DOI:
10.1039/D0TA05827K
(Paper)
J. Mater. Chem. A, 2020,
8, 19573-19587
Realizing a high-performance LiNi0.6Mn0.2Co0.2O2/silicon–graphite full lithium ion battery cell via a designer electrolyte additive
Received
11th June 2020
, Accepted 28th August 2020
First published on 17th September 2020
Abstract
An optimized dosage of (2-cyanoethyl)triethoxysilane (TEOSCN), is investigated as the electrode/electrolyte interface (EEI) modulating electrolyte additive to improve electrochemical performance of LiN0.6Mn0.2Co0.2O2(NMC622)/silicon(Si)–graphite(Gr) battery cells at a high temperature (45 °C). The addition of 1 wt% of TEOSCN to 1 M LiPF6 in EC:DEC + 5 wt% FEC/2 wt% VC electrolyte is found to significantly improve the long-term cyclability, capacity retention and coulombic efficiency of NMC622/Si–Gr cells at 45 °C. Pouch cells cycled in a nitrile-functionalized silane bearing electrolyte show superior capacity retention (∼75.95%) compared to those with FEC/VC (∼8.05%) and without additives (EC:DEC, ∼19.23%) electrolytes at the 364th cycle. Chemical mimicking and X-ray Photoelectron Spectroscopy (XPS) analysis proved that the enhanced electrochemical performance is attributed to the formation of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N reduction/oxidation induced robust EEI layers, both on the anode and cathode compartments, thus mitigating the escorted prevailing challenges. This work provides a highly promising electrolyte additive enabling the large-scale commercial deployment of Si-containing high-energy lithium-ion full cell batteries.
N reduction/oxidation induced robust EEI layers, both on the anode and cathode compartments, thus mitigating the escorted prevailing challenges. This work provides a highly promising electrolyte additive enabling the large-scale commercial deployment of Si-containing high-energy lithium-ion full cell batteries.
1. Introduction
The quest for more efficient and reliable electrochemical energy storage technologies that go beyond the specific energy density of the present-day Li-ion batteries (LIBs) is driven by the urge to replace internal combustion engine vehicles in cars, planes and other means of mobility.1 Incentivized by the high specific theoretical capacity (3590 mA h g−1 for Li15Si4 and 4212 mA h g−1 for Li22Si5 at 25 °C and 45 °C, respectively), suitable (i.e., low) operating potential (∼0.2 V–0.4 V vs. Li/Li+), high abundance and thus low cost, environmental benignity, and non-toxicity, silicon (Si) has attracted much attention as the most promising next-generation anode material for high-energy LIBs.2,3 Uniquely, the combination of high capacity Si with layered oxide-type intercalation cathode materials such as high-energy LiNxMnyCozO2 (NMC), is one of the most appealing strategies towards the development of high energy density practical full cell batteries. However, its anticipated large-scale commercialization is delayed due to several challenges such as cumbersome volume change (≥280%) during the charge/discharge process,4 low electrical conductivity,5,6 low coulombic and energy efficiencies, unstable solid electrolyte interphase (SEI), electrode swelling, electrolyte drying etc.3,7–11 Ever since the genesis of Si alloying anodes, numerous remedies have been proposed aiming at solving the encounters and thereby making the best use of Si anodes in practical LIBs.12 Amid others, the incorporation of a small dose of foreign molecules, called additives, into the bulk electrolyte is hailed as one of the most economical, effective and scalable approaches to circumvent the above-mentioned predicaments.3,13–22
The quality of the SEI at the interface between Si and liquid electrolytes is a critical factor, expressly with highly reactive and those experiencing large volume change electrode materials. The cathode-electrolyte interphase (CEI) is equally important in the cathode compartment. Therefore, they are represented to be the most ubiquitous and indispensable parts of the battery system, dictating the long-term cycling stability, obtainable capacity, rate capability, coulombic/energy efficiency, safety and cost linked to their formation stages.23 Among diverse electrolyte additives, reductive/oxidative-types (film forming/passivating) are of supreme importance to regulate the anode and/or cathode interphasial chemistries. The hunt for organic compounds as electrolyte additives is usually carried out randomly, and accordingly becomes a highly empirical one, making it risky and cost ineffective.24 In an attempt to systematically screen highly performing additives for Si-alloy based anodes, our group has thoroughly investigated various additives, making use of diverse analytical tools.25 Molecular additives bearing special moieties, such as unsaturated bonds and substituted electron withdrawing groups enlisting –F, –O–CN, –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO, S–CN, –CN etc. preferentially get reduced and/or oxidized, forming tailored electrode/electrolyte interphases, prior to the bulk electrolyte activity. This alleviates the detrimental features associated with Si anodes and high capacity cathode materials. Silane-type,26–30 fluoroethylene carbonate (FEC),17,19,31–35 vinylene carbonate (VC),36–38 carbon dioxide (CO2),39–41 pentafluorophenyl isocyanate (PFPI),15 and lithium fluoromalonato(difluoro)borate (LiFMDFB)42 are among the potential electrolyte additives investigated for Si based anode LIBs. However, all these have their own advantages and drawbacks. The high temperature-induced gassing and rapid consumption of FEC16 and formation of a highly resistive SEI (i.e. high impedance) in VC43 based electrolytes are among the critical issues associated with state-of-the-art additives. Hence, the search for more tailored electrolyte additives with multifunctional and/or single functional synergistic roles are of supreme interest.
CO, S–CN, –CN etc. preferentially get reduced and/or oxidized, forming tailored electrode/electrolyte interphases, prior to the bulk electrolyte activity. This alleviates the detrimental features associated with Si anodes and high capacity cathode materials. Silane-type,26–30 fluoroethylene carbonate (FEC),17,19,31–35 vinylene carbonate (VC),36–38 carbon dioxide (CO2),39–41 pentafluorophenyl isocyanate (PFPI),15 and lithium fluoromalonato(difluoro)borate (LiFMDFB)42 are among the potential electrolyte additives investigated for Si based anode LIBs. However, all these have their own advantages and drawbacks. The high temperature-induced gassing and rapid consumption of FEC16 and formation of a highly resistive SEI (i.e. high impedance) in VC43 based electrolytes are among the critical issues associated with state-of-the-art additives. Hence, the search for more tailored electrolyte additives with multifunctional and/or single functional synergistic roles are of supreme interest.
In our previous paper,25 we extensively investigated the role of different electrolyte additives enlisting vinylene carbonate (VC), fluoroethylene carbonate (FEC), tetraethoxysilane (TEOS), (2-cyanoethyl)triethoxysilane (TEOSCN) and their blends, focusing on half-cell configurations. X-ray Photoelectron Spectroscopy (XPS), Density Functional Theory (DFT) calculations and Differential Scanning Calorimetry (DSC) evaluation provided the role of the above-mentioned additives in the interphasial and thermal reactivity of Si-anode based half-cells. In our findings, the SEI formed on the Si-alloy electrode in the presence of 1 M LiPF6 in EC:DEC + 5 wt% FEC/VC/TEOSCN (1.67/1.67/1.67, wt%) was found to be of high quality with tuned beneficial features in terms of mechanical and chemical stabilities, ionic conductivity, and thickness.
Inspired by the obtained results, the authors continued with the optimization of the electrolyte, their validation in pouch-type full cell configuration and investigation of the corresponding governing mechanisms using chemical simulation and X-ray photoelectron spectroscopy (XPS) analysis of both the anode and cathode electrodes. In the present work, superior electrochemical performance on NMC622/Si–Gr pouch cells is obtained by further regulating the amount of TEOSCN and FEC/VC blends in the baseline electrolyte (1 M LiPF6 in EC:DEC). The incorporation of only 1 wt% TEOSCN has incredibly enhanced the high temperature long-term cyclability, obtainable capacity, and coulombic efficiency of the FEC/VC-containing base electrolyte. The improvement is attributed to the beneficial effect of the TEOSCN additive on regulating both the SEI and CEI chemistries.
2. Experimental section
2.1 Preparation of electrolyte solutions
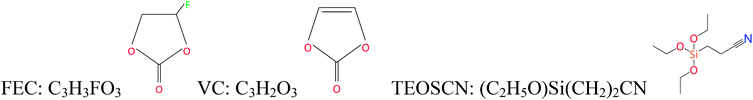
All investigated electrolytes consist of 1.0 M lithium hexafluorophosphate (LiPF6) dissolved in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC), (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as a baseline electrolyte (BE), and in mixtures of fluoroethylene carbonate (FEC) and vinylene carbonate (VC) (i.e. FEC/VC) and FEC, VC, and (2-cyanoethyl)triethoxysilane (TEOSCN) (i.e. FEC/VC/TEOSCN). High quality and battery grade (high purity) BE, VC (97%), and TEOSCN (97%) from Sigma Aldrich and FEC (99.9%) from Solvionic were obtained and used without further purification. The electrolytes were prepared inside an argon-filled glove box (O2 and H2O < 0.5 ppm). The electrolyte compositions used in this work are detailed in Table 1.
:1, v/v) as a baseline electrolyte (BE), and in mixtures of fluoroethylene carbonate (FEC) and vinylene carbonate (VC) (i.e. FEC/VC) and FEC, VC, and (2-cyanoethyl)triethoxysilane (TEOSCN) (i.e. FEC/VC/TEOSCN). High quality and battery grade (high purity) BE, VC (97%), and TEOSCN (97%) from Sigma Aldrich and FEC (99.9%) from Solvionic were obtained and used without further purification. The electrolytes were prepared inside an argon-filled glove box (O2 and H2O < 0.5 ppm). The electrolyte compositions used in this work are detailed in Table 1.
