
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel molybdenum oxide–Starbon catalyst for wastewater remediation†
Jaspreet
Kaur
 ab,
Khushwinder
Kaur
b,
Surinder K.
Mehta
b and
Avtar S.
Matharu
*a
ab,
Khushwinder
Kaur
b,
Surinder K.
Mehta
b and
Avtar S.
Matharu
*a
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, England YO10 5DD, UK. E-mail: avtar.matharu@york.ac.uk
bDepartment of Chemistry and Centre of Advanced Studies in Chemistry, Panjab University, Chandigarh, 160014, India
First published on 6th July 2020
Abstract
The exploration of novel media for environmental remediation, in particular wastewater treatment, is a global imperative. Herein, the in situ green synthesis, characterisation and application of a novel Starbon™ composite comprising molybdenum oxide nanoparticles are reported. Starbons™ are carbonaceous mesoporous materials derived from starch with applications ranging from chromatography to gas capture. The molybdenum (Mo) loading, evidenced by inductively coupled plasma-mass spectrometry (ICP-MS), was 179.337 mg g−1, and the molybdenum oxide nanoparticles were observed via transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDX). The Mo-containing composite was an efficient catalyst for the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in the presence of sodium borohydride, NaBH4 (k = 11.2 × 10−2 min−1). The Mo-composite showed superior 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical quenching activity with a low inhibitory concentration [IC50 = 1.006 mg ml−1] and ferric reducing power compared with other green synthesised composites and nanoparticles. The novel Mo-containing Starbon™ composite has real time applications in water treatment such as in catalysis, adsorption and filtration.
Introduction
With increasing global population, urbanization, industrialization and climate change, securing access to clean water is a global grand challenge. Water pollution is linked to environmental, social and economic development.1 Water pollution induces chronic illnesses such as cholera, typhoid, hepatitis, dysentery, diarrhea, jaundice and tuberculosis.2 Concerns have therefore been raised that multifarious chemical-containing effluents might move up the food chain entering human consumption without our perceived knowledge.3 Thus, it is imperative that these diverse chemicals be managed meticulously before discharging into watercourses. A wide range of technologies have been acknowledged for the removal of effluent from wastewater.4 These include physical (filtration, osmosis, electrodialysis), chemical (coagulation/flocculation, precipitation, oxidation) and biological (aerobic and anaerobic microbial degradation, enzymatic degradation) methods. However, these approaches are not without their disadvantages such as high cost, being chemically intensive, difficult to use and practically challenging.3Catalytic reduction is a new and promising water management technique that has been thoroughly researched both at the laboratory- and field-scale. The latter trials have produced impressive results for industrial wastewater and groundwater. Compared to traditional technologies, it exhibits robust operation and superior selectivity, and yields non-harmful or considerably less harmful and more readily biodegradable compounds.5 Hence there is a need to design new materials which could play an important role in the development of modern, quick, environmentally safe and cost-effective methods for the identification, degradation, reduction and removal of organic and inorganic toxic pollutants.6
In terms of sustainability, significant efforts are being directed towards creating environmentally benign materials using renewable resources or biomass.7 For example, biomass-derived hierarchically structured porous carbons have attracted great interest for environmental remediation.8 Polysaccharides such as starch, pectin and alginic acid, as a biomass source, can be used as precursors in materials synthesis to afford multiple levels of porosity such as micro (<2 nm), meso (2–50 nm) and macro (>50 nm).9 In fact, Starbons™ are a family of mesoporous carbonaceous materials derived from polysaccharides.10 Starbons™ possess high surface area, large accessible space, electrical conductivity, thermal stability, scalability, low density and interconnected hierarchical porosity of different length scales, which are very favorable for adsorption, photocatalysis, separation, and energy storage and conversion.11
One recent development in this field is directed towards the incorporation of metal oxide nanoparticles into porous matrices.12 Compared to their bulk counterparts, nanoparticles show significant potential for application in electronic, chemical, biological and catalytic fields.2 Amongst various semi-conducting metal oxides, molybdenum oxide (MoO3) is a promising material due to its range of functionality and, thus, diverse applications. MoO3 shows excellent photochromic, electrochromic, thermochromic and catalytic properties.13,14 Chithambararaj et al.15 reported that hexagonal MoO3 nanoparticles resulted in 6–8 times higher photocatalytic activity than other phase structures. To enhance its catalytic activity, MoO3 has been modified by doping with transition metals, such as Ni2+, Cu2+, Pb2+, and Pd, or fabrication with other metal oxides, such as TiO2, WO3, and reduced graphene oxide.16–18 Furthermore, MoO3 also shows good anti-microbial, anti-bacterial and anti-oxidant activity,19 and molybdenum is not listed as an endangered or critical element.20
Thus, herein the synthesis, characterization and application as a potential environmental remediation agent of a novel MoO3–Starbon composite are investigated (see Fig. 1). The salient features of this composite are as follows: (i) it employs biomass as a renewable resource; (ii) HF and/or high temperature carbonization is not required as the materials are processed in water and alcohol and carbonized at 350 °C; (iii) templating agents are not required as the inherent hierarchical structure of the polysaccharide is used to control porosity; (iv) the MoO3 nanoparticles act as catalytically active sites for degradation of noxious effluents, and (v) the mesoporous matrix facilitates the adsorption of chemicals through an array of intermolecular interactions enabling fast accessibility to surface active sites and mass transport.
 | ||
| Fig. 1 Graphical representation of the synthetic procedure for Starbon-350 (control, without Mo) and CMO-350 (with Mo). | ||
Combining the inherent properties of Starbon™ (mesoporous and high surface area) with the excellent properties of MoO3 nanoparticles, this novel composite is explored as a catalyst for the abatement of 4-nitrophenol (4-AP), a common water pollutant and a known carcinogen, teratogen and mutagen. Furthermore, the radical quenching activity of the synthesized composite is assessed via DPPH and FRAP assays. The structure–property relationship, morphology, composition, and surface functionality with respect to catalytic and radical quenching activity are also developed.
