
Efficient and stable charge transfer channels for photocatalytic water splitting activity of CdS without sacrificial agents†
 a
Jian
Zhang
*bc
a
Jian
Zhang
*bc
Abstract
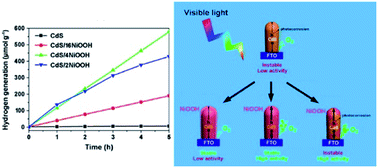
Semiconductor-based durable photocatalysts for efficient water splitting have attracted much attention for the development of sustainable hydrogen energy production, but it is challenging for CdS to achieve the expectation of the absence of hole scavengers. Herein, it is reported that uniform CdS nanorods coated with ultrathin NiOOH were prepared as a photocatalyst for high-efficiency photocatalytic water splitting without using any hole sacrificial agents. For the novel surface dynamics features, electron accumulation on CdS was detected with in situ irradiated X-ray photoelectron spectroscopy and accelerated hole transfer was recorded as 49.6 ± 9.2 ps by femtosecond time-resolved transient absorption spectroscopy. The thickness dependence of the NiOOH wrapper with an ultimate continuous thickness of ∼4 nm not only achieved the record high value for photocatalytic hydrogen generation rate (118.6 μmol h−1 g−1) among CdS-based heterojunction photocatalysts without any scavengers, but also exhibited good photostability (over 25 h of cycling measurements). This work provides valuable guidelines for the design of next-generation, high performance CdS-based photocatalysts.
 Please wait while we load your content...
Please wait while we load your content...

