
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSugar-alcohol@ZIF nanocomposites display suppressed phase-change temperatures†
Lukas
Hackl‡
ab,
Chih-Hao
Hsu‡
a,
Madeleine P.
Gordon
ac,
Kelly
Chou
ad,
Canghai
Ma
a,
Matthew
Kolaczkowski
ad,
Christopher L.
Anderson
 ad,
Yi-Sheng
Liu
e,
Jinghua
Guo
e,
Peter
Ercius
a and
Jeffrey J.
Urban
*a
ad,
Yi-Sheng
Liu
e,
Jinghua
Guo
e,
Peter
Ercius
a and
Jeffrey J.
Urban
*a
aThe Molecular Foundry, Lawrence Berkeley National Lab, 1 Cyclotron Road, Berkeley, CA 94720, USA. E-mail: jjurban@lbl.gov
bDepartment of Civil and Environmental Engineering, University of California Berkeley, 760 Davis Hall, Berkeley, CA 94720, USA
cApplied Science and Technology Graduate Group, University of California Berkeley, 210 Hearst Memorial Mining Building, Berkeley, CA 94720, USA
dDepartment of Chemistry, University of California Berkeley, 419 Latimer Hall, CA 94720, USA
eAdvanced Light Source, Lawrence Berkeley National Lab, 1 Cyclotron Road, Berkeley, CA 94720, USA
First published on 16th November 2020
Abstract
For the sake of water and energy conservation, development of latent heat cooling and thermal storage systems that minimize water consumption and operate with higher efficacy than their water-driven counterparts is a crucial task. Phase change materials (PCMs) present a potential solution, but their integration into real-world systems abounds with scientific challenges such as material toxicity, flammability, low thermal performance and lack of tunable phase-change temperatures. In this study we report on a first-in-class nanocomposite PCM that leverages non-flammable, non-toxic, high latent heat sugar alcohols (SAs) encapsulated within easy-to-synthesize zeolitic imidazolate framework (ZIF) crystals. We also outline a practical route for surface functionalization with hydrophilic and hydrophobic moieties. The SA@ZIF composites display suppressed phase-change temperatures which, together with alterable surface functionality, broadens their applicability to a plethora of working environments. Direct synthesis of the SA@ZIF composite generates nanoconfined SAs with phase-change temperatures as low as 19.8 °C and latent heats as high as 285 J g−1. This nanoconfinement-induced thermal phenomenon is conserved even after functionalization of the SA@ZIF crystal surface. We believe this study will lay the groundwork as a platform for next generation high performing, tunable PCMs to aid in the realization of waterless cooling systems.
1. Introduction
Water and energy are deeply intertwined resources. In the U.S, thermoelectric power generation consumes over 40% of fresh water (primarily used in cooling towers to regulate reactor temperatures), more than any other industrial sector.1 Re-injection of cooling waters from power plants into rivers and lakes has been shown to raise temperatures of aquatic ecosystems, often with detrimental effects referred to as thermal pollution.2 For the sake of both energy efficiency and environmental sustainability, there has therefore been increasing effort within the scientific community to develop materials that reduce or eliminate the need to pump, treat, and consume water for cooling.Phase change materials (PCMs) have received increasing attention for their ability to store thermal energy in the form of latent heat when undergoing a phase transition.3 Numerous studies have shown how this property can be leveraged for thermal management applications by encapsulating the PCM to serve as a functional additive.4 PCMs have been integrated into solar thermal storage systems,5 pumped coolants,6 and heat exchangers7 to varying degrees of success.8 To be of relevance, a given PCM must not only display a latent heat of fusion (ΔHL) that is as high as possible, its phase-change temperature (TPC) must also correlate the operating range of the application in question. As such, being able to control and tailor a PCM's TPC is highly desirable.
Organic, hydrocarbon derived materials like paraffin waxes are commonly used PCMs due to their suitable thermal properties (3 °C < TPC < 75 °C), low vapor pressure and chemical stability.9 However, paraffins suffer from technical shortcomings such as low volumetric latent heats, flammability and low thermal conductivity, vastly limiting their applicability.9 Inorganic salt hydrates (e.g. sodium sulphate), the other commonly used class of PCMs, exhibit a different set of undesirable material properties: despite their sharp melting points, these materials undergo large volume changes upon crystallization, can corrode cooling hardware and are susceptible to super cooling, thus requiring the use of nucleating agents to induce phase change.10 Sugar alcohols (SAs) on the other hand, like erythritol and xylitol, display some of the highest latent heats (ΔHL > 300 J g−1) amongst all organic and inorganic PCMs, are non-corrosive, non-toxic, inexpensive and their chemical stability over multiple solid/liquid cycles has been demonstrated, for example, by Akiyama et al.11 What limits their integration into real-world cooling systems is their relatively high phase change temperature (>100 °C) – a problem we address in this study.