Results and discussion
Molybdenum incorporation and characterisation
The incorporation of molybdenum into expanded starch was evidenced initially by the appearance of the bluish colour characteristic of MoO3 nanoparticles. In the presence of starch, the H+ and OH− ions present in the water molecules bind strongly to nuclei (MoO6, OH− and NH4+) and help enhance the surface coverage.14 The hydroxyl ions aid the hydrolysis reaction and enhance the dissolution–crystallization process, providing ample time for collision and for the diffusion of ions at the crystal surface.21 This helps in rearrangement and steric shielding of nucleation, which in turn helps in the growth of nanoparticles. In the absence of starch, directional growth of nuclei occurs, which leads to the formation of MoO3 nanorods (see the ESI (Fig. S1†)). Hence, starch serves two purposes: (i) it acts as a templating agent and impedes the directional growth of nanoparticles and forms spherical structures and (ii) the natural pH of aqueous starch solution is 5, and thus it maintains the necessary acidic conditions for the formation of nanoparticles.The resultant bluish starch was carbonised at 350 °C to afford the desired molybdenum containing Starbon composite, CMO-350, with a Mo loading of 179.337 mg g−1 as determined by ICP-MS.
Infrared spectroscopy
To confirm the synthetic procedure and changes occurring in the chemical composition, the fabricated material was analysed by FT-IR spectroscopy (Fig. 2). Pure Expst and Mo-Expst show two prominent absorption bands at 3300 and 1000 cm−1 which are due to the O–H and C–O stretching vibrations, respectively, present in the starch precursor.22 The intensity of these bands diminishes as a result of carbonization. The IR spectra of Starbon-350 and CMO-350 exhibit two intense bands at approx. 1606 cm−1 and 1714 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO stretching vibrations, respectively.23 Importantly, in the FT-IR of CMO-350 two prominent bands at 951 and 912 cm−1 are observed and ascribed to the MoO stretching vibration. A broad and complex band centred at 791 cm−1 corresponds to the Mo–O vibration of MoO3.24
C and CO stretching vibrations, respectively.23 Importantly, in the FT-IR of CMO-350 two prominent bands at 951 and 912 cm−1 are observed and ascribed to the MoO stretching vibration. A broad and complex band centred at 791 cm−1 corresponds to the Mo–O vibration of MoO3.24
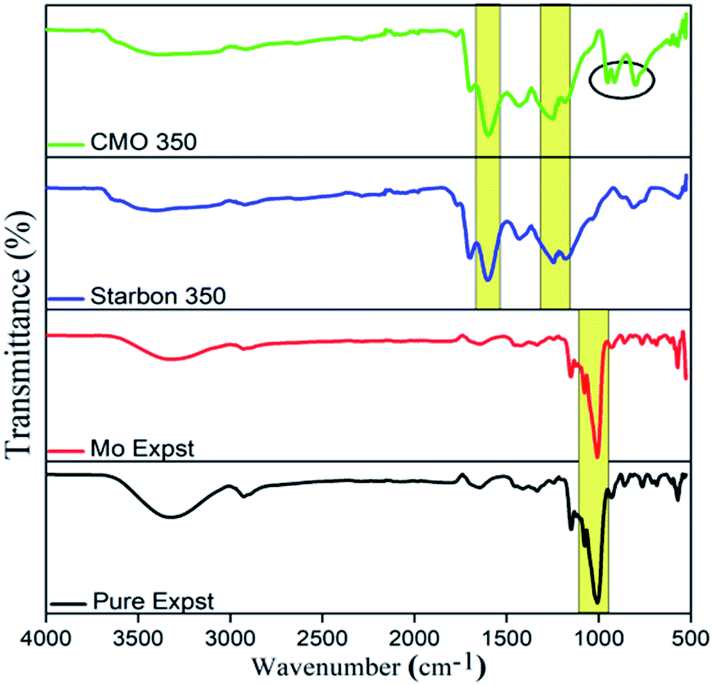 | ||
| Fig. 2 The comparative analysis of the IR spectra of the pure and metal loaded composite. | ||
X-ray diffraction (XRD)
The distinguishable small peaks (2θ = 15°, 18°, 20° and 22°) in the XRD patterns of Mo-Expst and Pure Expst (see Fig. 3) are associated with crystalline starch granules within a backdrop of highly amorphous character.25 The latter is more evident in the XRD pattern of Starbon-350, which shows the absence of any marked crystallinity but instead displays a broad diffraction signal from 15 to 30° (2θ) centred around 20°.26 Interestingly, upon carbonization of Mo-Expst to afford CMO-350, this broad pattern shifts and is centred around 25° (2θ). This could be attributed to the presence of MoO3 which is known to give characteristic peaks around 2θ = 20–30° (see Fig. 3).13,27 | ||
| Fig. 3 XRD patterns of the as-synthesized materials. | ||
N2 adsorption porosimetry
The desorption–adsorption isotherms of Pure Expst and Mo-Expst and their respective counterparts, Starbon-350 and CMO-350, are shown in the ESI (Fig. S2†), and the porosity data are listed in Table 1. Upon carbonisation, porosity develops as evidenced by an increase in the BET surface area and total pore volume. The materials are mesoporous in nature (BJH diameter, 14.64–23.11 nm), with negligible microporosity. However, the data reported are computed during desorption and as full hysteresis (loop closure) was not observed for Starbon-350 and CMO-350, this may be associated with filled micropores and negligible microporosity.| Sample | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Micropore volume (cm3 g−1) | Mesopore volume (cm3 g−1) | BJH adsorption pore diameter (nm) |
|---|---|---|---|---|---|
| Pure Expst | 74.96 | 0.3099 | 0.0002 | 0.3096 | 16.53 |