Table 1 Electrolytes compositions used in this study
| Electrolyte ID |
Formulation |
| BE |
1.0 M LiPF6 in EC : DEC (1:1, v/v) |
| BE + FEC/VC |
BE + 5 wt% FEC + 2 wt% VC |
| BE + FEC/VC/TEOSCN |
BE + 5 wt% FEC + 2 wt% VC + 1 wt% TEOSCN |
|
2.2 Electrochemical performance of pouch cells
2.2.1 Pouch cell design.
Sealed and dry machine wound pouch cells were used for the electrochemical long-term evaluation of the above stated electrolyte formulations. The ratio of the first lithiation capacity of the negative electrode and first delithiation capacity of the positive electrode of the pouch cells was 1.13 (i.e. N/P, negative:positive). This was optimized to ensure that no lithium plating would occur at the highest upper cut-off potential (i.e., 4.35 V). The composition of the negative electrode was 17.5 wt% 3 M Si-alloy, 76.6 wt% artificial graphite (BTR-918-II), 3 wt% KS6 (Imerys), 1.7 wt% styrene-butadiene rubber, and 1.2 wt% carboxymethyl cellulose (CMC) as a binder. The pure alloy has a reversible capacity of 1020 mA h g−1 with a first cycle efficiency of 80%, a density of 3.3 g cm−3, a surface area of 6 m2 g−1, and a median particle size of 6 μm. The positive electrode was LiN0.6Mn0.2Co0.2O2 (NMC622). Table 2 shows the general electrode parameters.
Table 2 Full cell information
| Parameter |
Anode |
Cathode |
| Formulation |
76.6% BTR-918-II (artificial graphite)/17.5% 3 M Si-alloy/3% KS6/1.7% styrene-butadiene (SBR)/1.2% carboxymethyl cellulose (CMC) |
95% NMC622/3% Super P/2% polyvinylidene fluoride (PVDF) |
| Areal capacity (mA h cm−2) |
4.2 |
3.7 |
| Coating weight (mg cm−2) |
11.1 |
21.1 |
| Collector |
Copper (Cu) |
Aluminum (Al) |
| Collector thickness (μm) |
15 |
20 |
Neware BTS-4000-5V6A source units, which supply currents in the range of 0.5 mA to 6 A and potential in the range of 25 mV to 5 V with an accuracy of ±0.05% in both cases, were used to charge and discharge the pouch cells. For coin cells, Neware BTS-4000-5 V 10 mA source units were used, which supply currents in the range of 5 μA to 10 mA and potential in the range of 25 mV to 5 V with an accuracy of ±0.05% in both cases.
2.2.2 Electrolyte filling, formation, and cycling protocols.
The cells were first opened inside an argon-filled glove box (LABstar, MBRAUN) with an inert environment (O2 and H2O < 0.5 ppm), followed by drying at 80 °C under vacuum overnight. Following that, the pouch cells were filled (by weight) with electrolyte inside the glove box. For this reason, the cells were placed on a high precision scale (MSA3203P-100_DU, Sartorius AG) and were filled with 3.2 g of electrolyte solution (see Table 1 above). To accelerate the wetting of the active material and separator with electrolyte and thereby removing residual gases out of the pores, the electrolyte filled pouch cells were transferred to an airlock, in which a negative pressure cycle was applied. The airlock was set carefully to a negative pressure of −0.6 atm. After 20 seconds of rest, the negative pressure was reduced again carefully. The build-up and release of the negative pressure must be slow and soft to ensure that the gas escaping from the pores does not blow the electrolyte away. For validation, the cell weight was measured before and after the negative pressure cycle was applied. Thereafter, the pouch cells were vacuum-sealed with a vacuum sealing machine (MSK-115A, MTI KJ Group). As a prior formation step, the cells were allowed to stand at open-circuit voltage (OCV) for 24 hours to ensure complete wetting of the electrodes and separators by the electrolyte. The protocol for the formation was as follows: cells were charged and discharged once with 0.05C for a full potential range of 4.35–2.5 V at 45 °C with a constant weight of 2 psi on top. Thereafter, the cells were taken back to an argon-filled glove box, in which they were opened and resealed under vacuum in order to remove any gas formed during the formation step. Next, the cells were cycled in the full potential range of 4.35–2.5 V using a constant-current constant-voltage charge (CCCV) and constant-current discharge protocol. Charging was performed at a current of 1C and held at the upper cut-off voltage (UCV) until the current decayed to a value of 0.05C. Discharge was done at a current of 0.5C to the lower cut-off voltage (LCV). For characterization of the battery performance, every 25th cycle the current was set to 0.1C with a constant potential step at the UCP until the current again dropped below 0.05C. Between charge and discharge, a rest time of 60 seconds was implemented. The cells were cycled at an elevated temperature of 45 °C in an oven (UFE 500, Memmert GmbH + Co. KG). To ensure a constant pressure on the cells, each pouch cell was weighed all the time with a mass of 2 psi on top.
2.3 Chemical mimicking
2.3.1 Electrode preparation.
Carbon-coated 3 M silicon alloy, MagE3 from Hitachi, SuperP carbon from TIMCAL, lithium polyacrylate (LiPAA), and carboxymethyl cellulose–sodium (CMC) were used as the anode electrode and NMC622 from Custom Cells, as the cathode electrode. The anode electrode slurry containing 88 wt% active material (20 wt% 3 M Si alloy and 68 wt% MagE3), 10 wt% binder (LiPAA and CMC), and 2 wt% SuperP and was coated onto a pretreated and clean copper foil (Electronics Inc.) current collector. LiPAA solution was prepared by mixing 25 wt% aqueous solution of poly(acrylic acid) (PAA, Mw = 240 K, purity 23−27% in solids, Acros Organics), deionized water and lithium hydroxide monohydrate (battery grade, Alfa Aesar) in a 0.401:0.550:0.049 weight ratio. The solution was then heated (<60 °C) with stirring for 7 h, resulting in a 10 wt% solution of LiPAA binder solution of a pH value between 7 and 8. CMC solution was prepared by mixing carboxymethyl cellulose (MTI Corporation) with deionized water in a 0.025:0.975 weight ratio. For the slurry mixing, 3 M Si alloy, MagE3, SuperP, LiPAA, and CMC were mixed in a 0.20:0.68:0.02:0.02:0.07 weight ratio. First, SuperP, LiPAA, and CMC were mixed for 20 min in a planetary micromill (Fritsch Pulverisette 7) at a speed of 800 rpm. Second, MagE3 was added and stirred for 20 min under similar conditions. Third, 3 M Si alloy was added for the last 20 min of the stirring process. The resulting slurry was coated onto 10 μm copper foil using a coating unit (CUF 5, Sumet Systems GmbH) with a 250 μm gap coating Rakel. After that, it was left to air dry for 24 h, out of which electrodes were punched using a high-precision electrode cutter (EL-CELL) of 14 mm diameter. Prior to further usage, the punched electrodes (anodes: 14 mm in diameter; cathodes: 16 mm in diameter) were dried under vacuum (<10−8 mbar) at 90 °C for 12 h.
One-half of the electrodes were assembled into 2332-type coin cells inside an argon-filled glove box using an electrolyte-impregnated borosilicate glass fiber (Whatman GF/D, Sigma Aldrich) as a separator. Previously, the separator (17 mm in diameter) was dried for 12 h under vacuum at 90 °C. 90 μL electrolyte solution (see Table 1) was injected to ensure enough electrolyte for wetting and later forming the passivation layers on both electrodes. Once the 2032-type coin cells were assembled, they were put at open circuit voltage (OCV) for a 24 h rest period to ensure complete electrode wetting. After this resting step, the cells were subjected to C/20 (NMC622 cathode (from Custom cells)) and our own coated anode, composed of 88% wt active material (20% wt% 3 M Si-alloy and 68% MagE3), were matched, with an estimated capacity of the coin cell and corresponding current for C/20. Finally, from the cycled coin cell and obtained real capacity, the real current, for C/20, 0.154 mA h or 0.1 mA h cm−2 (in which it is normalized to our coin cell active area of 1.54 cm2), was determined. The cycling condition were between 4.35 V and 2.5 V at 45 °C. Before the post mortem analysis, three cycles for each coin cell were performed. The other half of the electrodes were submerged into the electrolyte solutions and stored for 24 h at 45 °C. All recovered electrodes (from coin cells and dipping pretreatment) were washed carefully in extra dry dimethyl carbonate (DMC, Acros Organics B.V.B.A.) to remove all residual salt and were dried under vacuum (<10−8 mbar) at RT for 12 h.
2.3.2 XPS measurement.
The samples were transferred into an XPS instrument (Phi5000 VersaProbe II, ULVAC-Phi Inc., USA) in an air-proof sealed transfer vessel. Al Kα radiation with an energy of 1486.3 eV, pass energy of 23.5 eV and 15 kV filament voltage source energy was used, and the charge neutralizer was switched on to compensate the charging of the sample. The XPS spectra were fitted by using the CasaXPS software (Version 2.3.16 PR 1.6, Casa Software Ltd., U.K.). The calibration of the binding energy of the recorded spectra was conducted using the C 1s C–C peak (i.e., 284.5 eV) as an internal reference.