Traditional liquid cooling systems rely on pumped thermal storage liquids (often water) to transport heat from a heat source and to a heat sink. To serve as functional additives for such a system, PCMs must be encapsulated to avoid their liquid phase from leaching into the transport fluid. Routes for encapsulation cover a wide range of techniques such as confinement to core–shell polymer architectures,12 infusion into 3D porous frameworks,13–15 and aided diffusion into carbon nanotubes,16 nanofibers,17 and metal–organic frameworks (MOFs).18–23 Owing to enhanced surface chemical effects at the nanoscale, PCM confinement has in many cases been shown to favourably impact thermodynamic properties, offering more efficient heat transfer, suppressing supercooling and accommodating volumetric changes.4,19 Confinement can also involve complex, multi-step syntheses and expensive materials like carbon nanotubes that prohibit production at meaningful scale. As such, materials with a high degree of nanoporosity, like MOFs for example, are one of the few candidates that offer true, three-dimensional nanoconfinement. MOFs also can often be synthesized easily at scale and tend to remain stable even at high temperatures.24 A variety of MOF-PCM composites have been synthesized and often, as exemplified by the works of Atinafu,18,21 Tang20 and Luan et al.,19 rely on polyethylene glycol (PEG) or organic acids as the PCM. Amongst these studies, Tang et al. report the highest latent heats (ΔHL ∼ 180 J g−1).20 At least one study of a zeolitic imidazolate framework (ZIF) – PCM composite exists. Li et al. report that their cobalt-based, stearic acid infused ZIF-67 crystals on expanded graphite display a ΔHL of ∼ 213 J g−1.23 While impressive in performance, all these studies fail to test for cyclability of their materials in water and do not enable any degree of phase change temperature tunability.
In this study, we present a new class of encapsulated PCMs which leverage the uniformly porous morphology of a common type of ZIF to induce shifts in the TPC of two high latent heat SAs (erythritol and xylitol). ZIFs, a subclass of MOFs, were chosen to serve as the encapsulating scaffold due to their chemical and thermal stability,25 homogenous pore size distribution,26 and their inherent stability in water.27 Within this class of materials, zinc-methylimidazole based ZIFs (ZIF8, ZIF-L, etc.) are widely considered the prototypical ZIF due to their basic precursors, and importantly, their well-documented and facile synthesis under a variety of reaction conditions.27,28 As we are attempting to synthesize our materials in an atypical solvent (molten erythritol), high tolerance for different solvents was a particularly important consideration. We go on to characterize their thermal properties and reveal how confinement of the SAs leads to effective suppression of their phase-change temperatures while maintaining exceptionally high latent heats. Given these properties, this ZIF@SA composite material possesses great potential for integration into thermal management applications. Also, we present a facile methodology for grafting hydrophobic molecules and hydrophilic polymer chains to the surface of the SA-infused ZIF crystals. Unlike other studies, we prevent leaching of liquid phase SA and tune surface wettability.
