ was inserted into V2O3(SO4)2, according to the linear relationship shown in Fig. 9(c), LiV2O3(SO4)2 would have a unit cell volume of 836(1) Å3. This corresponds to a 3.1(1)% volume expansion relative to the pristine phase. This volume change is comparable with volume changes reported in the literature for other vanadium-based polyanionic electrode materials.13,14,18
The evolution of the unit cell volume after chemical lithiation is presented in Fig. 9(d). This shows the relationship between the Li+ content (xLi in LixV2O3(SO4)2) obtained from ICP-OES analysis and the unit cell volume obtained from the Rietveld refinements. Fig. 9(d) shows the unit cell volume decreased linearly with increasing Li+ content. This result coincides with the change in unit cell volume observed for the electrochemically lithiated materials, demonstrating that the structural changes during the electrochemical lithiation were replicated chemically.
Conclusions
This work has investigated for the first time the Li+ insertion process into V2O3(SO4)2via electrochemical and chemical routes. V2O3(SO4)2 delivers an initial discharge capacity of approximately 160 mA h g−1, corresponding to the insertion of approximately 2.0 Li+ per formula unit, associated with four redox processes at 4.09 V, 3.25 V, 2.83 V and 2.23 V attributed to the V4+/V5+ redox couple. XANES measurements of the electrochemically lithiated materials showed that 2.0 Li+ ions were incorporated to give Li2V2O3(SO4)2. As for the chemically lithiated materials, ICP-OES data and XANES measurements revealed that up to 4.0 Li+ ions can be inserted into V2O3(SO4)2, reducing V5+ to V3+. Structural studies by PXRD revealed that the monoclinic symmetry of V2O3(SO4)2 was retained with Li+ insertion and was accompanied by changes in the unit cell volume. This analysis showed that the structural transformation which occurred during electrochemical lithiation was replicated chemically and that 4.0 Li+ ions can be inserted into V2O3(SO4)2 while it maintained its framework structure.This material exploits the property of vanadium to adopt numerous oxidation states and offers structural stability. This highlights the potential of vanadium sulfates and the opportunities to explore new vanadium-based polyanionic compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank M. Nowosielska for ICP-OES measurements. The authors are grateful for the provision of beam time and assistance from Dr Giannantonio Cibin on B18 at the Diamond Light Source (as part of the Energy Materials Block Allocation Group SP14239) and Dr Ron Smith on the GEM diffractometer at ISIS Neutron and Muon Source at the Rutherford Appleton Laboratory. The authors also thank EPSRC for funding of SFL's PhD thesis (EP/N509759/1).References
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS.
- A. Manthiram and J. B. Goodenough, J. Power Sources, 1989, 26, 403–408 CrossRef CAS.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS.
- J. Wang and X. Sun, Energy Environ. Sci., 2012, 5, 5163–5185 RSC.
- P. Barpanda, M. Ati, B. C. Melot, G. Rousse, J.-N. Chotard, M.-L. Doublet, M. T. Sougrati, S. A. Corr, J.-C. Jumas and J.-M. Tarascon, Nat. Mater., 2011, 10, 772–779 CrossRef CAS.
- M. Kim, Y. Jung and B. Kang, J. Mater. Chem. A, 2015, 3, 7583–7590 RSC.
- R. K. B. Gover, P. Burns, A. Bryan, M. Y. Saidi, J. L. Swoyer and J. Barker, Solid State Ionics, 2006, 177, 2635–2638 CrossRef CAS.
- J. Barker, R. K. B. Gover, P. Burns, A. Bryan, M. Y. Saidi and J. L. Swoyer, J. Power Sources, 2005, 146, 516–520 CrossRef CAS.
- J. Barker, R. K. B. Gover, P. Burns, A. Bryan, M. Y. Saidi and J. L. Swoyer, J. Electrochem. Soc., 2005, 152, A1776–A1779 CrossRef CAS.
- J. Barker, R. K. B. Gover, P. Burns and A. Bryan, Electrochem. Solid-State Lett., 2005, 8, A285–A287 CrossRef CAS.
- J.-M. Ateba Mba, C. Masquelier, E. Suard and L. Croguennec, Chem. Mater., 2012, 24, 1223–1234 CrossRef CAS.
- M. Bianchini, J. M. Ateba-Mba, P. Dagault, E. Bogdan, D. Carlier, E. Suard, C. Masquelier and L. Croguennec, J. Mater. Chem. A, 2014, 2, 10182–10192 RSC.
- K. L. Harrison, C. A. Bridges, C. U. Segre, C. D. V. Jr, D. Applestone, C. W. Bielawski, M. P. Paranthaman and A. Manthiram, Chem. Mater., 2014, 26, 3849–3861 CrossRef CAS.