3. Results and discussion
3.1 Electrochemical performance of NMC/Si–Gr pouch cells
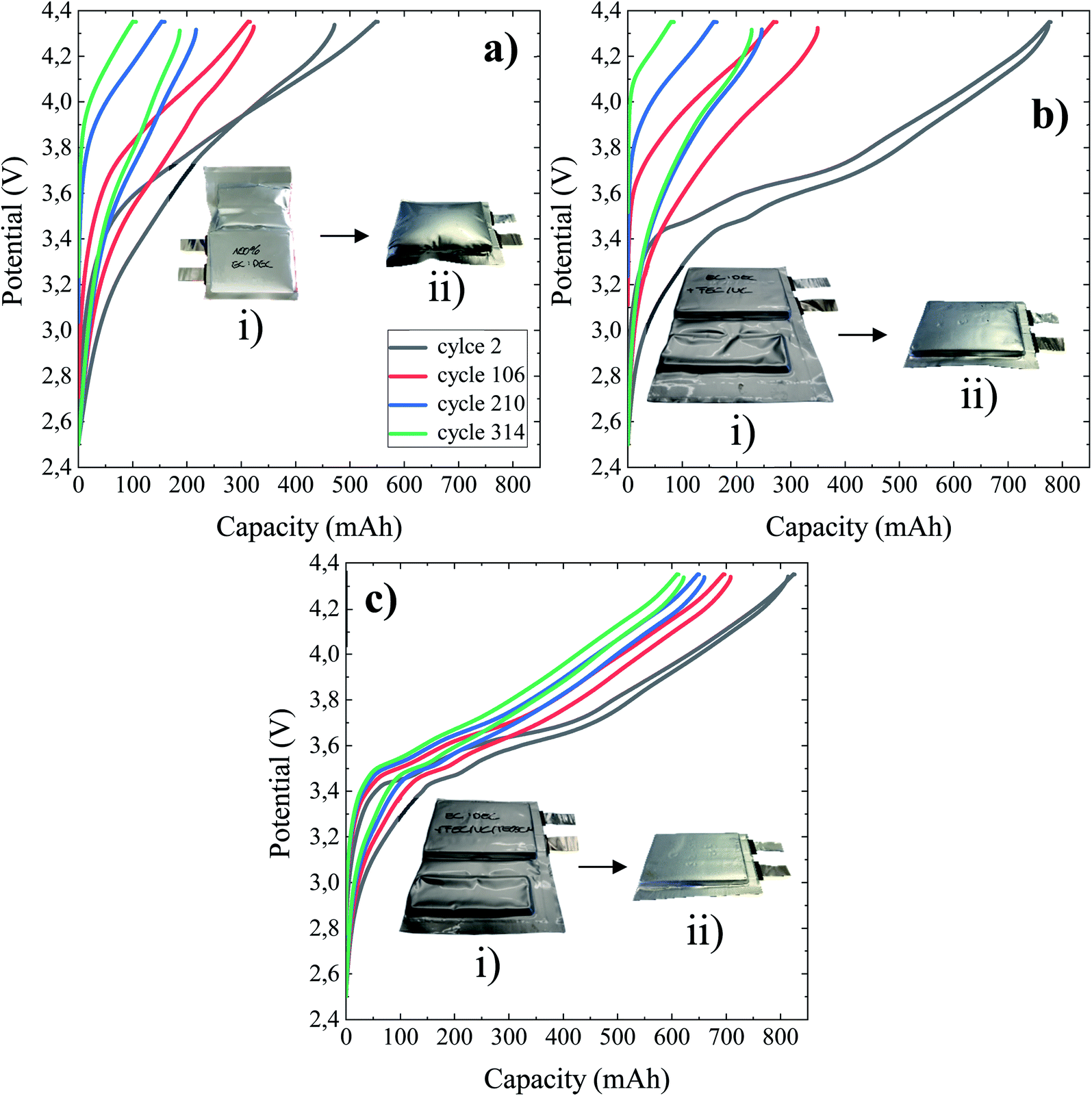
The potential versus capacity profiles of NMC/Si–Gr pouch cells at the 2nd, 106th, 210th, and 314th cycles using 1 M LiPF6 in EC:DEC (BE), BE + 5%FEC/2%VC and BE + 5% FEC/2% VC/1% TEOSCN as electrolytes at 45 °C are given in Fig. 1 of panels (a), (b), and (c), respectively. Inset figures refer to photographs of the pouch cells of the respective panels: (i) after the formation step and (ii) at the end of their cycling life (364th cycle). As can be clearly seen, the capacity decay and hysteresis between the cycles are highly pronounced for pouch cells cycled in the BE and BE + FEC/VC electrolytes. This could be linked to the formation of unstable and high impedance passivation layers in the electrolytes with and without the FEC/VC additive, respectively.43 To further substantiate the above differences, photographs of the pouch cells immediately after the formation step (Fig. 1a(i), b(i), and c(i)) and long-term cycling (Fig. 1a(ii), b(ii), and c(ii)) are given as inset figures. One can evidently notice that the pouch cell without additives is swelled with excess gassing in both cases, indicating an accelerated decomposition of the electrolyte, resulting in capacity decay. The gases generated during the formation step were degassed before the battery is sealed, and thus, further gas generation could be caused by additional electrolyte degradation as a result of the failure of the pre-formed SEI. Though not noticeable after the formation step, moderate swelling is seen after the end of cycling for the pouch cell cycled in the presence of the FEC/VC additive. This could be linked to the potential thickening and thereby increasing impedance of the SEI layer due to parasitic electrolyte reductions.43 Interestingly, no sign of swelling is observed for the cell cycled in the presence of 1 wt% TEOSCN, evidencing the formation of a high quality and stable SEI during the formation stage.25
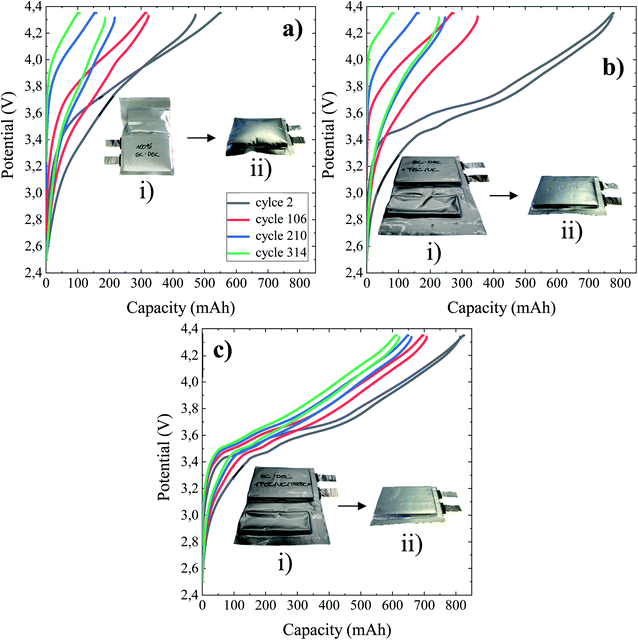 |
| | Fig. 1 Potential vs. capacity profiles of 790.53 mA h NMC622/Si–Gr pouch cells cycled at 45 °C between 4.35 V and 2.5 V in (a) BE, (b) BE + 5 wt% FEC/2 wt% VC, and (c) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. A current of 0.1C with a constant potential step was used and maintained at the highest potential until the current dropped below 0.05C. Inset figures are photographs of the respective pouch cells after formation (i) and end-of cycling life (ii). | |
The capacity performance of LIB cells evolves over time, usually crumbling at each consecutive cycle. Capacity loss in the full-cell configuration can occur at least via three routes: (i) a portion of Li supplied by the cathode electrode is irreversibly consumed at the anode side, which is followed by further Li loss due to SEI formation, (ii) diffusion limited Li trapping, and (iii) reduction of other battery elements such as binding agents. In view of this, the long-term cycling and accompanying coulombic/energy efficiency evaluation of the binary (FEC/VC) and ternary (FEC/VC/TEOSCN) electrolyte additives in pouch cells are of paramount importance. The charge/discharge cycling was conducted at 1C and 0.5C, respectively, and after every 25 cycles, a current corresponding to 0.1C was applied to access the real (actual) capacity. The discharge capacity vs. cycle, change in capacity of the high vs. low current cycling, coulombic efficiency, and accumulated coulombic inefficiencies (ACIE) of the NMC622/Si–Gr pouch cells of nominal capacity of 790.53 mA h are given in Fig. 2a–d. The cell cycled in the BE showed lower initial discharge capacity (471.43 mA h @ 0.1C), escorted with lower capacity retention upon cycling. The discharge capacity in the 2nd cycle at 0.5C is 454.9 mA h, which is about 57.54% of the nominal capacity. The lower capacity can be attributed to the rapid consumption of the electrolyte during SEI formation as also evidenced with the excess gassing, and thus leading to a large amount of dead Li. Though it has a higher initial capacity (∼97.96% of nominal value), FEC/VC-containing electrolyte also demonstrated rapid capacity decay, providing a capacity of ∼8.05% after 364 cycles, i.e. less than the control electrolyte (∼19.23%). Incredibly, the addition of 1 wt% TEOSCN to the binary (i.e. FEC/VC)-containing electrolyte resulted in a much more improved cycling performance, retaining ∼75.95% of its initial capacity at the 364th cycle. Though direct comparison due to different systems is not possible, the results of this study showed much more improved performances compared to those reported in the literature for full cell configurations.44–47Fig. 2b presents the difference in capacity between low (0.1C/0.1C) and high (1C/0.5C) current charge/discharge profiles for selected cycles (e.g., 106/107 refers to cycle at low/high current). After 100 cycles, visible dissimilarities can be perceived for the cells cycled with TEOSCN devoid electrolytes. The deviation could be related to an increase in the internal resistances of the different samples. While the difference remains constant at ∼2% for the ternary additive containing electrolyte, those cycled in the baseline and FEC/VC-containing electrolytes revealed significant differences, which increase with increasing number of cycles. This indicates that the internal resistance is lower for the –CN functionalized silane containing electrolyte, compared to others. Improvements linked to the incorporation of the TEOSCN electrolyte additive could be ascribed to its ability to form a high quality inorganic rich SEI layer, characterized by better chemical, mechanical and thermal properties, lower solubility, and higher shear strength, because of LiF.25 On the cathode side, the oxidative decomposition of nitrile groups is proved to result in the formation of tunable cathode/electrolyte interphase, which is responsible for high-voltage stability and thus improving the performance of –CN-containing electrolytes.48 It has to be accentuated that nitrile functional groups are intrinsically stable against oxidation in isolated states (i.e. in the absence of co-solvent(s), and salt anions, e.g., Li+ PF6−), however, they get decomposed (oxidized) before the solvent molecules in the presence of salt anions, leading to the formation N-containing robust interphases both on the highly polarized anode and cathode surfaces (cf. XPS section). Fig. 2c presents the coulombic efficiency of pouch cells cycled without and with the TEOSCN additive. Battery cells cycled in the FEC/VC/TEOSCN-containing electrolyte demonstrated an average charge efficiency of ∼99.46%, compared to ∼98.90% and ∼98.67% for the BE and with FEC/VC respectively at the end of the 364th cycle at high current, 1C/0.5C charge/discharge. Unlike BE and FEC/VC-based electrolytes, it shows less jumps during the actual capacity tracing cycles (i.e. every 25th cycle), all indicating the formation of robust CEI/SEI layers.