2. Results and discussion
2.1 SA@ZIF characterization
The ZIF based materials characterized in this study were synthesized in different molten SAs according to the general schematic shown in Fig. 1. The initial goal of this study was to synthesize ZIF-8 structures with SAs intercalated into the porous framework. To do so, a published ZIF-8 synthesis protocol was followed, using zinc hexahydrate (ZnH) and 2-methylimidazole (Mim) as precursors.29 Instead of methanol however, the reaction was carried out in molten SAs. Erythritol and xylitol were chosen due to their high latent heats (340 J g−1 and 280 J g−1) and comparatively low melting points (120 °C and 94 °C). The morphology of both of the resulting crystals, erythritol@ZIF (EZ) and xylitol@ZIF (XZ), were first assessed via scanning- (SEM) and scanning transmission electron microscopy (STEM). Fig. 2 depicts representative SEM and STEM images of the EZ and XZ samples, collected after 4 hours of reaction time. Neither sample was found to display the characteristic dodecahedral, “soccer ball” morphology typical of ZIF-8 crystals.29 | ||
Fig. 1 Schematic overview of the synthetic routes utilized in this study to generate both erythritol and xylitol infused ZIF crystals. The molar precursor ratios between the Mim and ZnH were adjusted (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 20:1) to induce ZIF-L or ZIF-8 crystal structures to emerge. :1 or 20:1) to induce ZIF-L or ZIF-8 crystal structures to emerge. | ||
 | ||
| Fig. 2 SEM micrographs at different magnifications of EZ (a and b) and XZ (c and d) crystals collected after 4 h at 230 °C and 200 °C respectively. Panel (e) depicts a STEM image of a six-armed, star-like EZ crystal (reaction carried out at 200 °C for 4 h). | ||
To better understand this unexpected crystal morphology, powder X-ray diffraction (XRD) spectra were collected and compared to the computer-simulated patterns of various ZIF polymorphs. The XRD patterns, shown in Fig. 3, reveal that by including SAs in the synthesis, the resulting crystalline morphology no longer corresponds to that of ZIF-8. Instead it matches the computer-generated XRD pattern of ZIF-L, a structurally similar 2D polymorph of ZIF-8. Particularly telling are the doublet peaks arising at low q spacings for both XZ and EZ.30 ZIF-L is a layered structure with ovular, “cushion-shaped” cavities, made up of the same building blocks as ZIF-8. Given the appearance of additional peaks, XZ and EZ XRD patterns match those reported for ZIF-L and further confirm the lower symmetry of the orthorhombic ZIF-L structure as compared to the cubic space group structure of ZIF-8.31,32 This not only enables the identification of the crystal structure of the material but also confirms that there is in fact a ZIF-based framework present in the system.
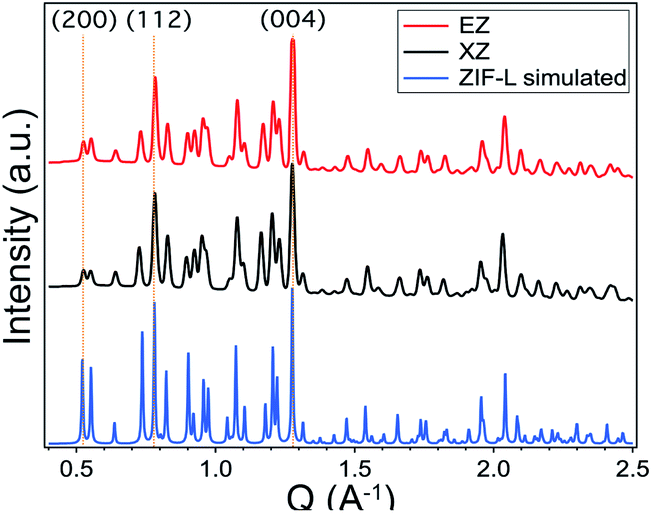 | ||
| Fig. 3 Powder X-ray diffraction patterns of EZ and XZ crystals in comparison to computer-generated diffraction patterns of ZIF-L. Both the EZ and the XZ samples display the doublet peaks around q = 0.5 Å−1 that correspond to the (200) and (111) planes and are indicative of ZIF-L structure. | ||
It is well known that synthesis conditions have a significant effect on ZIF crystal morphology, size, and topology.26 Solvent selection,33 precursor ratios,34 reaction temperature,35 and reaction time36 have all been shown to play a central role in the ZIF crystallization process. ZIF-L crystals made in DI water have for example been shown to convert into ZIF-8 crystals, simply by suspending them in ethanol for 8 hours at 60 °C.30 Any or all of these factors could be responsible for our reaction having yielded a ZIF-L rather than a ZIF-8 crystal structure. Given the high affinity of zinc and imidazole precursors to self-assemble in a variety of synthesis environments,25 we believe ligand replacement (i.e. imidazole substitution with SA) to be an unlikely explanation for the change in crystal structure.
It is also important to note that the samples depicted in Fig. 2 and collected after 4 hours of reaction time also appear to have different macroscale morphologies. While XZ crystals are star-shaped, the EZ crystals are platelets. Since the EZ synthesis was carried out at 230 °C rather than 200 °C, owing to erythritol's higher melting point, it is likely the two reactions progressed at different rates. To provide further clarification, the EZ synthesis was carried out at 200 °C and STEM and tomography images (Fig. S1†) show the sample displaying a star-like morphology similar to XZ. Aiming to incorporate these composite materials into real world cooling systems, their thermal properties were characterized via DSC.