| Mo-Expst | 102.77 | 0.2993 | 0.0010 | 0.2983 | 23.11 |
| Starbon-350 | 368.83 | 0.5065 | 0.0847 | 0.4218 | 20.08 |
| CMO-350 | 196.42 | 0.3502 | 0.0215 | 0.3286 | 14.64 |
After carbonization at 350 °C, much of the mesoporosity is retained but the presence of micropores also is now evidenced. Starbon-350 and CMO-350 exhibit BET surface areas of 368.83 m2 g−1 and 196.42 m2 g−1, respectively, with pore volume in the range of 0.3–0.5 cm3 g−1. The decrease in the surface area and pore diameter can be attributed to the accumulation of nanoparticles on the surface and within the pores (pore-filling and blocking).
Thermogravimetric analysis (TGA)
The thermal properties of Starbon-350 and CMO-350 were studied with TGA (see Fig. 4). The first mass loss occurs around 100 °C, which is attributed to the desorption of physically adsorbed water molecules from the surface and pores of the material.22 The characteristic decomposition profile of expanded starch (Pure Expst) is observed with a strong endothermic transition at approx. 195 °C. This transition occurs at approx. 210 °C in the presence of molybdate (Mo-Expst) showing the stabilising effect of molybdenum on the decomposition of starch (see the ESI (Fig. S3†)). Furthermore, an endothermic transition at approx. 720 °C is observed in the molybdenum-containing samples providing evidence for molybdenum. MoO3 decomposes in the presence of carbon to form MoO2 and CO2.28 | ||
| Fig. 4 TGA profile of the as-synthesized products. | ||
X-ray photoelectron spectroscopy (XPS)
To further validate the FT-IR, TGA and XRD results of the synthesized MoO3 nanoparticles, XPS analysis was undertaken (see the ESI (Fig. S4†)). CMO-350 shows binding energy peaks at 232.36 and 235.5 eV corresponding to Mo 3d (6+) present in MoO3.29 The O 1s spectra show four binding energy peaks out of which three are at 531.78, 533.39 and 530.75 eV assigned to CO, C–O and O–H bonds, respectively.22 The deconvoluted spectra of C 1s show binding energy peaks at 287.94, 286.21, and 288.92 eV corresponding to CO, C–O and O–CO, respectively. C (sp2) and C (sp3) have binding energies of 284.27 eV and 284.8 eV, and π–π* has binding energies at 290.78 and 293.02 eV.23 The C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O atomic concentration as determined by XPS is 2.75:1, which is lower than that of Starbon-350 (5.10:1).26 The presence of oxygen in the nanoparticles decreases the atomic ratio in CMO-350, accordingly.
:O atomic concentration as determined by XPS is 2.75:1, which is lower than that of Starbon-350 (5.10:1).26 The presence of oxygen in the nanoparticles decreases the atomic ratio in CMO-350, accordingly.
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM)
The TEM and SEM images of Pure Expst, Starbon-350, Mo-Expst and CMO-350 are shown in Fig. 5 and 6, respectively. The TEM image of Pure Expst (Fig. 5, image A) shows an almost homogeneous structure around the pores, whilst the image for Starbon-350 (Fig. 5, image B) shows a platelet-like mosaic texture with wormhole-like connections. Importantly, the TEM images of Mo-Expst (image C) and CMO-350 (image D) are significantly different compared to their molybdenum-free counterparts. The MoO3 nanoparticles are homogeneously dispersed in the expanded starch matrix (image C) with an average size of approximately 2 nm, whilst the TEM of CMO-350 clearly shows that the surface and pores are decorated with the small-sized nanoparticles. | ||
| Fig. 5 TEM images of (A) Pure Expst, (B) Starbon-350, (C) Mo-Expst and (D) CMO-350. | ||
 | ||
| Fig. 6 SEM images of (A) Pure Expst, (B) Starbon-350, (C) Mo-Expst and (D) CMO-350. | ||
The SEM images further confirm the trends observed in the porosity of the material due to the incorporation of nanoparticles (Fig. 6). Significant differences are seen in the SEM images of molybdenum samples compared to their molybdenum-free counterparts. The latter reveal a highly porous, interconnected network (Fig. 6A and B), whilst the former show dendritic, fern-like growth emanating from a single point (Fig. 6C) followed by entanglement of these motifs into thick rope-like structures (Fig. 6D). Small nanoparticles can be seen in a clustered form decorated on the surface and pores of CMO-350.
