
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation on the formation of Mg metal anode/electrolyte interfaces in Mg/S batteries with electrolyte additives†
Vinayan
Bhaghavathi Parambath
 *a,
Zhirong
Zhao-Karger
b,
Thomas
Diemant
c,
Markus
Jäckle
d,
Zhenyou
Li
a,
Torsten
Scherer
b,
Axel
Gross
ad,
R. Jürgen
Behm
ac and
Maximilian
Fichtner
*ab
*a,
Zhirong
Zhao-Karger
b,
Thomas
Diemant
c,
Markus
Jäckle
d,
Zhenyou
Li
a,
Torsten
Scherer
b,
Axel
Gross
ad,
R. Jürgen
Behm
ac and
Maximilian
Fichtner
*ab
aHelmholtz Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstr. 11, D-89081 Ulm, Germany. E-mail: vinayan.bp@gmail.com; vinayan.parambath@kit.edu; m.fichtner@kit.edu
bKarlsruhe Institute of Technology (KIT), Institute of Nanotechnology, P.O. Box 3640, D-76021 Karlsruhe, Germany
cInstitute of Surface Chemistry and Catalysis, Ulm University, Albert-Einstein-Allee 47, D-89081 Ulm, Germany
dInstitute of Theoretical Chemistry, Ulm University, Albert-Einstein-Allee 11, D-89081 Ulm, Germany
First published on 22nd October 2020
Abstract
The combination of sulfur (S) as cathode with a Mg anode is a promising approach for batteries because of the high theoretical capacity (1672 mA h g−1) as well as the abundance and relatively low cost of these elements. However, up to now Mg/S batteries face serious drawbacks like a large overpotential between charge/discharge cycles, rapid capacity fading, poor cycling efficiency and slow kinetics, etc. Most of these problems are strongly correlated with polysulfide dissolution in the electrolyte and their subsequent shuttling to the Mg anode side. In this work, we investigate the Mg2+ deposition/dissolution process on a Mg metal electrode using a non-nucleophilic magnesium electrolyte (0.4 M Mg[B(hfip)4]2/DME) in the presence or absence of polysulfides. The large overpotential increase of the Mg metal anode in the presence of polysulfide species is reduced by the addition of optimum concentration of iodine additives to the Mg electrolyte, which help in the formation of a favorable, less corrosive, uniform and stable interfacial layer. In addition to the electrochemical studies, this work examines the chemical composition, thickness and morphology of the interphase layer formed on Mg metal by using X-ray photoelectron spectroscopy (XPS) with depth profiling and focused ion beam-scanning electron microscopy (FIB-SEM) techniques. Furthermore, the stability of different MgX phases (X = S, I2 and F2) formed between the Mg metal anode and different additive containing electrolytes and Mg diffusion through these interphases are investigated by density functional theory (DFT) calculations.
1. Introduction
Although Li ion batteries are presently considered as the most promising technology for electric vehicle propulsion, the development of alternative and complementary battery chemistries and technologies is of great importance, especially aiming at large-scale applications, e.g., for stabilizing the power grid, where the cost (€ per (kW h)) and sustainability are crucial indicators. Indeed, the implementation of Li based technologies on a large scale are increasingly questioned, since the controversial debates on lithium, cobalt, and nickel availability and moreover the cost cannot be ignored.1 Amongst different battery chemistries, appealing alternatives involve the use of multivalent cations. Magnesium (Mg) is especially interesting due to the abundance of Mg raw materials, its low cost as compared to Li, high safety due to a lack of dendrite formation, non-toxicity, and stability.2 Performance wise, the standard reduction potential for Mg is −2.37 V vs. SHE (standard hydrogen electrode) and the two electron reduction process results in a high theoretical energy density (Mg: 3837 A h L−1 as compared to Li: 2046 A h L−1). Even though most of the experimental and theoretical studies demonstrated a dendrite free deposition of Mg,3,4 some recent reports showed the formation of uneven structures during Mg electrodeposition under certain circumstances.5,6 This hints at an impact of the electrochemical environment (electrolyte concentration, coordinating ligands, etc.) and the applied current density on the growth morphology resulting during Mg electrodeposition.The development of Mg batteries has been hindered for a long time by the lack of suitable electrolytes with high ionic conductivity and large voltage window, wherein Mg ions could reversibly deposit and dissolve.7 Earlier studies showed that Mg could not be reversibly deposited from the solutions of simple magnesium salts such as Mg(ClO4)2 in conventional organic solvents.7,8 The main reason for this is the formation of a dense blocking layer at the surface, due to the deposition of the reduction products formed by the reaction of Mg metal with various anionic electrolyte species, such as ClO4−, BF4−, etc.8 The use of electrolytes with Grignard reagents (R-MgX, R = alkyl; X = Br, Cl), which are intrinsically strong reducing agents with limited oxidative stability and a high nucleophilicity,9–11 requires cathode materials that are chemically inert, for example MgxMo3S4 Chevrel phase cathodes. However, the high molecular weight of Chevrel phase cathode materials results in a low energy density (∼60 W h kg−1) which makes it less attractive.12 The well-known Mg(TFSI)2 based electrolyte system is less interesting in rechargeable Mg batteries due to its large overpotential and unstable SEI formation resultant from the partial decomposition of the Mg(TFSI)2 salt.2,6,13,14