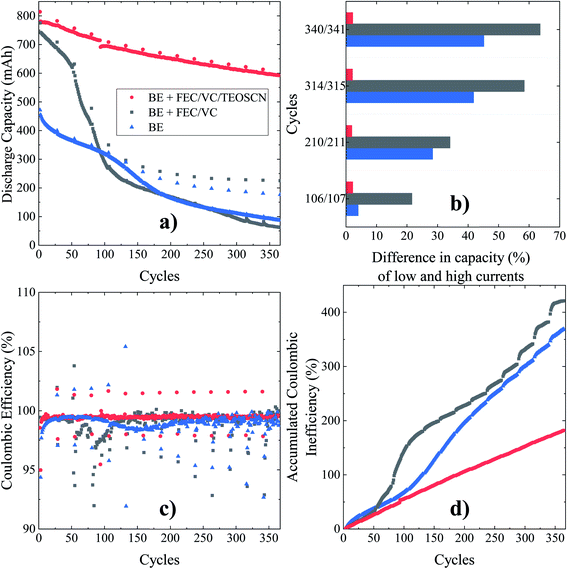 |
| | Fig. 2 Long-term electrochemical performance (a) discharge capacity vs. cycles, (b) difference in capacity of low and high current cycles, (c) coulombic efficiency, and (d) accumulated coulombic inefficiencies of 790.5 mA h NMC622/Si–Gr pouch cells cycled at 45 °C between 4.35 V and 2.5 V at charge and discharge currents of 1C and 0.5C, respectively. A constant potential step at top of charge was applied until the current dropped below 0.05C. A 0.1C cycle with the same constant potential step was performed every 25th cycle. | |
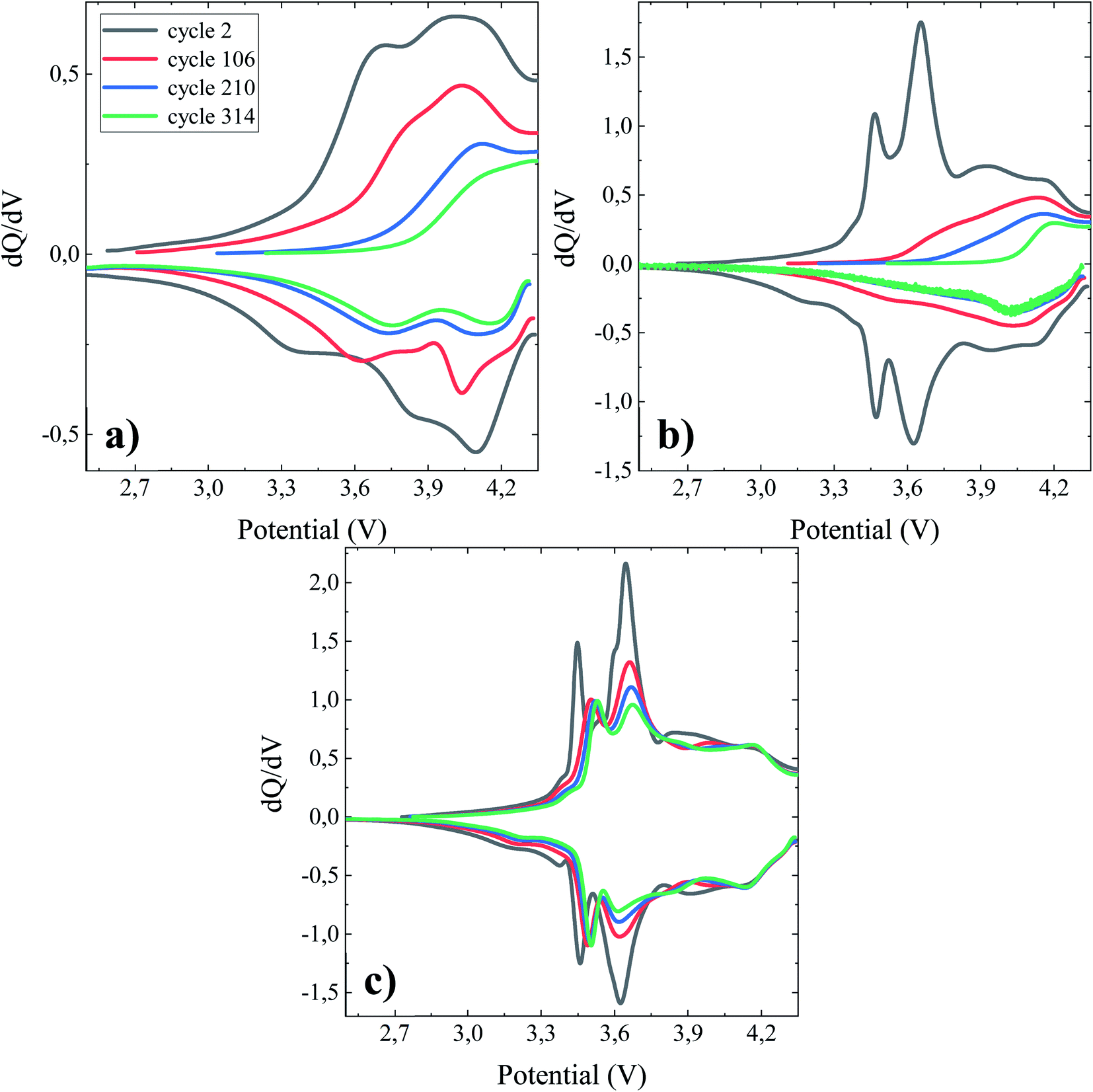
For a full cell configuration using highly reactive anode materials (ca. Li, Si), the accumulated coulombic inefficiency (ACIE), i.e. the sum of the coulombic inefficiencies over a number of cycles, is predominantly attributed to SEI formation.49 Accordingly, it can be considered an indicator of the active Li loss, or the degree of the inactive or dead Li accumulation linked to the SEI formation. In agreement with the data presented in Fig. 2d, the cell containing the TEOSCN additive exhibited the lowest ACIE (∼181.75%) compared to the baseline (∼369.00%) and FEC/VC (∼421.31%) containing one at the end of the 364th cycle. The less steady slope seen with TEOSCN containing electrolyte once again proves the formation of a robust SEI and thereby high coulombic efficiency, compared to the other electrolyte formulations. Fig. 3 demonstrates the differential capacity (dQ/dV) vs. potential (V) curves for NMC622/Si–Gr pouch cells cycled at 45 °C, between 4.35 V and 2.5 V. For full cells, evaluation of the contributions of each electrode, i.e. changes in the negative and positive electrode peaks or the relative electrode slippages, are crucial. However, analyzing these features is not straightforward, instead, the dQ/dV plot can provide insight into accessing some distinguishable characteristics, enlisting changes in the peak position and peak width, which can give rise to invaluable information about the prevailing parasitic reactions and material loss.50 Based on Ohm's law (U = I × R), shifting in position, decreasing in intensity and widening of the peaks are related to an increase in the internal resistance, decrease in the activity of the electrochemical process and chemical changes in the electrode materials respectively.51 For the BE, the electrochemical activity is weak as can be seen from the broad dQ/dV plot (Fig. 3a. Except for the 2nd cycle, the pouch cell cycled in the presence of FEC/VC also showed a sharp decrease in intensity, corroborating the decrease in the obtainable capacity. The cell cycled with the TEOSCN-containing electrolyte, on the other hand, presented well-defined peaks after the 314th (even after the 364th) cycle. The potential differences (i.e. peak shift) between the 2nd and the 314th cycles are found to be 0.54 V, 0.71 V and, 0.08 V for the pouch cells cycled in the BE, FEC/VC and FEC/VC/TEOSCN containing electrolytes respectively (Fig. 3a–c),). This again confirms that the highest and lowest internal resistances are found for FEC/VC and its TEOSCN bearing derivative electrolyte additives respectively. The intensity of the first peak is in the ratio of 1:1.2:4.13 for the BE and electrolytes containing FEC/VC and FEC/VC/TEOSCN respectively (Fig. 3a–c), clearly depicting the difference in the electrochemical activities of the electrolytes and electrode materials.
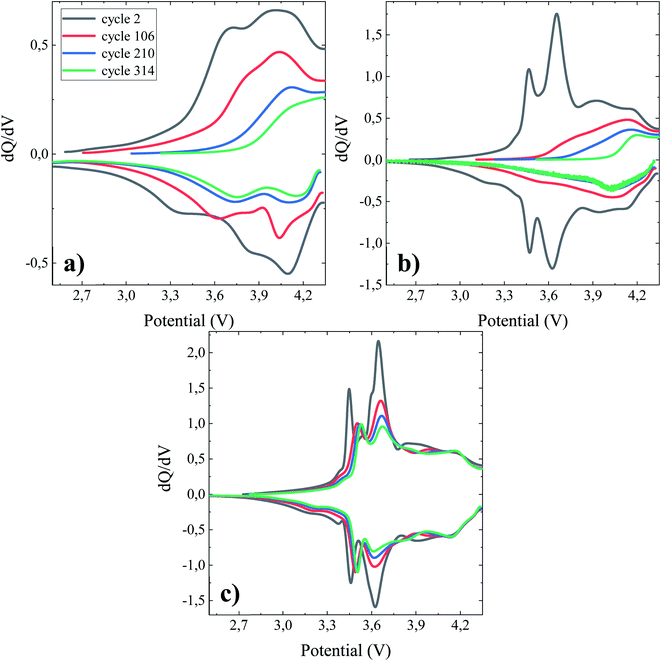 |
| | Fig. 3 dQ/dV vs. potential profiles of 790.53 mA h NMC622/Si–Gr pouch cells cycled at 45 °C between 4.35 V and 2.5 V at a current of 0.1C with a constant potential step until the current dropped below 0.05C. Pouch cells were filled with different electrolyte formulations (a) BE, (b) BE + 5 wt% FEC/2 wt% VC, and (c) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. | |
3.2 Postmortem analysis: mechanistic understanding
Aiming at gaining deeper insight and understanding of the role of the additives, XPS analysis of the surface chemistry of both Si–Gr anode and NMC622 cathode electrodes, both recovered from cycled NMC622/Si–Gr full cells, has been conducted. Moreover, in order to differentiate the origin of the electrolyte decomposition, measurements on simply mounted (without passing current) and electrochemically cycled electrode samples were performed.