As seen in Fig. 4c and d, heat flow into the SA samples starts to deviate from baseline at ∼120 °C and ∼93 °C, defined as a material's phase change temperature (TPC). These values correspond closely to the literature reported phase change temperatures of erythritol and xylitol, respectively.37
 | ||
| Fig. 4 DSC heat flow (mW) into (a) EZ, (b) bulk erythritol, (c) XZ and (d) bulk xylitol upon heating. TPCs for EZ and XZ are clearly depressed compared to their bulk SA counterparts. When normalized by weight of contained SA the latent heats of fusion are 150 J g−1 for the XZ and 285 J g−1 for the EZ sample. | ||
More interesting are the results for the XZ and EZ samples shown in Fig. 4a and b. The phase change temperatures, suppressed significantly as compared to their bulk counterparts, were determined to be 19.8 °C for EZ and 23.3 °C for XZ. The better-performing EZ sample was heated repeatedly to confirm the reproducibility of TPC depression phenomenon (Fig. S8†). XANES and XRD spectra collected for both samples at room temperature and 240 °C look virtually identical in terms of peak positions and shape for each sample (Fig. S9†). This suggests that these materials undergo little to no structural or chemical change in response to heating. Per TGA, the amount of SA contained in each sample as a fraction of total mass is 19 wt% for EZ and 28 wt% for XZ crystals (Fig. S2†).
When normalized by the respective mass of SA, the latent heats of fusion (ΔHL) were determined to be 150 J g−1 for the xylitol-infused ZIFs. At a latent heat of 285 J g−1, erythritol containing crystals displayed superior thermal performance compared to materials produced in other studies. For comparison, Li et al. measured a ΔHL of 216 J g−1 for their stearic acid infused ZIF-67 crystals,23 whereas Tang et al.'s PEG-infused MOF-5s displayed a ΔHL of 181.9 J g−1 (ref. 20) and the stearic acid loaded MOFs (MIL-101-NH2) by Luan et al. came in around 120 J g−1 (ref. 19).
Further evidence of the successful encapsulation of SAs within the ZIF architecture can be gathered from X-ray absorption spectra in Fig. 5. Specific attention was paid to the oxygen 1s K-edge absorption (usually seen at 532 eV) as the SAs present in the system are the only oxygen containing compounds. The surface of a sample can be differentiated from the bulk by probing at the oxygen edge absorption in the total electron yield (TEY) and total fluorescence yield (TFY), respectively. The TEY spectra of XZ and EZ (Fig. 5a) show no difference in the pre-edge peak position (532 eV) compared to their counterpart bulk SAs. This indicates that the SAs close to the surface of ZIF exist in the same chemical environment as they do in the bulk state. In Fig. 5b where the bulk is probed in TFY, there is a clear shift in pre-edge peak position from 532 eV to 534 eV for the SAs trapped inside the ZIF pores. This blue shift is indicative of an increase in electron density that is likely a result of orbital overlap between the host ZIF architecture and the SAs.38 This data further provides evidence that the SAs are in fact confined to the ZIF-L framework.
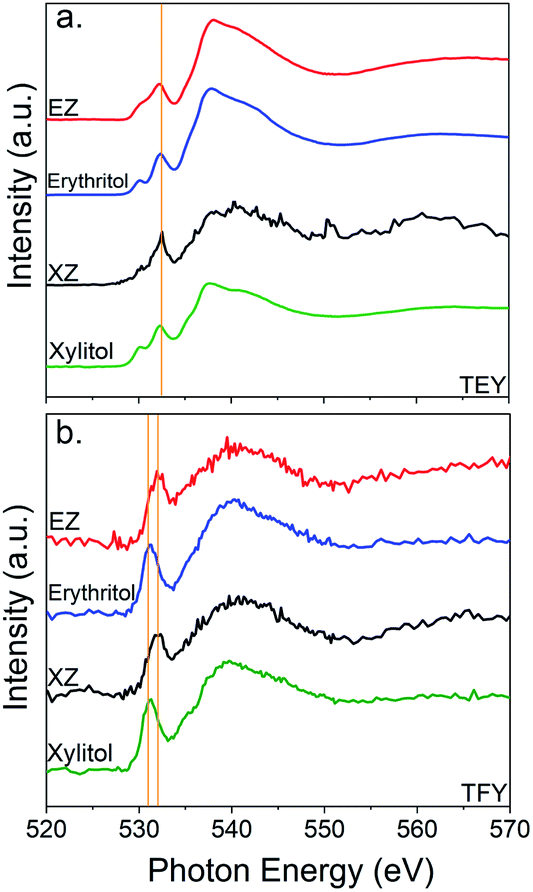 | ||
| Fig. 5 (a) Total electron yield (TEY) and (b) total fluorescence yield (TFY) of the oxygen K-edge spectra of EZ and XZ samples alongside bulk erythritol and xylitol. Erythritol and xylitol are the only oxygen containing molecules in the SA@ZIF crystal assemblies. In the both cases, the TFY signal is shifted from 531 eV to 533 eV when comparing pure SA to encapsulated SA, indicating a shift in the hybrid orbital structure of oxygen of the SAs upon encapsulation. | ||
2.2 SA@ZIF surface functionalization and robustness