Furthermore, the EDX surface analysis (Fig. 7D) provides clear evidence that nanoparticles are not aggregated in the form of clusters but are homogeneously dispersed on the interconnected framework of the composite.
 | ||
| Fig. 7 The elemental distribution: (A) the grey image of the area under focus, (B) carbon, (C) oxygen and (D) molybdenum. | ||
Catalytic study
The borohydride reduction of 4-NP to 4-AP, in the presence of CMO-350, as monitored by UV-vis spectroscopy is shown in Fig. 8A. The spectrum of the neutral aqueous solution of 4-NP exhibits a strong absorption peak with the wavelength maxima at 317 nm. Upon addition of an excess amount of NaBH4, the alkalinity of the solution increases to pH 9–10 and 4-NP transforms into 4-nitrophenolate ions evidenced by a bathochromic shift to 400 nm and a yellow-coloured solution.30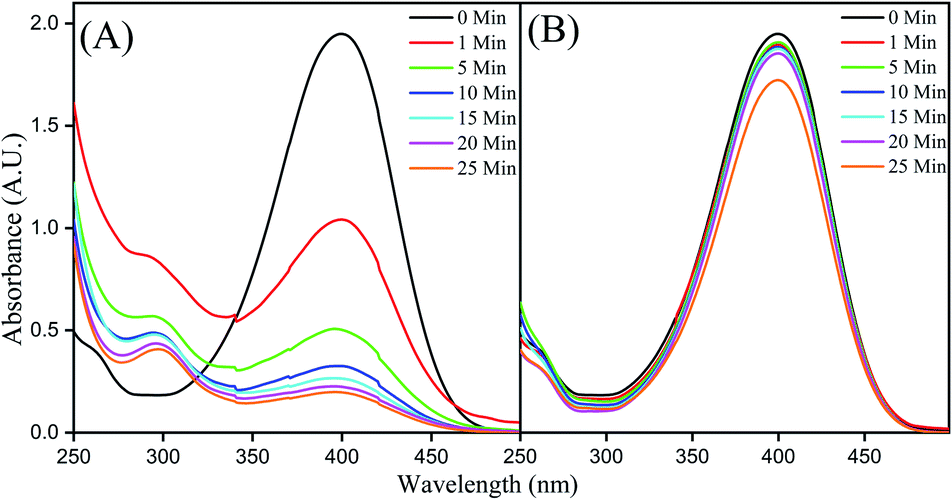 | ||
| Fig. 8 (A) UV-Vis spectra for the time study of reduction of 4-NP by NaBH4 on CMO-350 and (B) Starbon-350. | ||
In the presence of the catalyst, the intensity of the absorption band corresponding to 4-nitrophenolate anions decreases with the simultaneous appearance and growth of a weak absorption band at 290 nm corresponding to the formation of 4-AP.31 Over time, the yellow colour gradually faded and diminished along with the concomitant enhancement of the signal at ∼290nm.32 A control experiment with Starbon-350 was conducted which demonstrated no reduction activity, although a small decrease in the signal intensity due to surface adsorption could be observed (see Fig. 8B). The molybdenum nanoparticles were exerting a catalytic effect and not the mesoporous scaffold.
The catalytic reduction of 4-NP involves two steps: (i) initial diffusion and adsorption of 4-NP on the catalyst surface and (ii) catalyst-mediated electron transfer from NaBH4 to 4-NP.40,41 The large exposed surface area of the composite leads to instantaneous adsorption of 4-NP and promotes faster electron transfer.42,43 This leads to high initial catalytic reduction rate which is overall presented in Fig. 9. The apparent rate constant, kapp, for the conversion of 4-NP to 4-AP in the presence of CMO-350 was estimated to be 11.2 × 10−2 min−1.
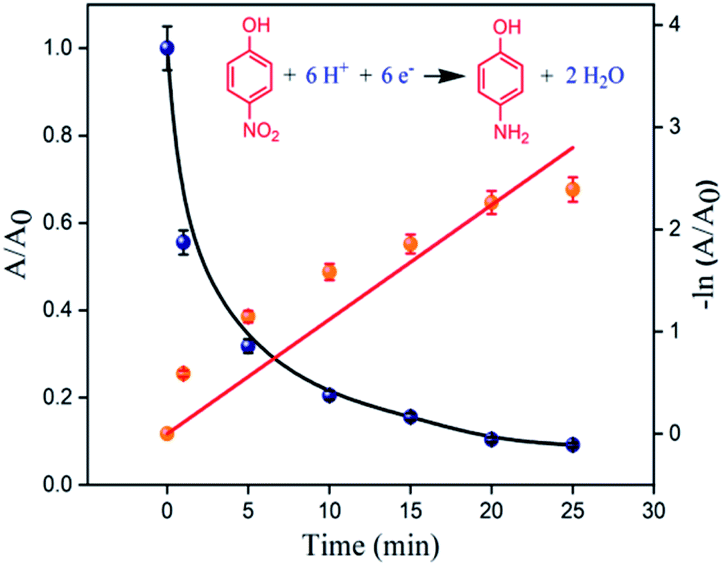 | ||
| Fig. 9 Comparison of the plots of absorbance ratio A/A0vs. t (blue spheres) and ln(A/A0) vs. t (orange spheres). | ||
In order to better contextualize kapp, the activity factor (κ), i.e., the ratio of the rate constant kapp to the total weight of the catalyst, was compared with that of literature reported catalysts for the borohydride reduction of 4-NP to 4-AP (see Table 2). Compared to other molybdenum catalysts (entries 2 and 3, Table 2), CMO-350 has a significantly better activity factor. For example, CMO-350 is almost 5-fold better than Ru/MoO3 (entry 2, 0.25 × 10−2 min−1 mg−1) and almost 3-fold better than CdS–3% MoS2/0.01rGO (entry 3, 0.42 × 10−2 min−1 mg−1).
| S. no. | Catalyst | k app (×10−2 min−1) | Quantity (mg) | Activity factor, κ (×10−2 min−1 mg−1) | Reference |
|---|---|---|---|---|---|
| 1 | CMO-350 | 11.2 | 10 | 1.12 | Present work |
| 2 | Ru/MoO3 | 6.3 | 25 | 0.25 | 33 |
| 3 | CdS–3% MoS2/0.01rGO | 8.44 | 20 | 0.422 | 34 |
| 4 | Ni/TiO2–C-2 | 9.0 | 3.4 | 2.65 | 35 |
| 5 | Ag/CB–CS | 10.4 | 15 | 0.69 | 36 |
| 6 | GNP(PEI/MWNTs) | 8.7 | NA | 2.88 | 37 |
| 7 | Pd NP/CNT-220 | 63.2 | 50 | 1.26 | 38 |
| 8 | Au@C | 2.88 | 2 | 1.44 | 39 |
DPPH radical quenching activity
Electron paramagnetic resonance (EPR) spectroscopy was conducted to determine the radical quenching activity of different concentrations of CMO-350 and Starbon-350 (Fig. 10). The results were further validated by complementary UV-Vis spectroscopy studies. The DPPH radical scavenging activity was estimated as a ratio of individual DPPH signal peak height to that of the control. | ||
| Fig. 10 Concentration dependent EPR graph of DPPH radicals for (A) CMO-350 and (B) Starbon-350; UV graph for (C) CMO-350 and (D) Starbon-350. | ||
As CMO-350 concentration increases, the relative peak height of DPPH radicals decreases. Complete radical quenching is observed at a concentration of 2.5 mg ml−1. However, pure Starbon-350 (no molybdenum, acting as a control) exhibits no significant changes in the peak intensity of DPPH radicals even at a high concentration (5 mg ml−1). The very small decrease in peak intensity can be attributed to the adsorption of DPPH molecules on the Starbon matrix. Thus, the molybdenum nanoparticles are responsible for DPPH quenching and not the Starbon matrix. For all the spectroscopic methods used, the IC50 values (the concentration which causes 50% quenching of free radicals) were in close agreement (Fig. 11: EPR, 1.006 mg ml−1; UV-vis, 1.066 mg ml−1).
 | ||
| Fig. 11 (A) EPR signal inhibition graph and (B) UV signal inhibition graph when using CMO-350. The inset shows the corresponding values of IC50. | ||
The radical quenching activity of CMO-350 was compared with that of other reported catalysts in the literature (see Table 3). For example, the IC50 value of CMO-350 was less than that for biologically-reduced graphene oxide (entries 4 and 5) and almost 8 times less than that of bio-Ag nanoparticles (entry 7). CMO-350 also shows good radical scavenging activity in comparison to the widely used magnetic iron NPs and CeO2 NPs (see entries 2 and 6).
FRAP assay
A FRAP assay was conducted to correlate the inferences obtained from DPPH quenching. Similarly, as the concentration of CMO-350 increased, the yellow colour of the solution changed from light to dark green (Fig. 12). Mechanistically, the reduction of the Fe3+/ferricyanide complex to the ferrous form in the presence of reducers (i.e. antioxidants) leads to the formation of Prussian blue, which can be used to monitor the concentration of Fe2+ at 700 nm. The onset of a plateau is noted at concentrations above 100 μl mg−1. The greater the intensity of the colour formed, the greater will be the absorption, consequently the greater will be the antioxidant activity.50 Again, no FRAP activity was detected for the control sample, Starbon-350.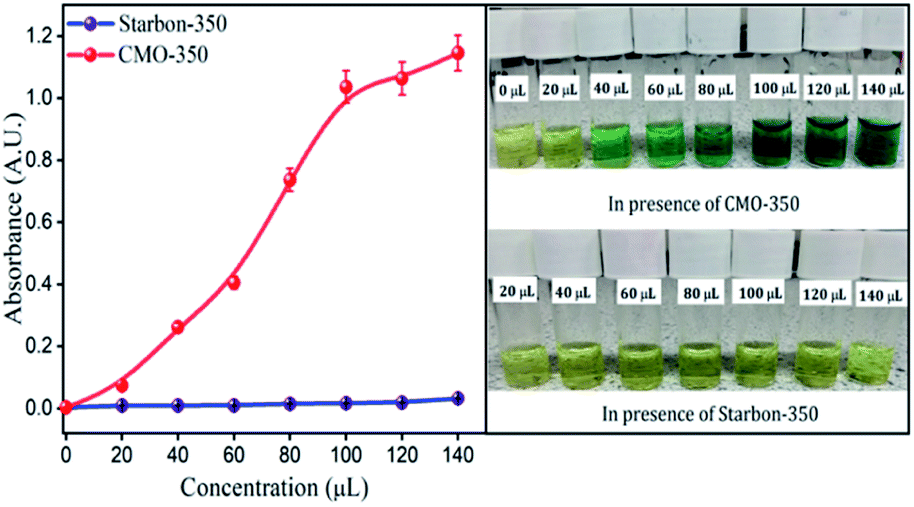 | ||
| Fig. 12 FRAP activity of CMO-350 and Starbon-350 measured at 700 nm. | ||
Conclusions
In summary, a novel composite containing molybdenum oxide nanoparticles embedded on a Starbon framework has been synthesized, characterized and evaluated for its potential as a novel material for wastewater remediation. The material is easy to fabricate, robust and derived from a renewable resource. The process follows an in situ green process without the use of any harsh chemicals. The structure has high mesoporosity with an interconnected pore network, which aids adsorption and transport to the active sites. The catalytic activity of CMO-350 was evaluated through the reduction of 4-NP in NaBH4 solution. The results demonstrated that the as-prepared composite shows a good apparent rate constant, kapp, of 11.2 × 10−2 min−1, in comparison to the literature reported catalysts. Furthermore, the composite shows very good antioxidant behaviour against DPPH free radicals as evidenced by EPR (IC50 = 1.006 mg ml−1) and UV-vis measurements (IC50 = 1.066 mg ml−1). This study was corroborated by the FRAP assay and showed that CMO-350 has high reducing capacity in comparison to Starbon-350.Thus, the study presented is expected to pave the way for large-scale production of a highly active and durable material for real world catalytic water treatment and abatement of 4-nitrophenol.