3.2.1 Mimicking chemical reactivity of the Si–Gr electrode towards electrolyte additives.
The surface of Si is always covered with an insulating native film of oxide layer (amorphous SiOx, x ≤ 2), terminated with active –OH (silanol) moieties.3,16,48,52 Such a reactive surface, in the presence of electrolyte solutions, leads to various electrochemical and chemical reactions (e.g., forming organosiloxane species). Moreover, the reduction of the –OH termination can result in the generation of a highly reactive surface ending with Si–H, further accelerating the electrolyte decomposition.53
At first, an XPS study was carried out aimed to check the chemical reactivity (if exist), between the Si–Gr surface/surface moieties and the corrosive electrolyte solutions containing the above-mentioned additives. To this purpose, Si–Gr electrodes in electrolytes with and without additives were mounted and stored without any electrochemical cycling at 45 °C for 24 h. Then after, the retrieved electrodes were washed with DMC to remove any residual salt and were vacuum dried before XPS analysis. Fig. 4 depicts the Si 2p spectra of the Si–Gr electrode dipped in an electrolyte solution of: the BE (Fig. 4(i)), BE + FEC/VC (Fig. 4(ii)), BE + TEOSCN (Fig. 4(iii)) and BE + FEC/VC/TEOSCN (Fig. 4(iv)). The Si 2p3/2 fitting peaks at 99.6, 101.4, and 103.5 eV are assigned to elemental bulk Si (Si0, Si–Si), SiOx sub-oxides, and a surface fully terminated with SiO2 (Si4+), respectively (see reactions (1), (2), and (3)). The SiO2/Si area ratio remains the same for the baseline and electrolyte solutions containing FEC/VC, and FEC/VC/TEOSCN, with a slightly lower value for the latter. However, the electrolyte containing TEOSCN (Fig. 4(iv)) presented a much lower amount of Si○ and native (sub-) oxides (i.e. SiOx and SiO2), which could be explained by the possible Si surface modification via chemical reaction with the –CN ending functionality. Distinctively, the electrode dipped in the electrolyte solution containing TEOSCN presents an additional peak at a higher binding energy (106.6 eV), possibly linked to the partially fluorinated silicon sub-oxides (SiOxFy, y ≤ 3)54,55 (see reaction (4)) and/or Si–F.56 The generation of SiOxFy is due to the reaction of surface oxide (i.e., SiO2) with hydrofluoric acid (HF), resulting from LiPF6 hydrolysis, in the presence of traces of water (see reaction (5)). The Si–F bond, on the other hand, can originate from the attack of HF on the Si–Si bond, resulting in the loss of active sites.
| | | Si + 2H2O → SiO2 + 2H2↑ | (1) |
| | | 2Si + O2 + 2H2O → 2SiO2 + 2H2↑ | (3) |
| | | SiO2 + HF → SiOxFy + H2O | (4) |
| | | LiPF6 + 2H2O → LiF + 2HF↑ + POF3↑ | (5) |
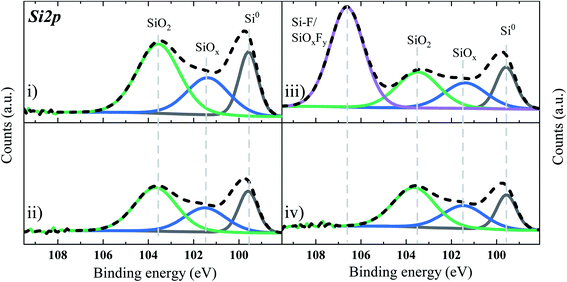 |
| | Fig. 4 Si 2p XPS core spectrum of the Si-based anode electrode dipped in four electrolyte solutions: (i) BE, (ii) BE + 5 wt% FEC/2 wt% VC, (iii) BE + 1 wt% TEOSCN, and (iv) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. | |
In conclusion, the chemical mimicking evidenced that TEOSCN additive based electrolyte solution resulted in a surface modification via chemical reaction between the Si surface and the –CN moieties.
3.2.2 Electrochemically cycled silicon-based anode electrode.
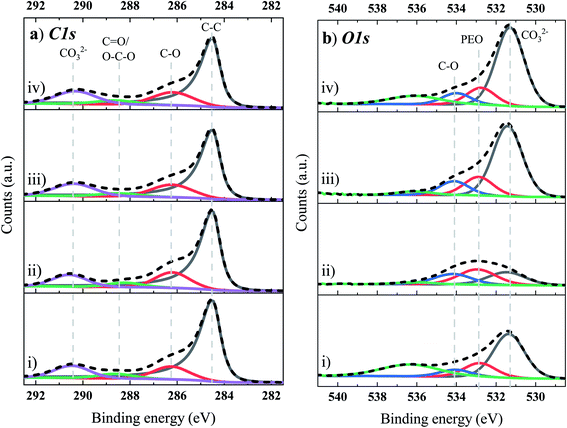
Fig. 5 presents the C 1s (Fig. 5a) and O 1s (Fig. 5b) XPS core spectra of the Si/Gr anode obtained from the NMC622/Si–Gr full cell, cycled in the range of 2.5 to 4.35 V, in the four different electrolyte systems. From the deconvolution of the C 1s peaks, a dominant peak for each electrolyte system is observed at ∼285 eV, originating from the C–C (sp2) hybridized graphite (C6). However, it can also likely include contributions from the C–C of SuperP (conductive carbon), C–N, C–Si, and a mix of alkane and alkyl groups linked with a variety of SEI functionalities on the lithiated Si surface (oligomers, side chains, and molecules) and that of the polymeric binders (CMC and LiPAA).57 The higher atomic concentration of the peak at 285 eV for TEOSCN and FEC/VC/TEOSCN containing electrolytes corroborates the presence of C–N and C–Si moieties. The peak at 283.1 eV is linked to lithiated graphite (i.e., LixC6, reaction (6) and/or lithium carbide (Li2C2) reaction (7)) species.57 As this peak refers to the graphite substrate and not to the SEI species, its existence indicates that the SEI layer, obtained from all electrolyte solutions, is not very thick or complete. As indicated from the atomic concentration (at%) of this peak, the thickness and/or degree of surface coverage increases in the order BE (3.8%) < BE + FEC/VC (2.7%) < BE + TEOSCN (2.0%) < BE + FEC/VC/TEOSCN (1.4%). The C 1s peak at 286.6 eV corresponds to a carbon atom surrounded by one oxygen, as in the LiOR, and a poly(ethylene oxide) oligomer (PEO, –CH2–CH2–O–)n, resulted from the ethylene carbonate (EC) ring-opening polymerization.57–59 Moreover, it could also be related to the ether carbon in (CH2OCOLi)2 and in other related environments.54 The binding energies at ∼288.5 eV and 290 eV correspond to the carboxylate carbon O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (O)R such as in lithium alkyl carbonates (LiCO3R, with R as different long-chain alkyl groups) and lithium carbonate (O–(CO)–O/CO3), respectively. The evaluation from the atomic concentration evidently shows lower amounts of PEO, ROCO2Li and Li2CO3 for the cells cycled in the TEOSCN containing electrolyte, evidencing the inhibition of EC and DEC reductions. Cells cycled in all of the four electrolyte systems revealed three distinct peaks in the O 1s spectrum (Fig. 5b), centered at 530 eV (CO), 531.8 eV (ROCO2Li/Li2CO3), and 533.7 eV (–(CH2–CH2–O)n–), all substantiating the above-mentioned C 1s spectra (Fig. 5a). Except for the electrolyte with TEOSCN, other electrolytes also presented an additional peak centered at 528.2 eV, linked to Li2O, resulting from the reduction of SiO during the first cycle (see reactions (8) and (9)).22,60–62
(O)R such as in lithium alkyl carbonates (LiCO3R, with R as different long-chain alkyl groups) and lithium carbonate (O–(CO)–O/CO3), respectively. The evaluation from the atomic concentration evidently shows lower amounts of PEO, ROCO2Li and Li2CO3 for the cells cycled in the TEOSCN containing electrolyte, evidencing the inhibition of EC and DEC reductions. Cells cycled in all of the four electrolyte systems revealed three distinct peaks in the O 1s spectrum (Fig. 5b), centered at 530 eV (CO), 531.8 eV (ROCO2Li/Li2CO3), and 533.7 eV (–(CH2–CH2–O)n–), all substantiating the above-mentioned C 1s spectra (Fig. 5a). Except for the electrolyte with TEOSCN, other electrolytes also presented an additional peak centered at 528.2 eV, linked to Li2O, resulting from the reduction of SiO during the first cycle (see reactions (8) and (9)).22,60–62| | | C6 + xLi+ + xe− → LixC6 | (6) |
| | | CO32− + 7Li+ + 5e− → 3Li2O + 0.5Li2C2 | (7) |
| | | SiO + 2Li+ + 2e− → Li2O + Si | (8) |
| | | SiO2 + 4Li+ + 2e− → 2Li2O + Si | (9) |
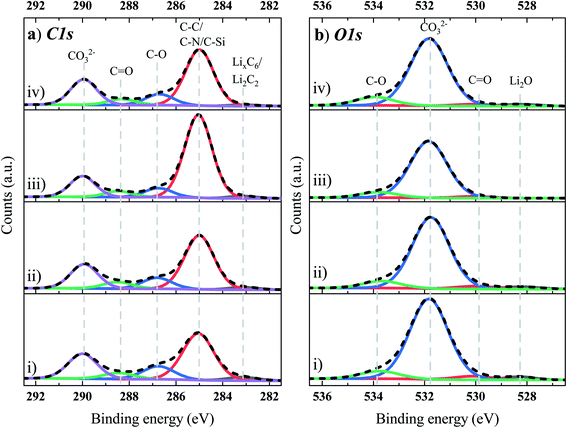 |
| | Fig. 5 (a) C 1s and (b) O 1s XPS core spectra of the Si-based anode electrode cycled in four electrolyte solutions: (i) BE, (ii) BE + 5 wt% FEC/2 wt% VC, (iii) BE + 1 wt% TEOSCN, and (iv) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. | |
The Li 1s core spectra (Fig. 6a) present two peaks at 54 eV and 55.6 eV for the BE (Fig. 6a–i) and the BE + FEC/VC (Fig. 6a(ii)) and one peak centered at 55.6 eV for the BE + TEOSCN (Fig. 6a(iii)) and the BE + FEC/VC/TEOSCN (Fig. 6a(iv)). While the weak intensity peak at 54 eV is associated with Li-lattice and/or Li2O, the more predominant one at 55.6 eV is linked to Li bound inorganic species such as LiF, Li2CO3etc. The absence of the peak at 54 eV for TEOSCN containing electrolytes once again confirms that the SEI built does not contain Li2O or if it is present, only in a tiny amount (i.e., below detection limit). In the Si 2p XPS core spectra (Fig. 6b), the fitting peaks in the range of 101–105.5 eV are attributed to LixSiOy, SiOx (Si2+/Si3+) and SiO2 (Si4+).25 The F 1s XPS core spectra (Fig. 6c) portray two characteristic peaks in the range of 685.1–687.5 eV and 687.1–687.4 eV, linked to LiF and LiPF6/LixPFy/LixPFyOz/OPF3−y(OR)y, respectively63 (see reactions (10), (11), and (12)). Among the tested additives, TEOSCN (followed by FEC/VC/TEOSCN) presented the highest amount of LiF. This could be explained due to the formation of strong nucleophiles, resulting from the reduction of the –CN, facilitating the stripping of –F to form LiF from LiPF6.25
| | | PF5 + 2xLi+ + 2xe− → LixPF5−x + xLiF | (10) |
| | | PF5 + H2O → OPF3 + 2HF | (11) |
| | | OPF3 + nLi+ + ne− → LixPOyFz + LiF | (12) |
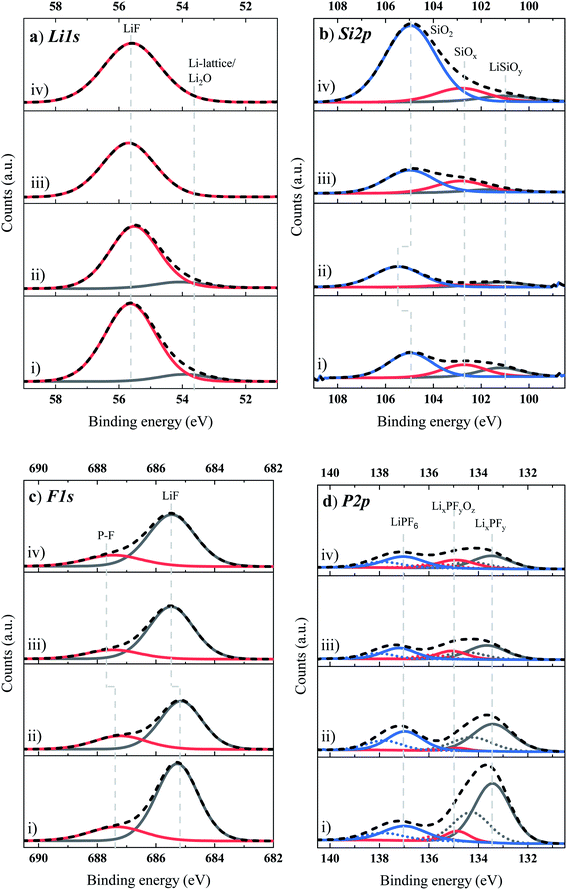 |
| | Fig. 6 (a) Li 1s, (b) Si 2p, (c) F 1s, and (d) P 2p XPS core spectra of the Si-based anode electrode cycled in four electrolyte solutions: (i) BE, (ii) BE + 5 wt% FEC/2 wt% VC, (iii) BE + 1 wt% TEOSCN, and (iv) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. | |
The P 2p XPS core spectra (Fig. 6d) reveal the existence of three distinct peaks at 133.4, 135, and 137 eV, associated with LixPOyFz, LixPFy and LiPF6, respectively. For the oxygenated species (LixPOyFz), TEOSCN containing electrolytes presented a lower amount compared to the BE and BE + FEC/VC, which could be linked to the inhibition of POF3 formation.
3.2.3 Electrochemically cycled NMC622 cathode electrode.
To shed light on the effect of the different electrolyte additives on the composition and structure of the cathode-electrolyte interphase (CEI), the XPS of pristine NMC622 composite electrodes was carried out, and surface chemistry of NMC622 electrodes, obtained from dipped NMC622 (in contact with electrolyte, but without cycling) and the cycled NMC622/Si–Gr cell, was evaluated. The XPS analysis was first conducted on a pristine NMC622 composite (combination of NMC622 active material, PVDF binder and C sp conductive carbon) electrodes and dipped/mounted NMC622 composite electrodes (i.e., NMC622 composite in contact with electrolyte solution). The obtained data represents a useful dataset for the analysis of the XPS spectra of all electrodes, permitting to evaluate the origin of the CEI film and thus to discriminate the impact of each component of the battery cell.
The binding energies of reference materials are reported in Table 3. For the pristine composite electrode (Table 3a), the C 1s peaks at 284.5, 286.2, 287.2, and 289/290.5 eV are in agreement with C–C/CC (carbon conductive), C–O residual, C–H (in PVDF), and C–F (in PVDF and shake-up satellite structure, π→π* transition, of carbon conductive),63–65 respectively. For the O 1 s spectra, binding energies of 529.6531.8, and 532.9/534.4 eV are attributed to the metal–oxygen lattice bond (M–O), CO, and C–O species, respectively.
Table 3 XPS binding energies of reference cathode material analyzed in this work
| Reference material |
Li 1s |
C 1s |
O 1s |
F 1s |
| (a) NMC622 (pristine) |
55.4 |
284.5 |
529.6 |
685.1 |
|
|
286.2 |
531.8 |
687.8 |
|
|
287.9 |
532.9 |
689.6 |
|
|
289.0 |
534.4 |
|
|
|
290.5 |
|
|
| (b) NMC622 (composite in contact with electrolyte) |
BE |
53.9 |
284.5 |
530.0 |
685.9 |
| 56.7 |
286.2 |
531.8 |
687.7 |
|
|
287.8 |
533.3 |
689.6 |
|
|
288.8 |
535.0 |
|
|
|
290.7 |
|
|
| BE + FEC/VC |
— |
284.5 |
529.7 |
684.9 |
|
|
286.1 |
531.4 |
687.7 |
|
|
287.8 |
532.9 |
689.5 |
|
|
288.4 |
534.2 |
691.4 |
|
|
290.6 |
|
|
| BE + TEOSCN |
56.7 |
284.4 |
529.9 |
685.8 |
|
|
286.2 |
531.7 |
687.7 |
|
|
288.2 |
533.2 |
689.5 |
|
|
290.6 |
534.9 |
|
| BE + FEC/VC/TEOSCN |
— |
284.5 |
529.6 |
685.0 |
|
|
286.2 |
531.2 |
687.9 |
|
|
288.0 |
532.9 |
698.7 |
|
|
289.1 |
534.3 |
691.7 |
|
|
290.6 |
536.1 |
|
In order to investigate the chemical reactivity of the electrolyte solutions with the pristine NMC622 composite electrode under OCV conditions, NMC622 electrodes with the electrolytes were stored for 24 h before being analyzed by XPS (Table 3b). This allows the distinction between chemical and electrochemical reactions contributing to the CEI chemistry. The XPS spectra of the composite NMC622 material that was stored for 24 h in contact with the electrolyte solutions presented a chemical composition similar to the pristine composite cathode. However, it is worth noting that the composite electrode in contact with the electrolyte displays a new peak at 56.7 eV (besides the Li-lattice at 55.4 eV) in the Li 1s region ascribed to the presence of LiF.63 In the F 1s spectra, species attributed to the PVDF binder, LiPF6 and its decomposition products are detected. In general, the XPS analysis of the pristine and dipped composite electrode materials shows the absence of significant reaction of the electrolyte components with electrode active materials.