Further work was done to explore possible post-processing techniques that would (1) permanently entrap the SA molecules and (2) make this material more amenable to application in different environments. For this purpose, a methodology was adopted from Ma et al. to adjust the surface wettability of the SA@ZIF assemblies by grafting hydrophilic polyethylene glycol (PEG) or hydrophobic perfluorodecanethiol (PFT) molecules to ZIF crystal surface.39 A hydrophilic surface coating was expected to enhance dispersibility in aqueous solutions. Hydrophobic surface functionalization on the other hand was anticipated to deter water intrusion and SA leaching. Erythritol was chosen as the SA for post-functionalization due to its superior thermal performance in the earlier experiments. While ZIF-L was successfully employed as a host matrix in the initial synthesis, ultimately the ZIF-8 structure is preferable due to its larger pore volume and enhanced surface area.40 To ensure that ZIF-8 was the structure produced, we increased the precursor ratio from 8:1 to 20:1 Mim:ZnH as outlined in the Experimental section. Generic click chemistry reactions were chosen as the route for surface functionalization. To facilitate this, 2-vinylimidazole was added halfway through the EZ crystal synthesis in order to provide reactive sites for the polymeric functionalization agents. The surface of the resulting crystals (denoted as “EZV”) was then functionalized with either hydrophilic PEG (EZV-PEG) or hydrophobic PFT (EZV-PFT) surface groups. A schematic of the functionalization methodology is shown in the ESI (Fig. S3†). SEM images (Fig. 6a1–3) show that the bare and surface-modified EZV samples all display the desired octahedral ZIF-8 morphology.
 | ||
| Fig. 6 SEM micrographs of (a1) bare EZV, (a2) EZV-PEG and (a3) EZV-PFT. No differences between functionalized and bare samples are apparent at this scale. Energy dispersive X-ray (EDX) surface scans of (b1) EZV, (b2) EZV-PEG and (b3) EZV-PFT samples. (C) Panels indicate that the contact angle between water and SA@ZIF materials decreases from 55° for unfunctionalized (c1) to 40° for PEG encapsulated (c2) sample and increases to 113° for the PFT encapsulated (c3) crystal assembly. | ||
The ZIF-8 crystal structure was further confirmed via XRD analysis (Fig. S4†). Under the SEM, the functionalized EZV crystals appear to have a coarser outer surface than those of bare EZV crystals, possibly due to the occurrence of a reaction on their exterior surface. Bolstering this point are EDX maps (Fig. 6b1–3) and corresponding EDX spectra (Fig. S6†) that clearly show the existence of fluorine containing compounds on the EZV-PFT surface. PEG does not contain any unique elements, making it difficult to differentiate from the EZV scaffold. To characterize the changes in surface wettability between the functionalized and non-functionalized samples, contact angle measurements were carried out. Fig. 6c1–3 shows contact angles of water droplets on the surface of pellet-pressed EZV, EZV-PEG and EZV-PFT crystals. The contact angle of water on EZV-PFT is as high as 113°, pointing to superhydrophobic surface features. This is followed by unfunctionalized EZV displaying a contact angle of 55°, and EZV-PEG at 40°. In addition, the DSC data shown in Fig. S5† demonstrates that surface functionalization does not interfere with the phase change temperature depression observed earlier. The phase change temperatures associated with bare and functionalized EZV samples remained depressed (between 27–32 °C) compared to bulk erythritol. TGA analysis (Fig. S14†) confirmed that the loading capacity of each crystal sample ranged from 17 wt% for EZV and EZV-PEG to 28 wt% for EZV-PFT. These results confirm that the surface of EZV crystals can be effectively coated to tune surface hydrophobicity, thus making them more ideally poised for real world industrial integration.