Experimental
Materials and chemicals
All chemicals used were of reagent grade and used without any further purification. Hylon®VII corn starch was purchased from National Starch and Chemical Limited with an amylose content of approx. 75%. Ammonium heptamolybdate tetrahydrate (AHM, Sigma Aldrich, 81–83% MoO3 basis), para-toluenesulfonic acid monohydrate (≥98.5%, Aldrich), tert-butanol (TBA, ≥99.0%, Aldrich), para-nitrophenol (4-NP, ≥99.0%, Sigma Aldrich), 2,2-diphenyl-1-picrylhydrazyl (DPPH, Aldrich), sodium hydroxide (NaOH, Fischer), hydrochloric acid (HCl, Fischer), ethanol and acetone were obtained from VWR Chemicals®. Deionised water was obtained in the laboratory using an ELGA Centra® system. All catalytic and radical quenching experiments were repeated thrice to maintain good reproducibility of the results.As control samples, expanded starch (Pure Expst) and Starbon-350 (undoped) were prepared according to a standard literature methodology.9,51 Full details of instruments used are given in the ESI.†
Synthesis of the MoO3 nanoparticles@mesoporous carbon composite (CMO)
AHM (1 wt%) was added to a mixture of Hylon® VII corn starch and deionised water (1:10 wt/wt) and stirred (800 rpm) for 5 min at room temperature. The mixture was transferred into a Teflon vessel and subjected to microwave irradiation (CEM Mars 6 instrument) at 140 °C for 10 min, 800 psi and 900 W. Thereafter, the mixture was placed at 4 °C for 48 h, and the final product was labelled Pure Expst. It was further macerated and p-toluenesulfonic acid (30 wt%) and tert-butanol (5 wt%) were added for solvent exchange. The above mixture was stirred at room temperature overnight and freeze dried to afford molybdenum doped expanded starch (Mo-Expst), as a light blue powder. The latter was subjected to controlled carbonization under vacuum at 350 °C to afford the desired molybdenum–Starbon-350 composite (CMO-350), as a black powder. A control experiment was also conducted in the absence of starch at pH < 5 under similar experimental conditions for the synthesis of nanocrystalline MoO3 (see ESI Fig. S1†).
4-NP adsorption study
Adsorption studies were investigated by UV-Vis spectroscopy (JASCO V-550). CMO-350 (2 mg) was added to an aqueous solution of 4-NP (5 ml, 0.1 mM) (see the ESI (Fig. S5†)).Catalytic reduction of 4-NP
The catalytic activity of CMO-350 was evaluated through the reduction of 4-NP to para-aminophenol (4-AP).30 NaBH4 (14.5 mg, 3.8 × 10−2 mol) was dissolved in an aqueous solution of 4-NP (5 ml, 0.1 mM) and stored in the dark for 1 h. CMO-350 (2.5 mg) was then added at room temperature and the mixture was subjected to UV-Vis spectroscopy. The absorbance of the solution was measured at different intervals from time t = 0 (with no catalyst addition) to 25 minutes (with addition of the catalyst) in the range of 250 to 500 nm. The kinetics of the 4-NP reduction was studied by monitoring the change in absorbance at 400 nm.DPPH radical quenching activity: ESR and UV study
Electron paramagnetic resonance (EPR) measurements were performed to determine the DPPH radical scavenging activity of CMO-350 and Starbon-350.52 A stock solution was prepared by dissolving DPPH (4 mg) in ethanol (100 ml) and stored in the dark at 25 °C until required. A range of ethanolic composite mixtures (10–100 μg ml−1) were prepared by mixing an appropriate amount of CMO-350 with a given volume of ethanol. An aliquot (1 ml) of the appropriate ethanolic composite mixture was mixed with the DPPH solution (1 ml). The resultant mixture was shaken well and incubated in the dark for 30 min at room temperature. The mixture was transferred to ESR tubes which were, in turn, placed in the cavity of an ESR spectrometer. The ethanolic DPPH solution was used as a control, i.e. without CMO-350. The DPPH radical scavenging activity was estimated as a ratio of individual DPPH signal peak height to that of the control (eqn (1)). | (1) |
Complementary information was also collected by UV-Vis study. The absorbance was measured at 517 nm where lower absorbance indicated higher radical scavenging activity (see ESI Fig. S6†). The scavenging activity against DPPH was calculated according to eqn (2) whilst % inhibition was determined using eqn (3):
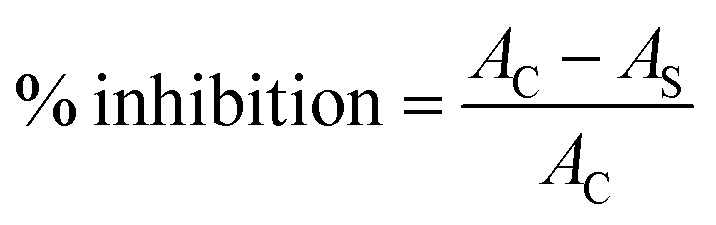 | (2) |
 | (3) |
FRAP assay
FRAP assay was performed by following the procedure of Oyaizu et al.,53 with some modifications. A mixture of 1% aqueous potassium ferricyanide (1 ml), aqueous sodium phosphate buffer (0.2 M, pH 6.6, 1 ml) and an appropriate ethanolic CMO-350 solution (1 ml) were mixed and incubated at 50 °C (water bath) for 20 min. After incubation, 10% aqueous trichloroacetic acid (2.5 ml) was added to each reaction mixture, centrifuged (3000 rpm, 10 min) and an aliquot (1.5 ml) of the supernatant was mixed with water (1.5 ml) and FeCl3 solution (0.1%, 0.1 ml). The resultant mixture was incubated for 10 min at room temperature and then transferred to a quartz cuvette. The absorbance of the mixture was measured at 700 nm with respect to the blank, which contained all the reagents except 1% potassium ferricyanide solution.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
ASM, SKM, KK and JK wish to acknowledge the Commonwealth Scholarship Commission in the UK for the award of a Commonwealth Split-Site Scholarship to JK (Ref No. INCN-2018-75). JK would also like to acknowledge GCCE, University of York, United Kingdom, and Panjab University, Chandigarh, for administrative and technical support. SKM and JK are also thankful to the CSIR for an open SRF Fellowship and for CSIR project grant (Ref No. 01(2951)/18/EMR-II). KK would like to thank the DST for funding and the INSPIRE faculty award.Notes and references
- G. Ghasemzadeh, M. Momenpour, F. Omidi, M. R. Hosseini, M. Ahani and A. Barzegari, Front. Environ. Sci. Eng., 2014, 8, 471–482 CrossRef CAS.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- H. Lu, J. Wang, M. Stoller, T. Wang, Y. Bao and H. Hao, Adv. Mater. Sci. Eng., 2016, 1–10 Search PubMed.
- I. Gehrke, A. Geiser and A. Somborn-Schulz, Nanotechnol., Sci. Appl., 2015, 1 CAS.
- M. Hu, Y. Liu, Z. Yao, L. Ma and X. Wang, Front. Environ. Sci. Eng., 2018, 12, 3 CrossRef.
- M. Anjum, R. Miandad, M. Waqas, F. Gehany and M. A. Barakat, Arabian J. Chem., 2019, 12, 4897–4919 CrossRef CAS.
- M. A. Tony, H. L. Parker and J. H. Clark, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2016, 51, 974–979 CrossRef CAS PubMed.
- A. S. Adeleye, J. R. Conway, K. Garner, Y. Huang, Y. Su and A. A. Keller, Chem. Eng., 2016, 286, 640–662 CrossRef CAS.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401 RSC.
- H. L. Parker, A. J. Hunt, V. L. Budarin, P. S. Shuttleworth, K. L. Miller and J. H. Clark, RSC Adv., 2012, 2, 8992 RSC.
- M. Ojeda, V. Budarin, P. Shuttleworth, J. Clark, A. Pineda, A. Balu, A. Romero and R. Luque, Materials, 2013, 6, 1891–1902 CrossRef CAS PubMed.
- H.-J. Lunk, H. Hartl, M. A. Hartl, M. J. G. Fait, I. G. Shenderovich, M. Feist, T. A. Frisk, L. L. Daemen, D. Mauder, R. Eckelt and A. A. Gurinov, Inorg. Chem., 2010, 49, 9400–9408 CrossRef CAS PubMed.
- V. A. Doss, A. Chithambararaj and A. C. Bose, AIP Conf. Proc., 2016, 050049 CrossRef.
- A. Chithambararaj, N. S. Sanjini, S. Velmathi and A. Chandra Bose, Phys. Chem. Chem. Phys., 2013, 15, 14761 RSC.
- D. Belanger and G. Laperriere, Chem. Mater., 1990, 2, 484–486 CrossRef CAS.
- R. Sundaram and K. S. Nagaraja, Sens. Actuators, B, 2004, 101, 353–360 CrossRef CAS.
- M. H. Yaacob, J. Yu, K. Latham, K. Kalantarzadeh and W. Wlodarski, Sens. Lett., 2011, 9, 16–20 CrossRef CAS.
- A. Fakhri and P. A. Nejad, J. Photochem. Photobiol., B, 2016, 159, 211–217 CrossRef CAS PubMed.
- A. J. Hunt, A. S. Matharu, A. H. King and J. H. Clark, Green Chem., 2015, 17, 1949–1950 RSC.
- A. Chithambararaj and A. Chandra Bose, CrystEngComm, 2014, 16, 6175–6186 RSC.
- A. S. Matharu, S. Ahmed, B. Almonthery, D. J. Macquarrie, Y.-S. Lee and Y. Kim, ChemSusChem, 2018, 11, 716–725 CrossRef CAS PubMed.
- S. Kim, A. M. Escamilla-Pérez, M. De bruyn, J. G. Alauzun, N. Louvain, N. Brun, D. Macquarrie, L. Stievano, B. Boury, L. Monconduit and P. H. Mutin, J. Mater. Chem. A, 2017, 5, 24380–24387 RSC.
- M. L. Bhaisare, K. H. Sharma, J.-Y. Lee, D.-R. Hang and H.-F. Wu, New J. Chem., 2016, 40, 8954–8960 RSC.
- Z. Zhang, D. J. Macquarrie, J. H. Clark and A. S. Matharu, RSC Adv., 2014, 4, 41947–41955 RSC.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- A. Chithambararaj and A. Chandra Bose, Adv. Mater. Res., 2012, 488–489, 940–944 CAS.
- P. A. Spevack and N. Mcintyre, J. Phys. Chem., 1992, 96, 9029–9035 CrossRef CAS.
- M. Anwar, C. A. Hogarth and R. Bulpett, J. Mater. Sci., 1990, 25, 1784–1788 CrossRef CAS.
- H. Hu, J. H. Xin, H. Hu and X. Wang, Nano Res., 2015, 8, 3992–4006 CrossRef CAS.
- Y. Feng, J. Yin, S. Liu, Y. Wang, B. Li and T. Jiao, ACS Omega, 2020, 5, 3725–3733 CrossRef CAS PubMed.
- F. Zhan, J. Yin, J. Zhou, T. Jiao, L. Zhang, M. Xia, Z. Bai and Q. Peng, Nanomaterials, 2020, 10, 6 CrossRef CAS PubMed.
- H. Yin, Y. Kuwahara, K. Mori, M. Che and H. Yamashita, J. Mater. Chem. A, 2019, 7, 3783–3789 RSC.
- W. Peng, Y. Chen and X. Li, J. Hazard. Mater., 2016, 309, 173–179 CrossRef CAS PubMed.
- W. Gao, W. Li, Z. Xue, M. Pal, Y. Liu, C. Wang, J. Wang, S. Wang, X. Wan, Y. Liu and D. Zhao, New J. Chem., 2016, 40, 4200–4205 RSC.
- F. Ali, S. B. Khan, T. Kamal, Y. Anwar, K. A. Alamry and A. M. Asiri, Chemosphere, 2017, 188, 588–598 CrossRef CAS PubMed.
- S. Liu, X.-L. Zhou, M.-M. Zhang, X. Lu, Y.-J. Qin, P. Zhang and Z.-X. Guo, Chin. Chem. Lett., 2016, 27, 843–846 CrossRef CAS.
- X. Gu, W. Qi, X. Xu, Z. Sun, L. Zhang, W. Liu, X. Pan and D. Su, Nanoscale, 2014, 6, 6609–6616 RSC.
- B. Guan, X. Wang, Y. Xiao, Y. Liu and Q. Huo, Nanoscale, 2013, 5, 2469 RSC.
- Md. Ahmaruzzaman, D. Mohanta and A. Nath, Sci. Rep., 2019, 9, 12935 CrossRef PubMed.
- L. Ge, M. Zhang, R. Wang, N. Li, L. Zhang, S. Liu and T. Jiao, RSC Adv., 2020, 10, 15091–15097 RSC.
- K. Sravanthi, D. Ayodhya and P. Y. Swamy, Mater. Sci. Energy Technol., 2019, 2, 298–307 Search PubMed.
- T. K. Das, P. Bhawal, S. Ganguly, S. Mondal and N. C. Das, Surf. Interface Sci., 2018, 13, 79–91 CrossRef CAS.
- S. T. Shah, W. A. Yehye, Z. Z. Chowdhury and K. Simarani, PeerJ, 2019, 7, e7651 CrossRef PubMed.
- S. Menon, H. Agarwal, S. R. Kumar and S. V. Kumar, Int. J. Appl. Pharm., 2017, 9, 42 CrossRef CAS.
- D. Suresh, P. C. Nethravathi, A. Udayabhanu, H. Nagabhushana and S. C. Sharma, Ceram. Int., 2015, 41, 4810–4813 CrossRef CAS.
- D. Suresh, A. Udayabhanu, H. Nagabhushana and S. C. Sharma, Mater. Lett., 2015, 142, 4–6 CrossRef CAS.
- I. Nurhasanah, W. Safitri, Z. Arifin, A. Subagio and T. Windarti, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 432, 012031 CrossRef.
- S. Paul, S. Sasikumar and M. Dakshinamoorthi, J. Complement. Med. Alt. Healthcare, 2018, 7(4), 555718 Search PubMed.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70–76 CrossRef CAS PubMed.
- P. S. Shuttleworth, J. Parker, V. L. Budarin, S. W. Breeden, D. J. Macquarrie, R. L. Luque, R. White and J. H. Clark, NSTI Nanotech., 2011, 3, 4 Search PubMed.
- Y. Deligiannakis, G. A. Sotiriou and S. E. Pratsinis, ACS Appl. Mater. Interfaces, 2012, 4, 6609–6617 CrossRef CAS PubMed.
- M. Oyaizu, Jpn. J. Nutr., 1986, 44(6), 307–315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05388k |
| This journal is © The Royal Society of Chemistry 2020 |