Fig. 7 shows the evolution of the C 1s and O 1s XPS core spectra of the CEI formed on NMC622 electrodes, cycled with or without the above-mentioned electrolyte additives. As evidenced in detail below, there are significant changes for the cycled electrodes compared to the pristine and dipped ones. The main peak in the C 1s XPS core spectra (Fig. 7a) at 284.4 eV originates from C–C (sp3)/CC (sp2) (i.e. conductive carbon), and C–F (i.e., PVDF of the NMC composite electrodes). The binding energies at 286.2, 288.4, and 290.5 eV correspond to the C–O/–CN/–CF2–, CO and –CF2/CO32−, respectively.64,66 The XPS spectrum in the O 1s region (Fig. 7b) depicts three characteristic peaks at 531.4, 532.8 (C![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) such as Li2CO3), and 534.1 eV, associated with the more oxidized oxygen state such as in peroxide-like O− ions, and (–CO), respectively. Moreover, the spectra at ∼532.8 eV and ∼533 eV are clearly reminiscent peaks of weakly adsorbed oxygen surface species. On the cathode, the presence and strength of the C–C/CC and M–O peaks estimates the thickness of the CEI layer. From the atomic concentration of the peak at 284.4 eV, the thickness follows the trend: BE + TEOSCN ≈ BE + FEC/VC/TEOSCN > BE + FEC/VC > BE. However, it is noteworthy that the absence of a peak in the range of 528–530 eV (which was observed for the pristine and NMC622 in contact with electrolyte) linked to the M–O bond indicates the formation of a surface film on the NMC622 electrode that has a thickness beyond the XPS penetration depth.
such as Li2CO3), and 534.1 eV, associated with the more oxidized oxygen state such as in peroxide-like O− ions, and (–CO), respectively. Moreover, the spectra at ∼532.8 eV and ∼533 eV are clearly reminiscent peaks of weakly adsorbed oxygen surface species. On the cathode, the presence and strength of the C–C/CC and M–O peaks estimates the thickness of the CEI layer. From the atomic concentration of the peak at 284.4 eV, the thickness follows the trend: BE + TEOSCN ≈ BE + FEC/VC/TEOSCN > BE + FEC/VC > BE. However, it is noteworthy that the absence of a peak in the range of 528–530 eV (which was observed for the pristine and NMC622 in contact with electrolyte) linked to the M–O bond indicates the formation of a surface film on the NMC622 electrode that has a thickness beyond the XPS penetration depth.
 |
| | Fig. 7 (a) C 1s and (b) O 1s XPS core spectra of the NMC cathode electrode cycled in four electrolyte solutions: (i) BE, (ii) BE + 5 wt% FEC/2 wt% VC, (iii) BE + 1 wt% TEOSCN, and (iv) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN. | |
The binding energies in the range of 55.0–60.5 eV in the Li 1s XPS core spectra (Fig. 8a) are ascribed to PF6− (i.e. LiF, LixPFy) and carbonate solvents, EC, and/or DEC (ROCO2Li and/or Li2CO3) decomposition products.67,68 The more number of peaks observed for BE and BE + FEC/VC based electrolytes depict a higher degradation rate of the salt and solvents compared to the TEOSCN and BE + FEC/VC/TEOSCN ones. The F 1s XPS core spectra (Fig. 8b) show two characteristic peaks, in the range of 684.7–685.3 eV and 687.5–687.6 eV, associated with LiF and LixPOyOz/CH2CF2, respectively. The amount of LiF is found to be in the order of BE + FEC/VC (33.9%) > BE (24.6%) > BE + FEC/VC/TEOSCN (5.0%) > BE + TEOCN (3.5%). This shows that the decomposition of the electrolyte salt is strongly inhibited by the presence of –CN functionalized silane additives. The P 2p XPS core spectra (Fig. 8c) reveals the existence of three distinct peaks at 133.4, 135, and 137 eV, associated with LixPOyFz, LixPFy and LiPF6, respectively.64 Various authors attributed the anodic stability of nitrile-containing electrolytes to the preferential chemisorption of nitrile molecules on the transition metal (TM) oxide surface.69–72 This generates a layer of (–CN–TM) complexes, which physically hinder the alkyl carbonate solvents from intimate contact with the cathode active material. However, Li and et al. recently argued that nitrile functionalities undergo rather an oxidative decomposition at high voltages, leading to the formation of a regulated new interphasial chemistry on the cathode surface.48 The authors proved that though the oxidation stability of isolated –CN moieties is higher than that of carbonate solvents, it is oxidized prior to carbonates in a solution containing Li+–PF6−.
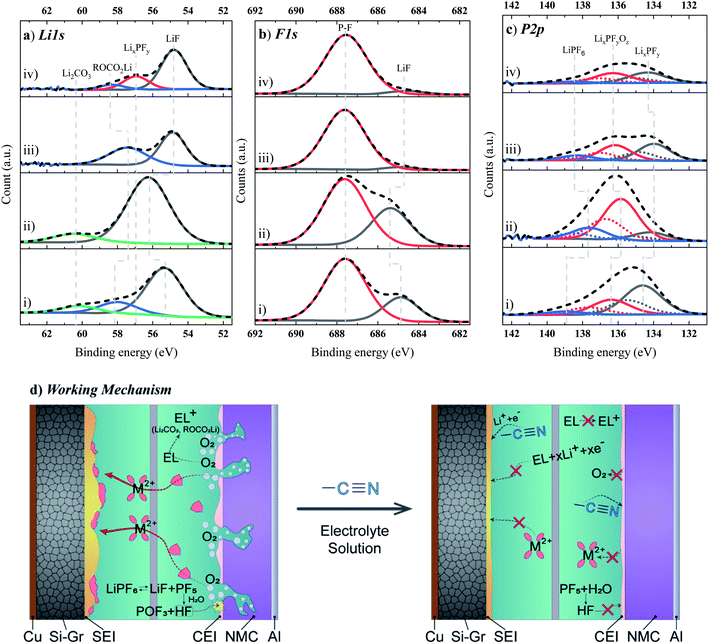 |
| | Fig. 8 (a) L 1s, (b) F 1s and (c) P 2p XPS core spectra of the NMC cathode electrode cycled in four electrolyte solutions: (i) BE, (ii) BE + 5 wt% FEC/2 wt% VC, (iii) BE + 1 wt% TEOSCN, and (iv) BE + 5 wt% FEC/2 wt% VC/1 wt% TEOSCN, and (d) working mechanism of the –CN–based additive. | |
In summary, the working mechanism of –CN-based additives is provided in Fig. 8d. On the basis of previous studies and the electrochemical performances evidenced in this study, the additive is found to have a synergistic beneficial effect by forming robust passivation layers both on the Si–Gr anode and NMC622 cathode materials via reduction and oxidation respectively, prior to the bulk electrolyte components. Thus, the improvement in the electrochemical performance of NMC622/Si–Gr full cells results from the combination of two functional moieties (i.e. silane and nitrile) within one molecule. The peculiar features of TEOSCN and its role as a precursor in the formation of highly tuned electrode/electrolyte interphases allows the inhibition of transition metal cation dissolution from the cathode electrodes (i.e. via limiting formation and direct reaction of acidic species such as (HF, and POF3), with NMC), oxygen evolution, structural disorder etc., which are known to accelerate the degradation phenomena in the Ni-rich NMC cathode.
4. Conclusion
In conclusion, the integration of nitrile (–CN)-functionalized silane ((2-cyanoethyl) triethoxysilane (TEOSCN)) into FEC/VC containing 1 M LiPF6 in an EC:DEC electrolyte solution proved to significantly boost the long-term cyclability, capacity retention, and coulombic efficiency of NMC622/Si–Gr pouch cells at a high temperature (i.e., 45 °C) and current density of 1C and 0.5C for charge and discharge, respectively. The additive significantly prevents NMC active material dissolution, which will otherwise produce resistive MFx as a side product on the surface of the NMC cathode and Si–Gr anode particles. Postmortem analysis using XPS measurements evidenced that the improvement is due to the –CN moieties of the designer TEOSCN additive, which leads to the formation of robust interphases on both the Si–Gr anode and NMC622 cathode electrodes. We hope that these findings will further facilitate the development of large-scale practical Si-containing high-energy LIBs.
Conflicts of interest
The authors declare that they have no competing financial interests.
Credit author statement
F. Aupperle: conducting experiment. F. Aupperle and G. G. Eshetu: writing original draft. G. G. Eshetu and E. Figgemeier: conceptualization, work design and supervision. G. G. Eshetu, E. Figgemeier, K. W. Ebermann, A. Xiao, and J-S. Bridel: revision and editing. E. Figgemeier: funding acquisition and project management.
Acknowledgements
E. Figgemeier acknowledges funding by the Federal Ministry of Education and Research through the project Meet HiEnD II (03XP0084B). G. G. Eshetu acknowledges the funding received from the European Commission via the H2020 IMAGE project (grant agreement number: 769929-IMAGE-H2020-GV-2016-2017/H2020-GV-2017). The authors specially thank Michel Armand for the for the fruitful scientific discussions, J. Hyunsang for drawing the Nitrile-based additive working principle, and N. Hamzelui for coating the Si–Gr anode electrodes.
References
- G. G. Eshetu, D. Mecerreyes, M. Forsyth, H. Zhang and M. Armand, Mol. Syst. Des. Eng., 2019, 4, 294–309 RSC.
- H. Tian, F. Xin, X. Wang, W. He and W. Han, J. Mater. Chem. A, 2015, 1, 153–169 Search PubMed.
- G. G. Eshetu and E. Figgemeier, ChemSusChem, 2019, 12, 2515–2539 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS.
- M. Pharr, K. Zhao, X. Wang, Z. Suo and J. J. Vlassak, Nano Lett., 2012, 12, 5039–5047 CrossRef CAS.
- E. Pollak, G. Salitra, V. Baranchugov and D. Aurbach, J. Phys. Chem. C, 2007, 111, 11437–11444 CrossRef CAS.
- X. Li, A. M. Colclasure, D. P. Finegan, D. Ren, Y. Shi, X. Feng, L. Cao, Y. Yang and K. Smith, Electrochim. Acta, 2019, 297, 1109–1120 CrossRef CAS.
- N. Yuca, Mater. Lett., 2020, 272, 127889 CrossRef CAS.
- S. Son, L. Cao, T. Yoon, A. Cresce, S. E. Hafner, J. Liu, M. Groner, K. Xu and C. Ban, Adv. Sci., 2019, 6, 1801007 CrossRef.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- D. Liu, Z. jiao Liu, X. Li, W. Xie, Q. Wang, Q. Liu, Y. Fu and D. He, Small, 2017, 13, 1702000 CrossRef.
- S. Chae, M. Ko, K. Kim, K. Ahn and J. Cho, Joule, 2017, 1, 47–60 CrossRef CAS.
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642–A646 CrossRef CAS.