Nitrogen sorption experiments were conducted to measure the surface area of the erythritol containing ZIF-8 samples in comparison to a ZIF-8 reference sample that had been prepared under identical reaction conditions except for the use of methanol as solvent. Isotherm data is shown in Fig. S10.† The differential pore volume distributed over nanopores with average diameters between 0.6–1.7 nm was calculated according to Hovarth–Kawazoe model with slit pore assumptions (Fig. 7a). While the ZIF-8 reference displayed a Brunauer–Emmet–Teller (BET) surface area of 1660 m2 g−1, that of the erythritol containing samples (EZV, EZV-PEG and EZV-PFT) was significantly lower, ranging only between 315–409 m2 g−1. The BET data for the ZIF-8 reference corresponds well with literature reported values.40 Erythritol-infused, surface-functionalized samples showed diminished nanopore volumes, with 0.7 nm, 1.0 nm and 1.3 nm diameter nanopores being particularly diminished in volume. This held true even for the unfunctionalized EZV sample, suggesting that it is not the surface coating but the erythritol that is confined to and filling these nanopores.
 | ||
| Fig. 7 Panel (a) shows volumetric pore size distributions in ZIF-8, EZV, EZV-PEG and EZV-PFT samples based on nitrogen sorption. Compared to ZIF-8 (synthesized in methanol), bare and functionalized EZV (synthesized in erythritol) display diminished or no porosity over the same range of average pore diameters. (b) Shows molar ratios of erythritol to methylimidazole in NMR spectral analysis of acid-digested SA@ZIF samples after soaking at 60 °C and 24 h. Each ratio was normalized to the ratio obtained for its analogous pristine sample. | ||
It should be noted that the nitrogen uptake of functionalized EZV-PEG is surprisingly similar to that of pristine the EZV parent, in the 0.6–0.8 nm pore range. EZV-PFT shows slightly different features over the same range, however. One possible explanation could be discerned from the molar ratios of methyl-to vinylimidazole groups in each sample (as determined via the NMR analysis explained below, raw data in Fig. S11–13 and Table S1†). Compared to EZV-PEG crystals, EZV-PFT was found to contain four times more vinyl groups, i.e. unused ‘docking’ sites for PFT/PEG molecules to attach. It is therefore likely that the PEG-ligation of the EZV material was less effective than the attachment of PFT, hence rendering a EZV-PEG sample with very similar pore size properties compared to EZV. This is also supported by the fact that, based on contact angle measurements (Fig. 6c1–3), PZV-PEG is less hydrophilic than EZV-PFT is hydrophobic, compared to EZV.
Lastly, the surface functionalized crystals were subjected to a simulated operations test to investigate their long-term stability. Long-term robustness is an important factor in the development of novel PCM assemblies as they should ideally last as long as the cooling hardware itself. Many studies report on the stability of their PCMs under continuous cycling in a dry state but fail to immerse them in a relevant transport fluid. Erythritol is highly soluble in water and so, to give better understanding of how they would behave in a real heat-transfer application, crystal samples were soaked in DI water at 60 °C for 24 hours while stirring vigorously. At the end of the test, particles were collected via centrifugation and dried under vacuum at room temperature. After drying, NMR spectra were recorded of the sulfuric acid digested (Fig. S11–S13†) and the molar ratios of methylimidazole to erythritol molecules could be determined for each sample. The same was done to pristine, non-soaked samples, the exact ratios of which are catalogued in Table S1.† Based on this analysis, we were able to compute the amount erythritol retained inside the ZIF scaffold by comparing the ratio of methylimidazole to erythritol molecules in the pristine and the soaked SA@ZIF crystal samples (Fig. 7b). All particle assemblies, whether surface functionalized or not, retained close to 90% of the entrapped erythritol over the course of the leach test. This evidence confirms the surface functionalization process to be a promising avenue for imparting the preferred surface properties while preserving material robustness to ensure optimal industrial implementation.
3. Conclusions
We present here the first successful encapsulation of SA PCMs within a MOF that demonstrates the potential for tunable phase-change temperatures and establishes straightforward techniques for effective functionalization. As such, this study introduces a new platform of inexpensive, non-flammable PCMs that maintain high degrees of latent heat performance and can be tailored to a variety of different working environments. We envision this material to be of relevance as a functional additive in thermal storage and heat exchange applications where water must be conserved or cannot be used at all. X-ray absorption and nitrogen sorption experiments suggest that the SA in our model system is confined to the nanopores of the host ZIF matrix. This confinement results in a clear suppression of phase-transition temperature. We have additionally shown that it is possible to functionalize the surface of this novel composite material with hydrophilic and hydrophobic moieties to effectively adjust surface wettability while preserving long-term robustness. Functionalized and unfunctionalized crystals were found to retain up to 90% of encapsulated SA species upon soaking in 60 °C water for 24 hours. To realize the full potential of a PCM with an entirely tunable phase-change temperature, further exploration into the interactions between the host matrix and guest molecules is necessary. Also, the long-term robustness of the PCM assemblies must be investigated via DSC cycling. Development of these materials is imperative to transition from water-based to water-free cooling and revolutionize the energy and water conservation landscape, ensuring resource sustainability for years to come. We believe our work paves the way for this new class of materials to be integrated into the next generation of highly efficient thermal management applications.4. Experimental
4.1 Synthesis
000 rpm. The final product was then dried overnight under vacuum at room temperature. Erythritol containing ZIFs are referred to as “EZ”, and xylitol containing ZIFs as “XZ”.