The combination of a Mg anode with conversion cathode materials such as sulfur (S) is promising due to their high theoretical capacity of 1671 mA h g−1 (3459 mA h cm−3).15 But the high energy density sulfur cathode (electrophilic in nature) in rechargeable Mg batteries necessitates a non-nucleophilic electrolyte.16 Previous electrochemical studies on rechargeable Mg/S batteries pointed out problems like large over-potentials, rapid capacity and voltage fading, slow kinetics, self-discharge,17etc. where most of them are connected to the Mg polysulfide (Mg PS) dissolution to the electrolyte.18,19 Past efforts to make rechargeable Mg/S batteries mostly focused on the development of non-nucleophilic electrolytes, leaving it necessary to prevent the oxidation of electrolyte when getting into contact with the electrophilic sulfur. Recently our group developed a non-corrosive, chlorine-free Mg electrolyte with high ionic conductivity at room temperature (∼11 mS cm−1 at ∼23 °C), which is based on a fluorinated alkoxyborate magnesium salt (Mg[B(hfip)4]2).20 With this electrolyte, an improved electrochemical performance was observed for a Mg/S cell with low cathode sulfur loading (<0.5 mgsulfur cm−2),19 while higher sulfur loading on the cathode severely affected the cell performance and augmented the dissolution of polysulfide to anode side.21
Even though there have been some recent reports on Mg anode/electrolyte interfaces, most of these studies were performed with electrolytes suitable for Mg-ion batteries rather than Mg/S batteries.22,23 Recently, we verified that the Mg metal anode/electrolyte interface has a higher contribution to the resistance of a Mg/S cell compared to that of the sulfur/carbon composite cathode, and this is also correlated with the polysulfide dissolution from sulfur cathode.21,24 Therefore, a thorough understanding of the interphase evolution and Mg ion dynamics at the Mg anode/electrolyte interfaces in the presence of sulfur species and different electrolyte additive species is important. Usually, the electro-deposition on metal anodes proceeds through a “nucleation and growth” mechanism, which consists of a series of consecutive steps: (i) diffusion/mass transport of the complexed cation to the metal anode side through a liquid electrolyte, (ii) adsorption of the complexed cation combined with partial de-solvation and charge transfer, (iii) ad-atom surface diffusion on the electrode surface, (iv) formation of adatom clusters (nucleation) and (v) subsequent two- and three-dimensional crystal growth.25 The activation energy barriers for each step in the metal deposition/dissolution process determine the kinetics of the whole process and the morphology of the resulting film. Compared with monovalent cations like Li+, the electro-deposition kinetics of multivalent ions like Mg2+ are expected to be more hindered due to higher energy barriers for diffusion in the electrolyte and desolvation.7 The electrolyte formulation can therefore play a major role in Mg batteries to achieve a fast cation mobility and high power density. Different ways to improve the Mg2+ mobility in electrolytes include (i) the use of cation complexing and anion encapsulating agents, and (ii) the proper selection of solvents used in the electrolytes.7 The tailoring of Mg metal/electrolyte interfaces is possible by (i) the addition of additives to the electrolyte, which should help in the formation of fast Mg2+ ion conductive and stable passivation layers,26,27 and (ii) the creation of Mg ion conductive artificial coatings on the Mg anode surface,22 which can protect the Mg metal from the detrimental attacks of various corrosive species. An electronic conductivity of the artificial coatings on Mg metal, which is higher than that of the electrolyte, is detrimental for the stability of the electrolyte. Hence, the protective coating should feature high Mg ionic but low electronic conductivity. Cui et al. recently demonstrated that the addition of the Li electrolyte salt (Li[B(hfip)4]) to the 0.5 M Mg[B(hfip)4]2/DME electrolyte, can reduce the parasitic side reactions on a Mg metal anode during the prolonged cycling.28 Li et al. illustrated the activation and stabilization of Mg anode by using a dual-salt Mg2+/Li+ electrolyte.29–31 Cao et al. improved the full cell performance of Mg ion batteries by creating a magnesium fluoride (MgF2) surface layer on a Mg anode.32 In another study, Chen et al. investigated various coating materials for both anode and cathode electrodes in Mg batteries using density functional theory (DFT) based calculations and found that Mg-halides like MgBr2 and MgI2 or MgSe are promising candidates for anode coating.33,34
In this work, we have initially investigated the Mg2+ deposition/dissolution electrochemical process on a Mg metal anode in a non-nucleophilic magnesium electrolyte (Mg[B(hfip)4]2/DME) in the presence and absence of polysulfide species. Later, the effect of the addition of small concentrations of iodine to this Mg electrolyte was investigated in the context of the large overpotential for Mg electrodeposition on Mg metal anodes in the presence of polysulfide species, where the addition of iodine may help for the formation of a stable Mg ion conductive interfacial layer. Along with the electrochemical studies, the present work examines the chemical composition and morphology of the passivation layer formed at the electrolyte/electrode interfaces in the presence of various additives. Finally, we theoretically checked the thermodynamic stability of the interphases, which would form with various electrolyte additive species, and the Mg diffusion barrier through the MgI2 interphase.
2. Results and discussion
In order to illustrate the effect of various additives, we first conducted cyclic voltammetry (CV) measurements using a 3-electrode cell (PAT-Cell from EL-CELL GmbH) at a scan rate of 25 mV s−1 with a Pt working electrode, a Mg counter electrode and a Mg ring as the reference electrode. Fig. 1 illustrates the CV profiles for (a) Mg electrolyte ((Mg[B(hfip)4]2) in dimethoxyethane (DME) solvent), (b) Mg electrolyte containing iodine as additive, (c) Mg electrolyte containing polysulfide species as additive, (d) Mg electrolyte containing both polysulfide and iodine as additive. The CVs show an increase of the oxidation/reduction peak current density from the initial to the 10th cycle and this may be at least partly due to self-conditioning of the electrolyte and activation of the Mg metal surface by modification of the passivation layer.35Table 1 illustrates the onset potentials and peak currents (Ip) from CVs for the initial and selected subsequent cycles of Mg plating process on the Pt working electrode using electrolytes with different additives. The initial onset potential for the pure Mg electrolyte was reduced from −383 mV to −227 mV after 10 cycles. The addition of iodine to the pure Mg electrolyte caused the onset potential to decrease to −280 mV in the initial cycle and further to −214 mV after 10 cycles. Similarly, the peak currents for plating also increased after the addition of iodine to the Mg electrolytes. The addition of polysulfide species to the electrolyte increased the onset potentials and decreased the peak currents considerably. The onset potentials for polysulfide containing electrolyte was −513 mV in the initial cycle and it further increased to −560 mV on the 10th cycle. The very low values of the peak currents (−6.2 mA cm−2 and −0.70 mA cm−2 for the 1st and 10th cycles) after the addition of PS to electrolytes prove that the presence of polysulfide species is highly detrimental for Mg stripping/plating. The addition of iodine to the polysulfide containing electrolyte reduced the onset potential to −480 mV for the initial cycle and further to −345 mV and −313 mV after 5th and 10th cycles, respectively. Meanwhile, the peak currents increased (−20 mA cm−2 and −24.1 mA cm−2 for the 1st and 10th cycles) after the addition of iodine to the PS containing electrolytes. This increase in peak current suggests that adding iodine to the PS containing electrolyte improves the Mg ionic conductivity of the interphase.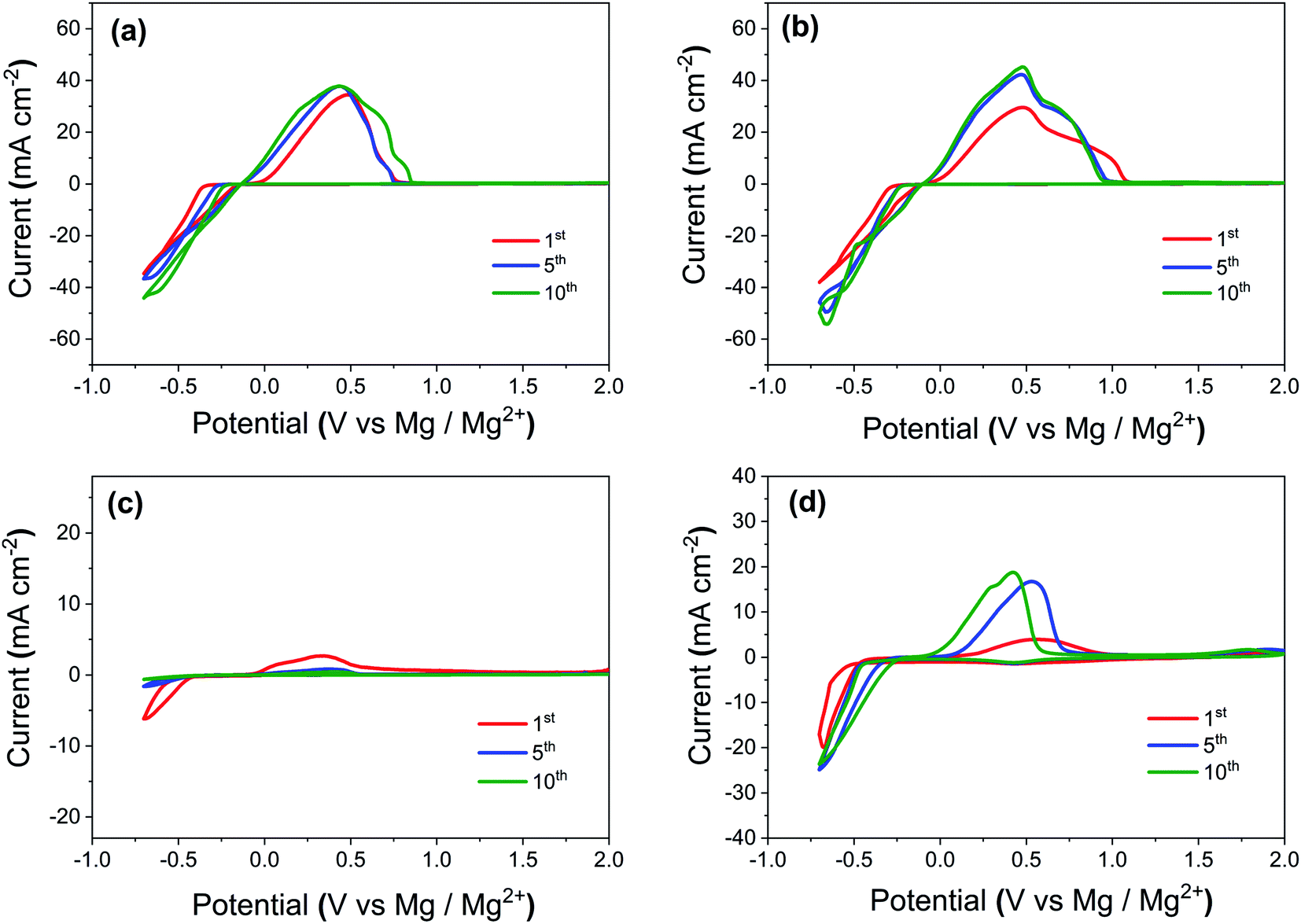 | ||
| Fig. 1 Cyclic voltammetry (CV) profiles for (a) pure Mg electrolyte, (b) Mg electrolyte containing iodine as additive, (c) Mg electrolyte containing polysulfide species as additive, (d) Mg electrolyte containing both polysulfide and iodine as additive. All the measurements were carried out using a 3-electrodes cell at a scan rate of 25 mV s−1 with Pt as the working electrode, Mg ring as the reference electrode, and Mg as the counter electrode. | ||
| Electrolyte with additives | Plating onset potentials (mV) for initial cycles | Plating peak currents (Ip) for initial cycles (mA cm−2) | ||||
|---|---|---|---|---|---|---|
| 1st | 5th | 10th | 1st | 5th | 10th | |
| Pure Mg electrolyte (0.4 M Mg[B(hfip)4]2 in dimethoxyethane solvent) | −383 | −265 | −227 | −34.7 | −36.7 | −44.0 |
| Mg electrolyte with iodine additive | −280 | −230 | −214 | −38.2 | −49.4 | −54.5 |
| Mg electrolyte with polysulfide additive | −513 | −524 | −560 | −6.20 | −1.60 | −0.70 |
| Mg electrolyte with polysulfide and iodine additive | −480 | −345 | −313 | −20.0 | −23.9 | −24.1 |
To investigate the long-term cycling performance of Mg electrolytes with additives, galvanostatic cycling was performed using symmetric Mg‖Mg cells. A constant current density of 100 μA cm−2 was applied for 30 minutes during the charge/discharge half cycle. These test conditions allow for an equal amount of charge shifted from one electrode to the other and give a better comparison of the cycling stability of the metallic Mg anode in various electrolytes. Fig. 2 shows the galvanostatic cycling profiles for (a and b) Mg electrolyte (0.4 M) and Mg electrolyte with iodine additive, (c and d) Mg electrolyte with a low amount of the polysulfide additive, (e and f) Mg electrolyte containing both polysulfides and iodine as an additive. The plating potentials during the 10th, 300th, 700th cycles for Mg electrolytes with various additives are given in Table 2.
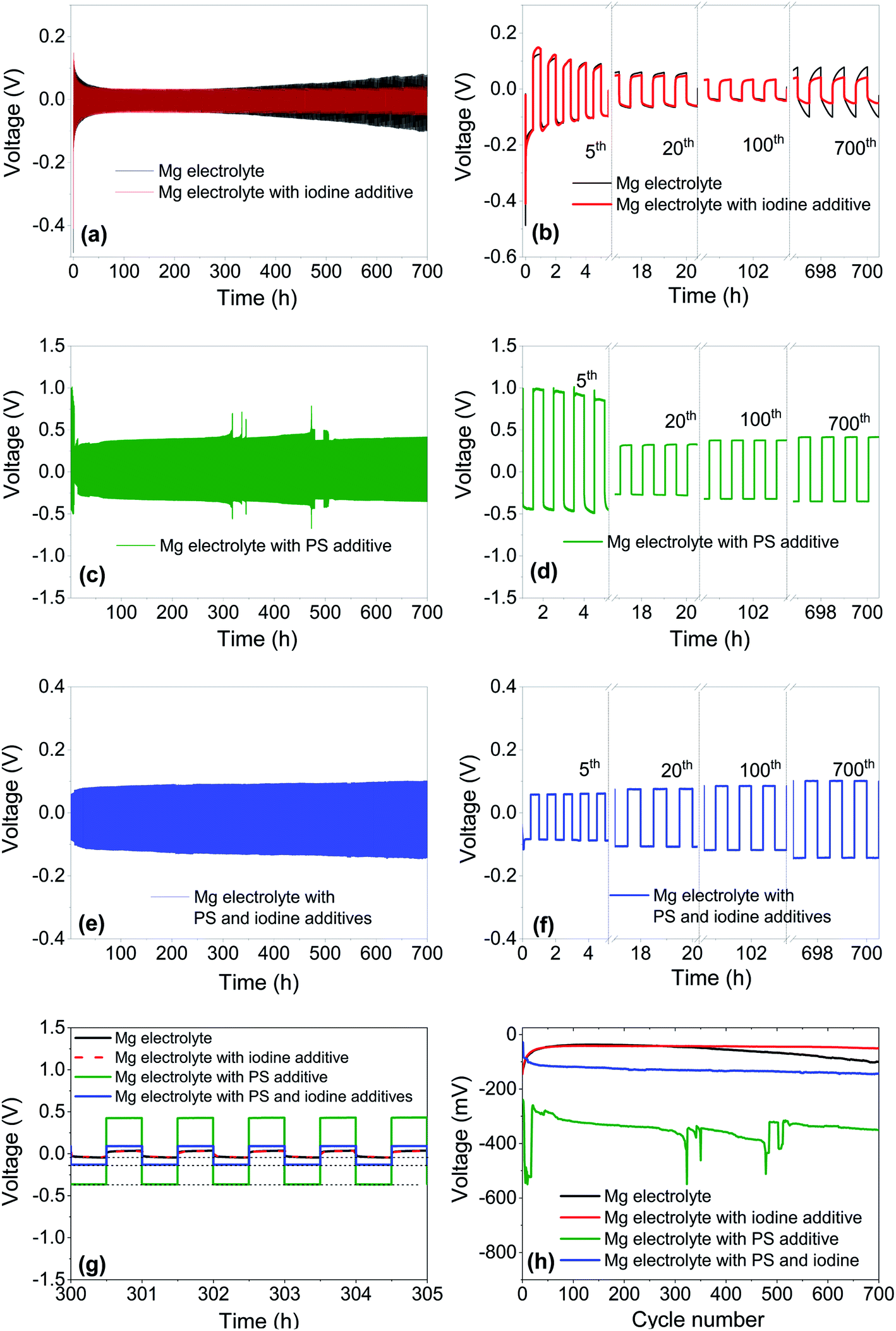 | ||
| Fig. 2 Galvanostatic cycling for 700 h using symmetric Mg‖Mg electrode cells with (a and b) Mg electrolyte and Mg electrolyte with iodine as additive, (c and d) Mg electrolyte with a polysulfide additive, (e and f) Mg electrolyte containing both polysulfides and iodine as an additive. (g) Comparison of stripping/plating profiles for Mg electrolytes with various additives after 300 h of cycling. (h) Comparison of the Mg plating potential over 700 cycles for Mg electrolytes with various additives based on galvanostatic measurements. The current density for these measurements is 100 μA cm−2, with 30 minutes stripping/plating time. | ||
| Electrolyte | Mg plating potential growth w.r.t cycle number (mV) | ||
|---|---|---|---|
| 10th | 300th | 700th | |
| Pure Mg electrolyte (0.4 M Mg[B(hfip)4]2 in DME) | −79.2 | −44.9 | −101.0 |
| Mg electrolyte with iodine additive | −76.5 | −42.5 | −50.2 |
| Mg electrolyte with polysulfide additive | −549.5 | −359.0 | −350.0 |
| Mg electrolyte with polysulfide and iodine additive | −102.0 | −130.2 | −143.2 |
The galvanostatic cycling profiles (Fig. 2) for pure Mg electrolyte show slightly higher plating potential for the first cycle, which subsequently decreases to −79 mV in the 10th cycle. This decrease hints to the formation of an interphase between the electrolyte and the Mg metal anode.14 After 300 cycles, the plating potential for the pure electrolyte was −45 mV, which further increased to −101 mV after 700 cycles. A similar trend with an increasing voltage hysteresis during the long-term cycling (>300 h) for Mg[B(hfip)4]2/DME electrolytes with higher concentrations (>0.35 M) was reported also previously.28,36 These studies concluded on a partial decomposition of the Mg[B(hfip)4]2 electrolyte salt on the Mg metal anode in higher concentrated electrolytes and the formation of an unstable SEI layer during prolonged cycling.28 After the addition of iodine additive to this 0.4 M Mg electrolyte, the plating potential was lower, even after long cycling (700 h) of the cell, as shown in Fig. 2a, b, g and h. After 300 and 700 cycles, plating potentials of −43 mV and −50 mV, respectively, were reached. The lower and stable potential proves that the addition of iodine to Mg electrolyte is beneficial and helps to form a stable interphase on the Mg metal electrode.
Adding a low amount of PS to the pure Mg electrolyte and cycling under the same experimental conditions showed a significant deterioration of the electrochemical responses of the Mg anode, as illustrated in the Fig. 2c, d, g and h. For the initial cycles, the plating potential considerably increased to 550 mV, which reduced in later cycles. The same trend was reported previously also on PS containing Mg electrolytes,24 and a probable cause for this is that prior to the cycling, Mg metal is reacting with polysulfide moieties to form a thin and fragile but highly resistive surface film.35 The thickness of the surface film depends on the amount of polysulfide species dissolved in the electrolyte. The formation of a PS containing surface film on the Mg anode requires a high overpotential to remove this insulating layer and to initiate an electrochemical Mg oxidation/reduction process at given constant current.24,37 The breaking of this resistive surface film during the plating/stripping process causes sudden fluctuations and bumps in the voltage hysteresis profile. The addition of more polysulfides to the Mg electrolyte further increased the plating overpotential from −0.58 V to −1 V for the initial cycles (Fig. S2, ESI†).
The addition of iodine to the polysulfide containing Mg electrolyte lowered the overpotential significantly, as shown in Fig. 2e–h. It was −102 mV after 10 cycles, increasing to −130 mV and −143 mV after 300 and 700 cycles, respectively. Hence, the iodine additive increased the cycling stability of the polysulfide containing electrolyte without any large increase of the overpotential . Furthermore, unlike the cells without iodine additive (Fig. 2c and d), the iodine containing cells did not show any pronounced overpotential spikes during cycling.
Electrochemical impedance spectroscopy (EIS) was used to characterize the resistances that develop at the interphases of the symmetric Mg‖Mg electrode cells in electrolytes containing different additives; the associated Nyquist plots are presented in Fig. 3. To reveal the underlying physical processes, the EIS data were fitted with equivalent electrical circuits as shown in the inset of Fig. 3b. In the fitted circuits, R1 indicates the electrolyte resistance and the electronic resistances of the current collectors. The high-frequency region semicircle, represented by a RSEI‖Q2 circuit, stands for the bulk resistance of the interphase layer. The low-frequency region semi-circle, represented by a RCT‖Q3 circuit, reflects the Mg ion migration through the interphase. Since the cells are symmetric, the value of RSEI and RCT for an individual interface is simply calculated by dividing the fitted resistances by a factor of 2. The resulting values are given in Table 3. Before any cycling at OCV, a more depressed semicircle was observed (Fig. 3a). The overall interfacial resistance (RSEI and RCT) of the Mg anode is very high; it drops significantly after initial cycling. This indicates the formation of an interphase between electrode and electrolyte during the initial cycling as supported by galvanostatic and voltammetric results. The total interfacial resistances of the Mg anode increased significantly when using a polysulfide containing electrolyte, which points to the formation of a thick, insulating passivation layer. On the other hand, the addition of iodine to the pure Mg electrolyte or to polysulfide containing electrolyte decreased the Mg metal anode resistances significantly due to the formation of a favorable Mg ion conductive interphase.
 | ||
| Fig. 3 (a) Electrochemical impedance spectroscopy (EIS) results of symmetric Mg‖Mg electrode cells using various additives containing Mg electrolytes under open circuit potential, before cycling, from 1 MHz to 50 mHz. (b) Electrochemical impedance spectroscopy (EIS) results of symmetric Mg‖Mg electrode cells using various additives containing Mg electrolytes after 50 cycles. The inset shows the impedance spectra in the higher frequency range and the associated equivalent circuit. | ||
| Electrolyte | Before cycling, under OCV (kΩ cm2) | After cycling, under OCV (Ω cm2) | ||
|---|---|---|---|---|
| R SEI | R CT | R SEI | R CT | |
| Pure Mg electrolyte (0.4 M Mg[B(hfip)4]2 in DME) | 4.8 | 19.5 | 47.5 | 7.4 |
| Mg electrolyte with iodine additive | 0.6 | 1.7 | 26.0 | 3.6 |
| Mg electrolyte with polysulfide additive | 9.5 | 27.6 | 227.8 | 158.2 |
| Mg electrolyte with polysulfide and iodine additives | 5.7 | 7.6 | 99.4 | 3.2 |
To gain information on the chemical surface composition of the Mg electrodes after cycling in electrolyte with different additives, X-ray photoelectron spectroscopy (XPS) measurements were carried out. They give the composition and the chemical state (oxidation state) of the elements in the surface layer of Mg electrodes after prolonged cycling under same test conditions. To compare with cycled state, XPS surface measurements were also conducted for fresh Mg metal electrodes after dipping (30 minutes) in Mg electrolytes with different additives (Fig. S3, ESI†). Prior to the measurements, all the electrodes were shortly washed with DME solvent and dried under vacuum overnight. Since XPS is a surface sensitive method, which probes only ∼3–5 nm of the outermost surface layer, measurements were also recorded after successive Ar+ ion sputter treatment in order to get a depth profile of the elemental composition in the surface-near region. XP spectra were recorded and analyzed for all Mg electrodes before sputtering and after sputter times of 3, 7, and 20 minutes, resulting in total sputter times of 0, 3, 10, and 30 min, respectively. Under the given conditions, the sputter rate is expected to be ∼1 nm min−1.
Fig. 4 displays the development of the elemental concentrations (Mg, C, O, S, F, and I) in the near-surface region of the Mg metal electrodes after cycling in different electrolytes, (a) with polysulfide additive or (b) with polysulfide and iodine additive, as a function of the sputtering time. For comparison, we show similar plots obtained for Mg electrodes cycled in pure Mg electrolyte and Mg electrolyte with iodine additive in Fig. S7 (ESI†). The quantitative analysis of the measurements of the surface layer on an Mg electrode cycled in polysulfide containing electrolyte (Fig. 4a, spectra shown in Fig. S6†) reveals an increase of the carbon concentration upon sputtering, arriving at ∼70 at% after 10 min sputtering (removal of ∼10 nm material) and staying constant after that. At the same time, the atomic concentrations of oxygen and fluorine decrease during the initial 10 minutes of sputtering and stay essentially stable after that. The Mg concentration remains rather low at all stages of the experiment (e.g., ∼5 at% after the last step).
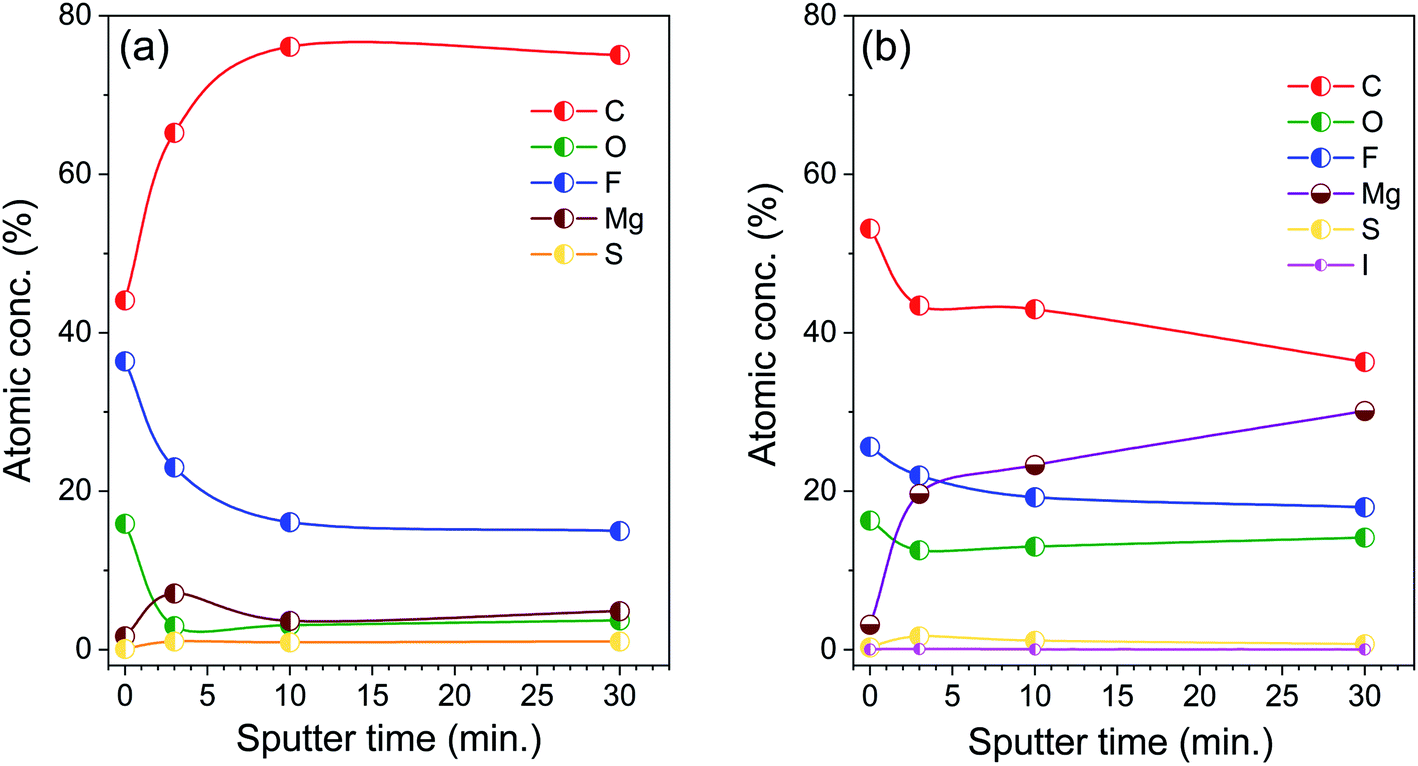 | ||
| Fig. 4 Development of the atomic concentrations of different elements in the near-surface region of Mg electrodes, which were cycled in electrolyte with (a) polysulfide additive or (b) polysulfide and iodine additive, as a function of the sputtering time. | ||
For Mg electrodes cycled in pure Mg electrolyte and Mg electrolyte with iodine additive (Fig. S4, S5 and S7†), we find an initial increase of the F concentration as well as a steady increase of the Mg and a concurrent reduction of the C concentration with increasing sputtering time. Finally, for the electrolyte containing polysulfide and iodine additive in combination (Fig. 4b, spectra shown in Fig. 5), we observe a continuous growth of the Mg concentration and simultaneous decrease of the C, O, and F concentrations with increasing sputtering time, i.e., from the surface to deeper layers.
 | ||
| Fig. 5 XP spectra recorded after different sputtering times on a Mg electrode after cycling for prolonged time in Mg electrolyte with polysulfide and iodine additive. | ||
Further information is obtained from the binding energies of the different elements recorded after prolonged cycling periods after different sputtering times. Here we will focus on the electrode cycled in Mg electrolyte containing polysulfide and iodine in combination (spectra in Fig. 5). The spectra recorded on the electrodes cycled in the other electrolytes, in (i) pure Mg electrolyte, (ii) in Mg electrolyte with iodine additive, and (iii) in polysulfide containing Mg electrolyte (Fig. S4–S6†), show qualitatively similar features, but with differences in the quantitative composition and in the trends during sputtering. Starting with the initial, non-sputtered surface, the C1s spectra show a dominant peak at 284.8 eV, which we attribute to C–C/C–H moieties. Furthermore, peaks at higher binding energies (287.4 eV and 289.0 eV) are assigned to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. CFx species from the hfip (hexafluoroisopropanolate) ligands of Mg[B(hfip)4]2 and its decomposition products lead to the signals at 293.0 eV and 294.2 eV, respectively. Peaks related to the C–O and CO groups were also observed in the O1s spectra at 532.0 eV (C–O) and 533.1 eV (CO), respectively. The spectra in the F1s region are initially dominated by a peak at 688.5 eV, which is due to the CFx groups of the hfip ligand. In addition, there is a small peak at 685.5 eV, which is attributed to MgF2. The sulfur concentration is comparatively low on the unsputtered surface. The position of the S2p peak doublet (162.5/163.7 eV), which is better visible after sputtering, indicates the presence of small amounts of disulfides (MgSx=2). Finally, in the I3d region we find a low-intensity peak at ∼619.3 eV, which according to the literature is typical for iodide species.38 From these XPS results, we believe that the topmost part of the interphase layer may contain some electrolyte species, with only small amounts of decomposed electrolyte salts.
O groups. CFx species from the hfip (hexafluoroisopropanolate) ligands of Mg[B(hfip)4]2 and its decomposition products lead to the signals at 293.0 eV and 294.2 eV, respectively. Peaks related to the C–O and CO groups were also observed in the O1s spectra at 532.0 eV (C–O) and 533.1 eV (CO), respectively. The spectra in the F1s region are initially dominated by a peak at 688.5 eV, which is due to the CFx groups of the hfip ligand. In addition, there is a small peak at 685.5 eV, which is attributed to MgF2. The sulfur concentration is comparatively low on the unsputtered surface. The position of the S2p peak doublet (162.5/163.7 eV), which is better visible after sputtering, indicates the presence of small amounts of disulfides (MgSx=2). Finally, in the I3d region we find a low-intensity peak at ∼619.3 eV, which according to the literature is typical for iodide species.38 From these XPS results, we believe that the topmost part of the interphase layer may contain some electrolyte species, with only small amounts of decomposed electrolyte salts.
In Fig. S8 (ESI†), we compare the Mg2s spectra recorded on Mg electrodes cycled in pure Mg electrolyte and in electrolytes with various additives, after different sputtering times. Since some of the cycled Mg electrodes contained iodine species, we chose to concentrate on the Mg2s peak region to avoid a possible interference in the Mg2p region with the I4p peak. For pure Mg electrolyte, the spectra in the Mg2s (Mg2p also) regions show initially the exclusive presence of oxidized Mg species (peaks at 89.3 eV and 50.6 eV). After the second sputter step, i.e., after removal of ∼10 nm material, additional peaks appear at lower binding energy (87.3 eV and 48.6 eV), which are characteristic of metallic Mg. After removal of ∼30 nm of material, metallic Mg amounts to ∼20%, while the rest still consists of oxidized species. We note that the peaks of metallic Mg appear at ∼1 eV lower binding energy in our measurements compared with what is usually reported in literature.39 We attribute this shift to a differential charging effect, which shifts the peaks of the non-conducting phase and thus also the binding energy reference, but does not affect the conducting Mg phase. For Mg electrodes cycled in electrolyte with iodine additive, the Mg2s spectra show the presence of metallic Mg even after the first sputter step (i.e., removal of ∼3 nm material) and the percentage of metallic Mg is ∼25% after removal of ∼30 nm of the surface layer. For the Mg electrode cycled in polysulfide containing electrolyte, the peak of metallic Mg appears only after 30 minutes sputtering (with ∼10% of the total Mg signal intensity). These results indicate the formation of a relatively thick passivation layer on polysulfide species containing Mg electrolyte. Finally, for the Mg electrode cycled in polysulfide and iodine containing electrolyte, the Mg2s spectra show the peak of metallic Mg even after the first sputter step (i.e., removal of ∼3 nm material), its intensity amounts to ∼45% after removal of ∼30 nm of the surface layer. It is interesting to note that the S2p peak doublet recorded on this electrode is shifted to lower binding energy (161.6/162.8 eV) compared with the sample without iodine, which could indicate the formation of mono-sulfide (MgS) instead of disulfide (MgS2) species in the presence of iodine.39 All in all, these XPS data indicate that the deeper regions of the interphase layer mainly consist of inorganic species, in particular of MgF2, MgI2, MgO and MgSx, etc.
Owing to the high ionic conductivity (∼10−5 S cm−1) and low electronic conductivity of MgI2 (∼10−9 S cm−1), its presence in the Mg interphase layer is highly beneficial for less resistive Mg stripping and plating.38 XPS studies already confirmed the formation of MgF2, MgS and MgI2 species in the interphase of cycled Mg metal electrodes. To better understand the Mg ion transport through these MgX (X = S, I2 and F2) species, DFT calculations (for more details please see the experimental section) were performed.40 The formation energy for the insertion (ΔEMgX,interstitial) of an Mg atom into the considered MgX phases (X = S, I2 and F2), as well as the energy required to create a vacancy (ΔEMgX,vacancy) that is necessary for the substitutional diffusion were calculated. For MgF2, MgS and MgI2 species, the ΔEMgX,interstitial values are 6.84 eV, 4.7 eV, and 1.78 eV, respectively. The related ΔEMgX,vacancy energies are 13.28 eV, 5.29 eV and 4.80 eV, respectively. Hence, the MgI2 species have significantly lower energies for the occupation of an interstitial site and the formation of vacancies, both of which are relevant for Mg ion transport, as compared to MgS and MgF2. In the interlayer region between the iodine layers, there are two adjacent local minima for the interstitial Mg atom corresponding to a tetrahedral and an octahedral site. The octahedral site is the energetically more favorable one; hence the diffusion path corresponds to the migration from the octahedral via the tetrahedral to the next symmetrically equivalent octahedral site. Hence there is only one possible pathway, as for example in many other materials such as spinel materials.41 Along this pathway we have performed a nudged elastic band calculation yielding a migration barrier of about 0.25 ± 0.05 eV between the octahedral and the tetrahedral site which thus yields the diffusion barrier along the minimum energy path, as shown in Fig. 6a. Earlier studies reported higher diffusion barriers for the transport of Mg ions in MgS (0.943 eV), MgF2 (1.123 eV) and MgO (1.851 eV) phases.34 It also needs to be mentioned that the calculated formation energies for the MgI2 species are still rather high, indicating that other magnesium iodide compounds might be present in the interphase layer contributing to the facile Mg transport; whose theoretical identification, however, is beyond the scope of the present paper.
 | ||
| Fig. 6 (a) Calculated minimum energy path (MEP) for the diffusion of an Mg atom (green in the MEP, otherwise grey) in between the iodine layers (yellow) in the MgI2 phase. Scanning electron microscopy (SEM) images of a (b) polished pristine Mg electrode, of (c) a Mg electrode after extensive cycling in Mg electrolyte with a low amount of polysulfide additive, and of (d) a Mg electrode after extensive cycling in Mg electrolyte with polysulfide and iodine additives. (e) FIB-SEM image of the Mg metal electrode cycled in electrolyte with polysulfide and iodine additives. Insets in (e) show the magnified image of the interphase and the EDXA spectrum at the interphase region. | ||
To gain information on the morphology of the surface and interphase of the cycled Mg electrodes, we performed electron microscopy (SEM) and focused ion beam (FIB)-SEM experiments. Fig. 6b–d shows electron microscopy images of the pristine polished Mg metal electrode, and of Mg metal electrodes after extensive cycling in polysulfide or polysulfide-plus-iodine containing electrolytes. The addition of polysulfide to the Mg electrolyte completely changed the surface morphology of the deposited Mg. The corrosion of the Mg surface was very obvious in the presence of polysulfide species and the extent of corrosion was found to strongly depend on the amount of polysulfide species in the electrolyte. Cycling in the electrolyte containing small amounts of polysulfide gave a surface with large and irregular deposits with thicker interphase (>500 nm) as shown in Fig. 6c and 7a. Increasing the polysulfide content in the Mg electrolyte led to the formation of a highly corroded Mg surface with rectangular nodules and pit-holes as shown in Fig. S9 (ESI†). The addition of iodine to the polysulfide containing electrolyte resulted in a much smoother Mg surface (see Fig. 6d, e and 7b), even after long cycling, indicative of a uniform Mg deposition/dissolution behavior. The FIB-SEM image (Fig. 6e) of the Mg metal electrode cycled in electrolyte with polysulfide and iodine additives illustrates the formation of a uniform passivation layer with an approximate thickness of ∼165 nm. Insets in Fig. 6e shows the energy dispersive X-ray analysis (EDXA) spectrum from the interphase region of Mg metal electrode cycled in iodine and polysulfide containing electrolyte. The spectrum clearly illustrates the presence of the chemical species like fluorine, iodine, sulfur, oxygen, etc. and these results are in accordance with the XPS results (Fig. 5).
 | ||
| Fig. 7 Schematic illustration of the interphase formation on the surface of Mg metal electrodes, which were cycled in electrolyte with (a) polysulfide additive or (b) polysulfide and iodine additive. Full cell performance of Mg–S battery tested with the optimized Mg electrolyte (0.4 M Mg[B(hfip)4]2/DME with iodine additive) and S/NC cathode at 0.05C-rate (25 °C) (c) galvanostatic charge/discharge voltage profiles of the 1st, 2nd, and 5th cycles, (d) cycle performance and coulombic efficiency at 0.05C-rate. | ||
In combination, the XPS and microscopy results indicate that the presence of polysulfides is highly detrimental and creates a thick non-uniform passivation layer that blocks the smooth deposition/dissolution of Mg ions and results in large overpotentials. In contrast, the addition of optimum concentration of iodine additives (MgI2 formation on the Mg surface) leads to a less corrosive, stable and Mg ion conducting interfacial layer with low thickness (cf. schematic depiction Fig. 7a and b). This interphase can support the uniform, smooth deposition/dissolution of Mg during extensive cycling with low overpotentials even in the presence of different chemical species such as sulfur, oxygen and fluorine species. In addition, the XPS sputter profiles demonstrate that the outermost part of the interphase of cycled Mg electrode consists mostly of organic species, while the deeper part, closer to the Mg electrode, mainly consists of inorganic species.
Finally, we tested the full cell performance of Mg/S battery with the optimized Mg electrolyte (0.4 M Mg[B(hfip)4]2/DME with iodine additive) using two electrode cell configuration at room temperature (25 °C) as shown in Fig. 7c and d. Sulfur impregnated nitrogen doped carbon matrix (S/NC) was used as the cathode electrode.21,42 The cells were cycled at a rate of 0.05C (1C = 1672 mA gsulfur−1) at 25 °C. Two plateau voltage profiles (at ∼1.74 V, and ∼1.43 V in Fig. 7c) indicate the conversion based reduction reaction of Mg with sulfur, which is associated with the S8 ring opening and the formation of high- and low-order Mg polysulfides (MgSx, x = 8–1). The discharge capacities for the 1st, 2nd, 5th, 50th, 100th cycles were ca. 899, 854, 701, 440, 330 mA h gsulfur−1, i.e., the cathode electrode retained 95%, 78%, 49% and 37% of its initial capacity after the 2nd, 5th, 50th, 100th cycles, respectively (Fig. 7d). For the initial 15 cycles, the cell gave a Coulombic efficiency of ≤ 97% and then after it increased to 98.5% for the following 85 cycles. Electrochemical performances of Mg/S full cell can be further improved by optimizing the sulfur cathodes.43,44
3. Conclusions
In summary, we have investigated the electrochemical Mg2+ deposition/dissolution on a Mg metal anode (a) in 0.4 M fluorinated alkoxyborate magnesium electrolyte, (b) in Mg electrolytes with either polysulfide or iodine additives, and (c) in Mg electrolyte contained both polysulfide and iodine additives. The presence of polysulfide species leads to a large overpotential at the Mg anode during cycling. Furthermore, microscopic and spectroscopic characterizations reveal significant corrosion of the Mg metal electrode in the presence of polysulfide species and the formation of a highly resistive thicker interphase layer, which increasingly blocks the Mg stripping/plating process. The large overpotential on Mg anodes observed in the presence of polysulfide species was considerably reduced upon the addition of optimum concentration of iodine to Mg electrolyte and this also eluded the time consuming electrolytic conditioning behavior. The formation of a MgI2 layer on the Mg anode helps (i) to reduce the detrimental side reactions of the polysulfide species on Mg metal anode and (ii) to form an electrochemically stable, thinner, and uniform Mg ion conductive interphase between the Mg metal and fluorinated alkoxyborate magnesium electrolyte. DFT calculations obtained lower vacancy/interstitial formation energies and a very low diffusion barrier of interstitial Mg in MgI2 compared to other MgX (X = F2, S, O) phases, which supports the high mobility of Mg in MgI2 phases in the interphase. This approach, the use of iodine additives, renders an easy way for the creation of a stable and ion conductive interphase in practical room temperature Mg/S batteries. The present studies furthermore underline that the proper choice of additives can strongly modify and even determine the composition of the solid electrolyte interphase and this way influence the performance on the Mg anode side. It also opens an avenue for the development of new electrolytes for Mg/S batteries operated with a Mg metal anode.4. Experimental section
Synthesis of materials and electrochemical measurements
0.4 M fluorinated alkoxyborate magnesium salt (Mg[B(hfip)4]2) in dimethoxyethane (DME) solvent was used as Mg electrolyte, it was synthesized according to our previous report.20 Since ether based solvents are more suitable for Mg battery electrolytes/electrodes as compared to organic carbonate solvents, due to their higher reduction stability, the electrolyte and all additives for the current study were prepared in DME solvent.45,46 Mg polysulfide (Mg PS) was used as sulfur additive, and synthesized according to our previous report.19 The concentration of sulfur species in the Mg PS solution was 68 mmol L−1 and determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). S2p X-ray photoelectron spectra (XPS) of the Mg PS (adsorbed on carbon matrix) showed the S2p peak doublets of bridging and terminal S atoms (163.4/164.6 eV & 161.5/162.7 eV), respectively, as compared to bulk sulfur (164.0/165.2 eV) (Fig. S1, ESI†). For sulfur cathode preparation, nitrogen doped (graphene–multiwall carbon nanotube) hybrid structure (NC) was used as the carbon host matrix. The sulfur was impregnated within the carbon host matrices (S/NC) by melt infiltration technique at 155 °C under an argon gas atmosphere. A detailed procedure for the synthesis of sulfur cathode was given in our previous report.21,42Cyclic voltammetry measurements (CVs) were performed using a three-electrode cell (PAT-Cell, EL-CELL GmbH) at a scan rate of 25 mV s−1 with a Pt disc as the working electrode, a Mg disc as counter electrode and a Mg ring as reference electrode. Galvanostatic cycling experiments for symmetric Mg‖Mg cells were carried out at a current density of ±100 μA cm−2 for 30 minutes with each charge/discharge half cycle. Electrochemical impedance spectroscopy (EIS) for symmetric Mg‖Mg cells was performed with an applied sinusoidal excitation voltage of 10 mV in the frequency range from 1 MHz to 50 mHz. All the cells were assembled inside an argon filled glove box (MBraun GmbH) and electrochemically tested using a Biologic VMP-3 multichannel potentiostats/galvanostats. Initial electrochemical experiments for Mg electrolyte were carried out without the presence of any polysulfide species, followed by measurements with the addition of different concentrations of polysulfide species, from low (∼14 mM) to high (∼50 mM) values. In a second series of experiments, we added optimum concentration of iodine-based additives (∼45 mM) to Mg electrolyte and performed the same electrochemical experiments in the absence and presence of polysulfide species.
For full cell measurements, the cathode slurry was prepared by mixing S/NC powders and the polyvinylidene fluoride binder (PVDF, received from Kynar) in the 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mass ratio using N-methyl-2-pyrrolidone (NMP) as solvent. This obtained slurry was cast onto aluminum foil by doctor blade techniques and thereafter dried at 60 °C for 24 h. The sulfur loading at the cathode electrode was ∼0.4 mgsulfur cm−2. 80 μL optimized Mg electrolyte (0.4 M Mg[B(hfip)4]2/DME with iodine additive) was used in each assembled cell with two electrode configurations (Swagelok). All the cells were assembled inside an argon-filled glove-box. The electrochemical performance of the full cells was tested by galvanostatic cycling with 0.05C (1C = 1672 mgsulfur cm−2) using an Arbin BT-2000 battery tester at room temperature (25 °C). The capacities were calculated based on the mass of sulfur in the cathode electrode.
:10 mass ratio using N-methyl-2-pyrrolidone (NMP) as solvent. This obtained slurry was cast onto aluminum foil by doctor blade techniques and thereafter dried at 60 °C for 24 h. The sulfur loading at the cathode electrode was ∼0.4 mgsulfur cm−2. 80 μL optimized Mg electrolyte (0.4 M Mg[B(hfip)4]2/DME with iodine additive) was used in each assembled cell with two electrode configurations (Swagelok). All the cells were assembled inside an argon-filled glove-box. The electrochemical performance of the full cells was tested by galvanostatic cycling with 0.05C (1C = 1672 mgsulfur cm−2) using an Arbin BT-2000 battery tester at room temperature (25 °C). The capacities were calculated based on the mass of sulfur in the cathode electrode.
:1 and a spin–orbit splitting 1.2 eV were used.
The crystalline systems were allowed to fully relax for the system energy calculations. In the case of the interstitial magnesium atom in MgI2, only the iodine and magnesium layers directly above and below the interstitial atom were allowed to relax. The system energies were determined within a 4 by 4 by 4 geometry for the MgI2 systems, a 3 by 3 by 3 geometry for the MgF2 system, and a 4 by 4 by 4 geometry for the MgS system. The diffusion barrier for interstitial diffusion in the MgI2 system was calculated in the same geometry as the system energies of this system. All systems were calculated using a 5 by 5 by 5 gamma-centered k-point grid. The diffusion path in the case of the interstitial diffusion in MgI2 was calculated employing the nudged elastic band (NEB) method,55–57 with nine images along the reaction path. The relaxation in the calculations has been performed until the forces were smaller than 0.01 eV Å−1 and the total free energy change was below 0.0001 eV.
The stability of different MgX (X = S, I2 and F2) phases with an interstitial Mg atom and with a Mg vacancy was calculated in analogy to the approach by Reddy et al.,40via
| ΔEMgX,interstitial = E(MgXwith interstitial Mg) − [E(MgXbulk) + E(Mgatom,bulk)] | (1) |
| ΔEMgX,vacancy = E(MgXwith Mg vacancy) − [E(MgXbulk) − E(Mgatom,bulk)] | (2) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study is supported by MagSiMal project (03XP0208H) of the German Ministry of Research and Education (BMBF). This work is partially supported by the European Union's Horizon 2020 Research and Innovation programme under grant agreement No. 824066. We also thank the Deutsche Forschungsgemeinschaft for the support under project ID 390874152 (POLiS Cluster of Excellence). This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe). This work was performed on the computational resource bwFORCluster JUSTUS funded by the Ministry of Science, Research and Arts and the Universities of the State of Baden-Württemberg, Germany, within the bwHPC framework program, supported by the German Research Foundation (DFG) through Grant No. INST 40/467-1 FUGG.References
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- P. Saha, M. K. Datta, O. I. Velikokhatnyi, A. Manivannan, D. Alman and P. N. Kumta, Prog. Mater. Sci., 2014, 66, 1 CrossRef CAS.
- Z. Ma, M. Forsyth, D. R. MacFarlane and M. Kar, Green Energy Environ., 2019, 4, 146–153 CrossRef.
- M. Jäckle and A. Groß, J. Chem. Phys., 2014, 141, 174710 CrossRef.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee, S. Banerjee, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS.
- M. S. Ding, T. Diemant, R. J. Behm, S. Passerini and G. A. Giffin, J. Electrochem. Soc., 2018, 165, A1983–A1990 CrossRef CAS.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- R. Mohtadi and F. Mizuno, Beilstein J. Nanotechnol., 2014, 5, 1291–1311 CrossRef.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS.
- W. V. Evans, F. H. Lee, C. H. Lee and I. Table, J. Am. Chem. Soc., 1935, 57, 489–490 CrossRef CAS.
- D. M. Overcash and F. C. Mathers, Trans. Electrochem. Soc., 1933, 64, 305 CrossRef.
- J. Muldoon, C. B. Bucur and T. Gregory, Angew. Chem., Int. Ed., 2017, 56, 12064–12084 CrossRef CAS.
- X. Li, T. Gao, F. Han, Z. Ma, S. Hou, N. Eidson, W. Li, X. Fan, S. Hou, N. Eidson, W. Li and C. Wang, Adv. Energy Mater., 2018, 8, 1701728 CrossRef.
- S.-J. Kang, H. Kim, S. Hwang, M. Jo, M. Jang, C. Park, S.-T. Hong and H. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 517–524 CrossRef CAS.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef.
- Z. Zhao-Karger, X. Zhao, D. Wang, T. Diemant, R. J. Behm and M. Fichtner, Adv. Energy Mater., 2015, 5, 1401155 CrossRef.
- Z. Zhang, S. Dong, Z. Cui, A. Du, G. Li and G. Cui, Small Methods, 2018, 2, 1800020 CrossRef.
- B. P. Vinayan, Z. Zhao-Karger, T. Diemant, V. S. K. Chakravadhanula, N. I. Schwarzburger, M. A. Cambaz, R. J. Behm, C. Kübel and M. Fichtner, Nanoscale, 2016, 8, 3296–3306 RSC.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- B. P. Vinayan, H. Euchner, Z. Zhao-Karger, M. A. Cambaz, Z. Li, T. Diemant, R. J. Behm, A. Gross and M. Fichtner, J. Mater. Chem. A, 2019, 7, 25490–25502 RSC.
- S. B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- M. Salama, R. Attias, B. Hirsch, R. Yemini, Y. Gofer, M. Noked and D. Aurbach, ACS Appl. Mater. Interfaces, 2018, 10, 36910–36917 CrossRef CAS.
- J.-G. Zhang, X. Wu and W. A. Henderson, Springer Series in Materials Science: Lithium Metal Anodes and Rechargeable Lithium Metal Batteries, 2017, vol. 249, DOI:10.1007/978-3-319-44054-5.
- B. Pan, J. Huang, N. Sa, S. M. Brombosz, J. T. Vaughey, L. Zhang, A. K. Burrell, Z. Zhang and C. Liao, J. Electrochem. Soc., 2016, 163, A1672 CrossRef CAS.
- B. Pan, K.-C. Lau, J. T. Vaughey, L. Zhang, Z. Zhang and C. Liao, J. Electrochem. Soc., 2017, 164, A902 CrossRef CAS.
- K. Tang, A. Du, S. Dong, Z. Cui, X. Liu, C. Lu, J. Zhao, X. Zhou and G. Cui, Adv. Mater., 2020, 32, 1904987 CrossRef CAS.
- Y. Zhang, J. Xie, Y. Han and C. Li, Adv. Funct. Mater., 2015, 25, 7300–7308 CrossRef CAS.
- X. Zhou, J. Tian, J. Hu and C. Li, Adv. Mater., 2018, 30, 1704166 CrossRef.
- J. Tian, X. Zhou, Q. Wu and C. Li, Energy Storage Mater., 2019, 22, 218–227 CrossRef.
- B. Li, R. Masse, C. Liu, Y. Hu, W. Li, G. Zhang and G. Cao, Energy Storage Mater., 2019, 22, 96–104 CrossRef.
- T. Chen, G. Ceder, G. S. Gautam and P. Canepa, Front. Chem., 2019, 7, 24 CrossRef CAS.
- T. Chen, G. Sai Gautam and P. Canepa, Chem. Mater., 2015, 27, 6016–6021 CrossRef.
- A. Du, Z. Zhang, H. Qu, Z. Cui, L. Qiao, L. Wang, J. Chai, T. Lu, S. Dong, T. Dong, H. Xu, X. Zhou and G. Cui, Energy Environ. Sci., 2017, 10, 2616–2625 RSC.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics Division, Perkin-Elmer Corp, 1992, ISBN: 0962702625 Search PubMed.
- M. Anji Reddy, M. Helen, A. Groß, M. Fichtner and H. Euchner, ACS Energy Lett., 2018, 3, 2851–2857 CrossRef CAS.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS.
- S. A. Pervez, B. P. Vinayan, M. A. Cambaz, G. Melinte, T. Diemant, T. Braun, G. Karkera, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2020, 8, 16451–16462 RSC.
- P. Wang and M. R. Buchmeiser, Adv. Funct. Mater., 2019, 29, 1905248 CrossRef CAS.
- X. Yu and A. Manthiram, ACS Energy Lett., 2016, 1, 431–437 CrossRef CAS.
- N. Kumar and D. J. Siegel, J. Phys. Chem. Lett., 2016, 7, 874–881 CrossRef CAS.
- A. Ericson and I. Persson, J. Organomet. Chem., 1987, 326, 151–158 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- G. I. Csonka, J. P. Perdew, A. Ruzsinszky, P. H. T. Philipsen, S. Lebègue, J. Paier, O. A. Vydrov and J. G. Ángyán, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155107 CrossRef.
- A. Groß, J. Phys.: Condens. Matter, 2009, 21, 084205 CrossRef.
- A. Groß, J. Comput. Theor. Nanosci., 2008, 5, 894–922 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05762b |
| This journal is © The Royal Society of Chemistry 2020 |