- T. Jaumann, J. Balach, U. Langklotz, V. Sauchuk, M. Fritsch, A. Michaelis, V. Teltevskij, D. Mikhailova, S. Oswald, M. Klose, G. Stephani, R. Hauser, J. Eckert and L. Giebeler, Energy Storage Mater., 2017, 6, 26–35 CrossRef.
- R. Nölle, A. J. Achazi, P. Kaghazchi, M. Winter and T. Placke, ACS Appl. Mater. Interfaces, 2018, 10, 28187–28198 CrossRef.
- J. Bareño, I. A. Shkrob, J. A. Gilbert, M. Klett and D. P. Abraham, J. Phys. Chem. C, 2017, 121, 20640–20649 CrossRef.
- Y. Horowitz, H. L. Han, F. A. Soto, W. T. Ralston, P. B. Balbuena and G. A. Somorjai, Nano Lett., 2018, 18, 1145–1151 CrossRef CAS.
- J. M. Park, S. Kim, J. H. Ha, S. W. Kim, J. Lee, S. Park, B. W. Cho and H. J. Choi, Chem. Phys. Lett., 2017, 684, 383–389 CrossRef CAS.
- A. Schiele, B. Breitung, T. Hatsukade, B. B. Berkes, P. Hartmann, J. Janek and T. Brezesinski, ACS Energy Lett., 2017, 2, 2228–2233 CrossRef CAS.
- H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798 CrossRef CAS.
- M. Klett, J. A. Gilbert, K. Z. Pupek, S. E. Trask and D. P. Abraham, J. Electrochem. Soc., 2016, 164, A6095–A6102 CrossRef.
- Y. Zhang, N. Du and D. Yang, Nanoscale, 2019, 11, 19086–19104 RSC.
- G. G. Eshetu, T. Diemant, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, ACS Appl. Mater. Interfaces, 2016, 8, 16087–16100 CrossRef CAS.
- C. C. Su, M. He, C. Peebles, L. Zeng, A. Tornheim, C. Liao, L. Zhang, J. Wang, Y. Wang and Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 30686–30695 CrossRef CAS.
- F. Aupperle, N. von Aspern, D. Berghus, F. Weber, G. G. Eshetu, M. Winter and E. Figgemeier, ACS Appl. Energy Mater., 2019, 2, 6513–6527 CrossRef CAS.
- G. Schroeder, B. Gierczyk, D. Waszak and M. Walkowiak, Electrochem. Commun., 2006, 8, 1583–1587 CrossRef CAS.
- Q. Xia, B. Wang, Y. P. Wu, H. J. Luo, S. Y. Zhao and T. van Ree, J. Power Sources, 2008, 180, 602–606 CrossRef CAS.
- M. Walkowiak, D. Waszak, B. Gierczyk and G. Schroeder, Cent. Eur. J. Chem., 2008, 6, 600–606 CAS.
- Y.-G. Ryu, S. Lee, S. Mah, D. J. Lee, K. Kwon, S. Hwang and S. Doo, J. Electrochem. Soc., 2008, 155, A583 CrossRef CAS.
- C. C. Nguyen, H. Choi and S.-W. Song, J. Electrochem. Soc., 2013, 160, A906–A914 CrossRef CAS.
- W. Wang and S. Yang, J. Alloys Compd., 2017, 695, 3249–3255 CrossRef CAS.
- K. Kim, I. Park, S.-Y. Ha, Y. Kim, M.-H. Woo, M.-H. Jeong, W. C. Shin, M. Ue, S. Y. Hong and N.-S. Choi, Electrochim. Acta, 2017, 225, 358–368 CrossRef CAS.
- Y. Horowitz, H. L. Han, W. T. Ralston, J. R. de Araujo, E. Kreidler, C. Brooks and G. A. Somorjai, Adv. Energy Mater., 2017, 7, 1–8 Search PubMed.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2011, 28, 965–976 CrossRef.
- S. Seko, H. Nara, M. Jeong, T. Yokoshima, T. Momma and T. Osaka, Electrochim. Acta, 2017, 243, 65–71 CrossRef CAS.
- A. Rezqita, M. Sauer, A. Foelske, H. Kronberger and A. Trifonova, Electrochim. Acta, 2017, 247, 600–609 CrossRef CAS.
- M. Haruta, T. Okubo, Y. Masuo, S. Yoshida, A. Tomita, T. Takenaka, T. Doi and M. Inaba, Electrochim. Acta, 2017, 224, 186–193 CrossRef CAS.
- S. Hy, Y. H. Chen, H. M. Cheng, C. J. Pan, J. H. Cheng, J. Rick and B. J. Hwang, ACS Appl. Mater. Interfaces, 2015, 7, 13801–13807 CrossRef CAS.
- R. Petibon, V. L. Chevrier, C. P. Aiken, D. S. Hall, S. R. Hyatt, R. Shunmugasundaram and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A1146–A1156 CrossRef CAS.
- B. Strehle, S. Solchenbach, M. Metzger, K. U. Schwenke and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2513–A2526 CrossRef CAS.
- L. J. Krause, V. L. Chevrier, L. D. Jensen and T. Brandt, J. Electrochem. Soc., 2017, 164, A2527–A2533 CrossRef CAS.
- J. G. Han, J. Bin Lee, A. Cha, T. K. Lee, W. Cho, S. Chae, S. J. Kang, S. K. Kwak, J. Cho, S. Y. Hong and N. S. Choi, Energy Environ. Sci., 2018, 11, 1552–1562 RSC.
- Y. S. Hu, R. Demir-Cakan, M. M. Titirici, J. O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645–1649 CrossRef CAS.
- K. Eom, T. Joshi, A. Bordes, I. Do and T. F. Fuller, J. Power Sources, 2014, 249, 118–124 CrossRef CAS.
- N. Dupré, P. Moreau, E. De Vito, L. Quazuguel, M. Boniface, H. Kren, P. Bayle-Guillemaud and D. Guyomard, Chem. Mater., 2017, 29, 8132–8146 CrossRef.
- D.-T. Nguyen, J. Kang, K.-M. Nam, Y. Paik and S.-W. Song, J. Power Sources, 2016, 303, 150–158 CrossRef CAS.
- A. Bordes, K. Eom and T. F. Fuller, J. Power Sources, 2014, 257, 163–169 CrossRef CAS.
- H. Zhi, L. Xing, X. Zheng, K. Xu and W. Li, J. Phys. Chem. Lett., 2017, 8, 6048–6052 CrossRef CAS.
- F. Holtstiege, A. Wilken, M. Winter and T. Placke, Phys. Chem. Chem. Phys., 2017, 19, 25905–25918 RSC.
- M. Klett, J. A. Gilbert, S. E. Trask, B. J. Polzin, A. N. Jansen, D. W. Dees and D. P. Abraham, J. Electrochem. Soc., 2016, 163, A875–A887 CrossRef CAS.
- A. Rezqita, A.-R. Kathribail, J. Kahr and M. Jahn, J. Electrochem. Soc., 2019, 166, A5483–A5488 CrossRef.
- E. Björklund, D. Brandell, M. Hahlin, K. Edström and R. Younesi, J. Electrochem. Soc., 2017, 164, A3054–A3059 CrossRef.
- R. T. Pekarek, A. Affolter, L. L. Baranowski, J. Coyle, T. Hou, E. Sivonxay, B. A. Smith, R. D. McAuliffe, K. A. Persson, B. Key, C. Apblett, G. M. Veith and N. R. Neale, J. Mater. Chem. A, 2020, 7897–7906 RSC.
- B. Philippe, R. Dedryveère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2013, 25, 394–404 CrossRef CAS.
- B. Philippe, R. Dedryveìre, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS.
- C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef CAS.
- G. G. Eshetu, T. Diemant, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, ACS Appl. Mater. Interfaces, 2016, 8, 16087–16100 CrossRef CAS.
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Nano Energy, 2019, 55, 327–340 CrossRef CAS.
- G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS.
- Z. Liu, Q. Yu, Y. Zhao, R. He, M. Xu, S. Feng, S. Li, L. Zhou and L. Mai, Chem. Soc. Rev., 2019, 48, 285–309 RSC.
- M. R. Babaa, A. Moldabayeva, M. Karim, A. Zhexembekova, Y. Zhang, Z. Bakenov, A. Molkenova and I. Taniguchi, Mater. Today: Proc., 2017, 4, 4542–4547 Search PubMed.
- K. W. Schroder, H. Celio, L. J. Webb and K. J. Stevenson, J. Phys. Chem. C, 2012, 116, 19737–19747 CrossRef CAS.
- M. Hekmatfar, A. Kazzazi, G. G. Eshetu, I. Hasa and S. Passerini, ACS Appl. Mater. Interfaces, 2019, 11, 43166–43179 CrossRef CAS.
- B. Deng, H. Wang, W. Ge, X. Li, X. Yan, T. Chen, M. Qu and G. Peng, Electrochim. Acta, 2017, 236, 61–71 CrossRef CAS.
- L. Liu, S. Gu, S. Wang, X. Zhang and S. Chen, RSC Adv., 2020, 10, 1704–1710 RSC.
- Y. Dong, B. T. Young, Y. Zhang, T. Yoon, D. R. Heskett, Y. Hu and B. L. Lucht, ACS Appl. Mater. Interfaces, 2017, 9, 20467–20475 CrossRef CAS.
- A. Schechter, D. Aurbach and H. Cohen, Langmuir, 1999, 15, 3334–3342 CrossRef CAS.
- D. Lv, Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1–7 Search PubMed.
- G.-Y. Kim and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A437–A447 CrossRef CAS.
- Y. S. Kim, H. Lee and H. K. Song, ACS Appl. Mater. Interfaces, 2014, 6, 8913–8920 CrossRef CAS.
- Y. Abu-Lebdeh and I. Davidson, J. Power Sources, 2009, 189, 576–579 CrossRef CAS.
- Y. Abu-Lebdeh and I. Davidson, J. Electrochem. Soc., 2009, 156, A60 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Gebrekidan Gebresilassie
Eshetu
a,
Gebrekidan Gebresilassie
Eshetu