EZV crystals were subsequently encapsulated relying on thiol–ene ‘click’ reactions to occur between the vinyl groups on the EZV surface and the terminal thiol groups of the PFT and PEG polymer chains. The procedure was inspired partially by a methodology outlined by Ma et al. In a 100 mL round bottom flask equipped with a stir bar, the previously synthesized EZV compound (0.3 g) was combined with either PFT in (trifluoromethyl)benzene (10 v/v%, 30 mL) or PEG-thiol in ortho-dichlorobenzene (0.3 g in 30 mL), along with a catalytic amount of AIBN (0.0003 g). The reaction vessels were heated to 60 °C and stirred continuously for 6 hours under a N2 atmosphere. The resulting mixtures were washed with diethyl ether (2 × 40 mL) and dried under vacuum to yield EZV-PFT or EZV-PEG as off-white, powdered solids.
4.2 Characterization
Conflicts of interest
The authors declare no conflicts of interest.Author contributions
L. Hackl, C. H. Hsu, K. Chou and C. Ma synthesized the materials presented here, carried out thermal characterizations and wrote the manuscript. M. Gordon carried out XRD analyses and helped edit. C. Anderson and M. Kolaczkowski helped with NMR characterization and encapsulation. P. Ercius was involved in collecting TEM and STEM tomography images. Y. S. Liu and J. Guo supported the XANES experiments at LBNL's ALS. J. J. Urban supervised the project at large.Acknowledgements
This work was performed at the Molecular Foundry and Advanced Light Source, Lawrence Berkeley National Laboratory, and was supported by the Department of Energy, Office of Science, Office of Basic Energy Sciences, Scientific User Facilities Division of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Partial support was also provided by the U.S./China Clean Energy Research Center for Water-Energy Technologies (CERC-WET, contract no. DEIA0000018). M. P. G. gratefully acknowledges the National Science Foundation for fellowship support under the National Science Foundation Graduate Research Fellowship Program.References
- J. J. Urban, Joule, 2017, 1, 665–688 CrossRef.
- T. M. Letcher and D. A. Vallero, Waste: a handbook for management, 2019 Search PubMed.
- M. M. Farid, A. M. Khudhair, S. A. K. Razack and S. Al-Hallaj, Energy Convers. Manag., 2004, 45, 1597–1615 CrossRef CAS.
- W. Aftab, X. Huang, W. Wu, Z. Liang, A. Mahmood and R. Zou, Energy Environ. Sci., 2018, 11, 1392–1424 RSC.
- M. Kenisarin and K. Mahkamov, Renew. Sust. Energ. Rev., 2007, 11, 1913–1965 CrossRef CAS.
- Y. Yamagishi, H. Takeuchi, A. T. Pyatenko and N. Kayukawa, AIChE J., 1999, 45, 696–707 CrossRef CAS.
- S. Boskovic, B. V. Reddy and P. Basu, Int. J. Energy Res., 2002, 26, 159–171 CrossRef.
- M. J. Li, B. Jin, Z. Ma and F. Yuan, Appl. Energy, 2018, 221, 1–15 CrossRef CAS.
- D. C. Hyun, N. S. Levinson, U. Jeong and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 3780–3795 CrossRef CAS.
- A. Solé, L. Miró, C. Barreneche, I. Martorell and L. F. Cabeza, Renewable Energy, 2015, 75, 519–523 CrossRef.
- T. Nomura, C. Zhu, A. Sagara, N. Okinaka and T. Akiyama, Appl. Therm. Eng., 2015, 75, 481–486 CrossRef CAS.
- Y. Yoo, C. Martinez and J. P. Youngblood, ACS Appl. Mater. Interfaces, 2017, 9, 31763–31776 CrossRef CAS.
- Z. Xiangfa, X. Hanning, F. Jian, Z. Changrui and J. Yonggang, J. Exp. Nanosci., 2012, 7, 17–26 CrossRef.