- J. Barker, M. Y. Saidi and J. L. Swoyer, J. Electrochem. Soc., 2004, 151, A796–A800 CrossRef CAS.
- M. S. Kishore, V. Pralong, V. Caignaert, U. V. Varadaraju and B. Raveau, Electrochem. Commun., 2006, 8, 1558–1562 CrossRef CAS.
- M. Satya Kishore, V. Pralong, V. Caignaert, S. Malo, S. Hebert, U. V. Varadaraju and B. Raveau, J. Solid State Chem., 2008, 181, 976–982 CrossRef CAS.
- M. Sun, G. Rousse, M. Saubanère, M.-L. Doublet, D. Dalla Corte and J.-M. Tarascon, Chem. Mater., 2016, 28, 6637–6643 CrossRef CAS.
- J. Gaubicher, Y. Chabre, J. Angenault, A. Lautié and M. Quarton, J. Alloys Compd., 1997, 262–263, 34–38 CrossRef CAS.
- K. S. Nanjundaswamya, H. Araib, J. Yamakib, S. Okadab and H. Ohtsukab, Solid State Ionics, 1996, 92, 1–10 CrossRef.
- J. Gaubicher, J. Angenaut, Y. Chabre, T. Le Mercier and M. Quarton, Mol. Cryst. Liq. Cryst., 1998, 311, 45–50 CrossRef.
- L. Lander, J.-M. Tarascon and A. Yamada, Chem. Rec., 2018, 18, 1394–1408 CrossRef CAS.
- K.-L. Richter and R. Mattes, Z. Anorg. Allg. Chem., 1992, 611, 158–164 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, GSAS - General Structure Analysis System, LANL Report, Los Alamos National Laboratory, LAUR, Los Alamos, USA, 2001, pp. 86–748 Search PubMed.
- B. Ravel and M. Newville, Phys. Scr., 2005, 115, 1007–1010 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- T. Ohzuku, J. Kato, K. Sawai and T. Hirai, J. Electrochem. Soc., 1991, 138, 2556–2560 CrossRef CAS.
- H. Björk, T. Gustafsson, J. O. Thomas, S. Lidin and V. Petříček, J. Mater. Chem., 2003, 13, 585–589 RSC.
- Y. J. Lee, F. Wang, S. Mukerjee, J. McBreen and C. P. Grey, J. Electrochem. Soc., 2000, 147, 803–812 CrossRef CAS.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 5596–5610 CrossRef CAS.
- C. J. Allen, Q. Jia, C. N. Chinnasamy, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2011, 158, 1250–1259 CrossRef.
- A. N. Mansour, P. H. Smith, W. M. Baker, M. Balasubramanian and J. McBreen, Electrochim. Acta, 2002, 47, 3151–3161 CrossRef CAS.
- T. Tsunehiro, Y. Hiromi, T. Risa, F. Takuzo and Y. Satohiro, J. Chem. Soc., 1988, 84, 2987–2999 Search PubMed.
- G. A. Horrocks, E. J. Braham, Y. Liang, L. R. De Jesus, J. Jude, J. M. Velázquez, D. Prendergast and S. Banerjee, J. Phys. Chem. C, 2016, 120, 23922–23932 CrossRef CAS.
- Y. U. Park, D. H. Seo, H. S. Kwon, B. Kim, J. Kim, H. Kim, I. Kim, H. I. Yoo and K. Kang, J. Am. Chem. Soc., 2013, 135, 13870–13878 CrossRef CAS.
- A. P. Tyutyunnik, V. N. Krasil, V. G. Zubkov, L. A. Perelyaeva and I. V Baklanova, Russ. J. Inorg. Chem., 2010, 55, 554–560 CrossRef.
- M. Schindler, F. C. Hawthorne and W. H. Baur, Chem. Mater., 2000, 12, 1248–1259 CrossRef CAS.
- A. P. Tyutyunnik, V. G. Zubkov, I. F. Berger, V. N. Krasil, L. A. Perelyaeva and I. V Baklanova, Russ. J. Inorg. Chem., 2007, 52, 1521–1526 CrossRef CAS.
- V. N. Krasil'nikov, A. P. Tyutyunnik, L. A. Perelyaeva and I. V Baklanova, Russ. J. Inorg. Chem., 2013, 58, 161–167 Search PubMed.
- J. Krakowiak, D. Lundberg and I. Persson, J. Inorg. Chem., 2012, 51, 9598–9609 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: More Rietveld refinements and Rietveld refinement results, additional electrochemical data, and EXAFS analysis. See DOI: 10.1039/d0ta06608g |
| This journal is © The Royal Society of Chemistry 2020 |