- Y. Li, X. Huang, Y. Li, Z. Xi, G. Hai, Z. Tao and G. Wang, Sustain. Energy Fuels, 2020, 4, 1764–1772 RSC.
- Y. Chen, X. Zhang, B. Wang, M. Lv, Y. Zhu and J. Gao, RSC Adv., 2017, 7, 15625–15631 RSC.
- L. Chen, R. Zou, W. Xia, Z. Liu, Y. Shang, J. Zhu, Y. Wang, J. Lin, D. Xia and A. Cao, ACS Nano, 2012, 6, 10884–10892 CrossRef CAS.
- Y. Li, S. Yu, P. Chen, R. Rojas, A. Hajian and L. Berglund, Nano Energy, 2017, 34, 541–548 CrossRef CAS.
- D. G. Atinafu, S. J. Chang, K. H. Kim, W. Dong and S. Kim, Chem. Eng. J., 2020, 389, 124430 CrossRef CAS.
- Y. Luan, M. Yang, Q. Ma, Y. Qi, H. Gao, Z. Wu and G. Wang, J. Mater. Chem. A, 2016, 4, 7641–7649 RSC.
- J. Tang, M. Yang, W. Dong, M. Yang, H. Zhang, S. Fan, J. Wang, L. Tan and G. Wang, RSC Adv., 2016, 6, 40106–40114 CAS.
- D. G. Atinafu, W. Dong, C. Hou, R. S. Andriamitantsoa, J. Wang, X. Huang, H. Gao and G. Wang, Mater. Today Energy, 2019, 12, 239–249 Search PubMed.
- L. Xu and R. Yang, Molecules, 2019, 24, 1482 CrossRef CAS.
- D. Li, X. Cheng, Y. Li, H. Zou, G. Yu, G. Li and Y. Huang, Sol. Energy, 2018, 171, 142–149 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 RSC.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- J. Cravillon, S. Münzer, S. J. Lohmeier, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2009, 21, 1410–1412 CrossRef CAS.
- A. Schejn, L. Balan, V. Falk, L. Aranda, G. Medjahdi and R. Schneider, CrystEngComm, 2014, 16, 4493–4500 RSC.
- Z. X. Low, J. Yao, Q. Liu, M. He, Z. Wang, A. K. Suresh, J. Bellare and H. Wang, Cryst. Growth Des., 2014, 14, 6589–6598 CrossRef CAS.
- R. Chen, J. Yao, Q. Gu, S. Smeets, C. Baerlocher, H. Gu, D. Zhu, W. Morris, O. M. Yaghi and H. Wang, Chem. Commun., 2013, 49, 9500–9502 RSC.
- A. Knebel, B. Geppert, K. Volgmann, D. I. Kolokolov, A. G. Stepanov, J. Twiefel, P. Heitjans, D. Volkmer and J. Caro, Science, 2017, 358, 347–351 CrossRef CAS.
- X. Li, Z. Li, L. Lu, L. Huang, L. Xiang, J. Shen, S. Liu and D. R. Xiao, Chem.–Eur. J., 2017, 23, 10638–10643 CrossRef CAS.
- H. Jiang, S. Xue, Y. Liu, R. Chen and W. Xing, RSC Adv., 2016, 6, 21337–21344 RSC.
- C. W. Tsai and E. H. G. Langner, Microporous Mesoporous Mater., 2016, 221, 8–13 CrossRef CAS.
- J. J. Beh, J. K. Lim, E. P. Ng and B. S. Ooi, Mater. Chem. Phys., 2018, 216, 393–401 CrossRef CAS.
- S. N. Gunasekara, M. Ignatowicz, J. N. W. Chiu and V. Martin, Int. J. Energy Res., 2019, 43, 1785–1801 CrossRef CAS.
- G. Y. Jeong, A. K. Singh, M. G. Kim, K. W. Gyak, U. J. Ryu, K. M. Choi and D. P. Kim, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- Q. Sun, H. He, W.-Y. Gao, B. Aguila, L. Wojtas, Z. Dai, J. Li, Y.-S. Chen, F.-S. Xiao and S. Ma, Nat. Commun., 2016, 7, 13300 CrossRef CAS.
- Y. Lo, C. H. Lam, C. W. Chang, A. C. Yang and D. Y. Kang, RSC Adv., 2016, 6, 89148–89156 RSC.
- P. Gabbott, in Principles and Applications of Thermal Analysis, Blackwell Publishing Ltd, Oxford, UK, 2008, pp. 1–50 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05019a |
| ‡ Denoting equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |