
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalyst Z-scheme system composed of a linear conjugated polymer and BiVO4 for overall water splitting under visible light†
Yang
Bai
 a,
Keita
Nakagawa
b,
Alexander J.
Cowan
c,
Catherine M.
Aitchison
a,
Yuichi
Yamaguchi
b,
Martijn A.
Zwijnenburg
d,
Akihiko
Kudo
*b,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*a
a,
Keita
Nakagawa
b,
Alexander J.
Cowan
c,
Catherine M.
Aitchison
a,
Yuichi
Yamaguchi
b,
Martijn A.
Zwijnenburg
d,
Akihiko
Kudo
*b,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*a
aDepartment of Chemistry, Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool, L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk; ssprick@liverpool.ac.uk; sebastian.sprick@strath.ac.uk
bDepartment of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo, Japan. E-mail: a-kudo@rs.tus.ac.jp
cStephenson Institute for Renewable Energy, University of Liverpool, Peach Street, Liverpool L69 7ZF, UK
dDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 22nd July 2020
Abstract
Linear conjugated polymers have potential as photocatalysts for hydrogen production from water but so far, most studies have involved non-scalable sacrificial reagents. Z-schemes comprising more than one semiconductor are a potential solution, but it is challenging to design these systems because multiple components must work together synergistically. Here, we show that a conjugated polymer photocatalyst for proton reduction can be coupled in a Z-scheme with an inorganic water oxidation photocatalyst to promote overall water splitting without any sacrificial reagents. First, a promising combination of an organic catalyst, an inorganic catalyst, and a redox mediator was identified by using high-throughput screening of a library of components. A Z-scheme system composed of P10 (homopolymer of dibenzo[b,d]thiophene sulfone)–Fe2+/Fe3+–BiVO4 was then constructed for overall water splitting under visible light irradiation. Transient absorption spectroscopy was used to assign timescales to the various steps in the photocatalytic process. While the overall solar-to-hydrogen efficiency of this first example is low, it provides proof of concept for other hybrid organic–inorganic Z-scheme architectures in the future.
Introduction
The photocatalytic production of hydrogen from water using solar energy has been studied extensively because it promises the sustainable production of renewable fuels from abundant resources.1–3 Photoelectrocatalysis4–6 and direct photocatalysis using catalyst suspensions have both been studied in detail.5 Conceptually, direct hydrogen production using photocatalyst suspensions is the simplest water-splitting approach in technological terms and it is potentially amenable to large-scale deployment.7–17 Recently, several examples of photocatalysis has been reported as a one-step overall water splitting.18–20 However, recombination of electron–hole pairs tends to decrease the photocatalytic reaction efficiency. Moreover, relatively few materials are known that both efficiently absorb visible light and have suitable valence and conduction band energetics. As such, a number of research teams have investigated systems that mimic nature using a two-step excitation process for overall water splitting, typically by coupling together two different photocatalysts with a redox mediator to form a ‘Z-scheme’.21–24Most photocatalysts so far have been inorganic materials, but organic photocatalysts have attracted growing attention25 because they can be prepared from earth-abundant elements and their properties—and in particular their light absorption spectrum—can be tuned easily and continuously by co-polymerisation.26–34 However, most polymer studies have been confined to the sacrificial half-reaction that produces hydrogen only, and few organic photocatalysts have been developed for overall water splitting. Carbon nitrides have been coupled with WO3, which acts as an O2 evolution photocatalyst in a Z-scheme system for overall water splitting,26,27,35 but the efficiencies were limited by the commonly observed back reaction. Other composites that are reported to facilitate overall water splitting are carbon nanodot–carbon nitride nanocomposites,36 Pt/PtOx/CoOx-loaded carbon nitrides,37 and Pt/CoP-loaded carbon nitrides.38 All of these materials are based on carbon nitride, which limits the potential for structural diversity and control over fundamental physical properties, such as optical gap. Also, the use of high synthesis temperatures for carbon nitrides yields relatively poorly defined bulk materials whose precise structure and composition can be hard to elucidate: this in turn makes it hard to establish structure–property relationships.
Here, we couple organic polymer photocatalysts with an inorganic semiconductor using a redox mediator. The polymer produces H2 and the inorganic catalyst produces O2 in a Z-scheme for overall water splitting. This is the first example of such a Z-scheme that uses an organic polymer that is prepared by low-temperature chemical synthesis, opening up a wide variety of possible two-component systems, leveraging the synthetic diversity that is intrinsic to polymer chemistry.
Results and discussion
First, we explored a range of polymer photocatalysts for the hydrogen evolution half-reaction and various metal oxide materials as photocatalysts for oxygen production. To do this, we performed high-throughput screening whereby the photocatalysts (5 mg) were added to water (5 mL) containing a redox mediator and dispersed by ultrasonication. The samples were then illuminated with a solar simulator for 5 hours before measuring the amount of hydrogen or oxygen produced using an automated gas chromatograph equipped with a pulsed discharge detector.We explored various polymers for the hydrogen evolution half-reaction, that is; P10 (homopolymer of dibenzo[b,d]thiophene sulfone),28 P34 (poly[9,9-dimethyl-9H-fluorene-2,7-diyl]),39 P64 (dibenzo[b,d]thiophene sulfone dibenzo[b,d]thiophene co-polymer),31,34 P74 (2,1,3-benzothiadiazole dibenzo[b,d]thiophene sulfone co-polymer), and S-CMP3 (conjugated microporous polymer based on dibenzo[b,d]thiophene sulfone and 2,2′,7,7′-linked 9,9′-spirobifluorene)39 (Fig. 1a and S-1†).
 | ||
| Fig. 1 High-throughput photocatalysis screening of (a) hydrogen evolution half-reaction of polymers; (b) oxygen evolution half-reaction of metal oxide and redox shuttles, irradiated by a solar simulator (AM1.5G, Class AAA, IEC/JIS/ASTM, 1440 W xenon, 12 × 12 in, MODEL: 94123A, see Fig. S-18† for output spectrum, illumination time: 5 hours). | ||
For the oxygen producing half-reaction, we considered various metal oxides that were reported previously, such as BiVO4 and WO3 (Fig. 1b). The polymers were prepared using Pd(0)-catalysed cross-coupling reactions and characterised using UV-vis spectroscopy, powder X-ray diffraction (PXRD), Fourier-transform infrared spectroscopy (FT-IR), photoluminescence spectroscopy (PL), scanning electron microscope (SEM), static light scattering, and time-resolved single photon counting (TRSPC) (Fig. S-2 to S-16†). All polymers contained residual metallic palladium particles, as evident from electron paramagnetic resonance results (Fig. S-17†), which remained in the materials after work-up; this residual metal acts as a co-catalyst for hydrogen production, instead of the more commonly used platinum.30,39,40 Both the polymers and the inorganic materials were tested against a range of electron donors and acceptors (i.e., Fe2+/Fe3+,41 I−/IO3−,42 Ce3+/Ce4+,43 NO2−/NO3−,44 [Co(phen)3]3+/2+, and [Co(bpy)3]3+/2+)22 to identify candidate redox pair combinations that could be taken forward in a Z-scheme for overall water-splitting.
Under these screening conditions, we found that the highest hydrogen evolution activity was obtained for polymer P10 with FeCl2 at pH 2.7 acting as the electron donor (hydrogen evolution rate (HER) of 1.4 μmol h−1 for 5 mg photocatalyst under solar simulator illumination AM1.5G, irradiation area = 4 cm2; pressure = 1 bar, N2). This rate was significantly higher than for the other polymers (P74, P64, P34 and S-CMP3), as tested under the same conditions (Fig. 1a). The catalytic rate for P10 with FeCl2 was more than 10 times lower than for triethylamine under the equivalent conditions (17.2 μmol h−1). The latter involves the irreversible oxidation of an organic donor, but the Fe2+-catalysed rate was sufficiently high to offer promise as a potential partner in a Z-scheme. An oxygen production screen showed that BiVO4 coupled with FeCl3 as the electron donor, again at pH 2.7, gave the highest oxygen evolution rates (Fig. 1b; OER, 0.32 μmol h−1, 5 mg photocatalyst), suggesting P10/BiVO4/Fe2+/Fe3+ as a potential Z-scheme.
The ionisation potential of P10, as previously predicted by DFT,28 indicates that it is possible to oxidise Fe2+ to Fe3+; likewise, the experimental band positions for BiVO4 (ref. 46) allow for the reduction of Fe3+ to Fe2+ (Fig. 2). The resulting holes in BiVO4 have a large driving force for water oxidation and the electrons in P10 have a large driving force for proton reduction. Hence, the combination of high-throughput photocatalysis screening and the predicted and measured potentials of the charge carriers in P10 and BiVO4, respectively, prompted us to explore Z-schemes comprising P10 for hydrogen production and BiVO4 for oxygen production with a Fe3+/Fe2+ redox couple.
 | ||
| Fig. 2 (a) Structures of the two photocatalysts, BiVO4 and P10; (b) alignment of the potentials of P10 (HOMO, ionization potential; LUMO, electron affinity) and bands (VB, valance band; CB, conduction band) of BiVO4 with the solution potential of the Fe2+/Fe3+ redox couple. P10 and BiVO4 data taken from ref. 28 and 46, respectively. | ||
We tested a Z-scheme for overall water splitting using different ratios of P10 and BiVO4 and different amounts of residual Pd, as shown in Table 1. For P10 containing 160 ppm of Pd, a low H2 evolution rate was observed (0.05 μmol h−1, entry 1), along with a much higher, non-stoichiometric O2 evolution rate (1 μmol h−1, entry 1). In line with previous observations on threshold values for metal incorporation for optimum H2 production in sacrificial systems,30,47 we found significantly increased H2 evolution rates (4 μmol h−1, entry 3) when P10 containing 3300 ppm residual Pd. The accompanying O2 evolution rate was 0.93 μmol h−1. Ruthenium, which is often used as a co-catalyst for hydrogen evolution catalysts in Z-schemes,22,48 acted here as a poor cocatalyst; for a Z-scheme with 2500 ppm Ru-loaded P10, we observed low, non-stoichiometric gas production rates (H2: 0.16 μmol h−1; O2: 0.76 μmol h−1, entry 2).
| Entry | Photocatalyst P10a | H2 evolution rateb (μmol h−1) | O2 evolution rateb (μmol h−1) | Kinetic data in ESI | |
|---|---|---|---|---|---|
| Amount (mg) | Residual Pd (ppm) | ||||
| a Reaction conditions: starting reactant solution, 50 mg BiVO4 and P10 with variation amounts, residual Pd or Ru loaded by photodeposition in 120 mL of an aqueous redox mediator solution (FeCl3, 2 mmol L−1, initial pH: 2.7); light source: 300 W xenon light source with a cut-off filter (λ > 420 nm, see Fig. S-18 for output spectrum); cell, top-irradiation, 70 Torr, Ar. b Rates of the equilibrated system. c FeCl2, 2 mmol L−1, initial pH: 2.7 was used. | |||||
| 1 | 50 | 160 | 0.05 | 1 | S-19 |
| 2 | 50 | 160 + 2500 ppm Ru | 0.16 | 0.76 | S-20 |
| 3 | 50 | 3300 | 4 | 0.93 | S-21 |
| 4 | 10 | 3300 | 0.95 | 0.52 | S-22 |
| 5 | 7 | 3300 | 3 | 1.29 | S-23 |
| 6 | 4 | 3300 | 3.55 | 1.76 | S-24 |
| 7 | 4c | 3300 | 5 | 2.7 | S-25 |
Decreasing the amount of the organic photocatalyst P10 (3300 ppm Pd) relative to BiVO4 from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 w/w to 7:50 w/w caused the water splitting reaction to proceed approximately stoichiometrically (H2: 3 μmol h−1; O2: 1.29 μmol h−1, entry 5). When the amount of polymer was reduced even further (4:50 w/w P10:BiVO4), the water splitting reaction still proceeded in a stoichiometric ratio (H2: 3.55 μmol h−1; O2: 1.76 μmol h−1, entry 6). The need for more BiVO4 than P10 for stoichiometric water splitting aligns with our observation that P10 evolves significantly more hydrogen under sacrificial conditions than BiVO4 does oxygen; as such, reducing the amount of the more active polymer photocatalyst lowers its competitive light absorption and light scattering, hence increasing the overall activity of the Z-scheme (for example, compare entries 4 and 5).
:50 w/w to 7:50 w/w caused the water splitting reaction to proceed approximately stoichiometrically (H2: 3 μmol h−1; O2: 1.29 μmol h−1, entry 5). When the amount of polymer was reduced even further (4:50 w/w P10:BiVO4), the water splitting reaction still proceeded in a stoichiometric ratio (H2: 3.55 μmol h−1; O2: 1.76 μmol h−1, entry 6). The need for more BiVO4 than P10 for stoichiometric water splitting aligns with our observation that P10 evolves significantly more hydrogen under sacrificial conditions than BiVO4 does oxygen; as such, reducing the amount of the more active polymer photocatalyst lowers its competitive light absorption and light scattering, hence increasing the overall activity of the Z-scheme (for example, compare entries 4 and 5).
In experiments where the redox mediator was initially Fe3+, we observed only oxygen production at the start of the reaction, as shown in Fig. 3a. This is consistent with the reduction of Fe3+ to Fe2+ by BiVO4. After 6 hours, we observed steady and simultaneous H2 and O2 production close to the expected stoichiometric ratio of 2:1 (H2: 3 μmol h−1 and O2: 1.29 μmol h−1) for 7 mg P10 in Z-scheme system under a visible light illumination (λ > 420 nm, 300 W Xe light source; Fig. S-23†). Under solar simulator irradiation, we observed rates of 0.66 μmol h−1 for H2, and 0.29 μmol h−1 for O2 (Fig. 3d). When Fe2+ was used as the initial species in the redox mediator solution, we observed both H2 and O2 production under a visible light illumination (λ > 420 nm, 300 W Xe light source; Fig. 3b) but the ratio was non-stoichiometric (5.4:1 H2:O2) at the start of the reaction. After a total of 10 hours, we saw stoichiometric production of H2 and O2 (H2: 5.0 μmol h−1; O2: 2.7 μmol h−1). Thus, overall water splitting proceeded in both cases, starting with either FeCl3 or FeCl2. As expected in either case the system has to equilibrate towards a mixture of Fe2+/Fe3+ which enables both catalysts to drive hydrogen and oxygen production in stoichiometric amounts after an initial period of non-stoichiometric water splitting. In the absence of a redox mediator, no overall water splitting proceeds, showing that electron and hole transfer indeed occurs via the redox mediator (Fig. S-29†).
 | ||
| Fig. 3 Time course of overall water splitting on P10 (4 mg) and BiVO4 (50 mg) under visible light illumination (300 W Xe light source, λ > 420 nm, see Fig. S-18† for output spectrum) with evacuation every 5 h (dashed line) using (a) an aqueous FeCl3 solution (2 mmol L−1, 120 mL, pH 2.7); (b) an aqueous FeCl2 solution (2 mmol L−1, 120 mL, pH 2.4). (c) Time course of overall water splitting on P10 (4 mg) and BiVO4 (50 mg) in an aqueous FeCl3 solution (2 mmol L−1, 120 mL, pH 2.7) under full arc light illumination (300 W Xe light source, full arc); (d) time course of overall water splitting on P10 (4 mg) and BiVO4 (50 mg) in an aqueous FeCl3 solution (2 mmol L−1, 120 mL, pH 2.7) under simulated sunlight (solar simulator AM1.5G filter, 100 mW cm−2); (e) wavelength dependence of the photocatalytic activity of P10 (4 mg) and BiVO4 (50 mg) in an aqueous FeCl3 solution (2 mmol L−1, 120 mL, Ph 2.7) using a Xe light source (300 W) with suitable cut-off filters; (f) time course of water splitting reaction of P10 (4 mg) and BiVO4 (50 mg) in an aqueous FeCl2 solution (2 mmol L−1, 120 mL, pH 2.4), under Ar flow using a Xe light source (300 W, λ > 420 nm). | ||
As might be expected, higher rates were observed under broadband illumination (full arc, 300 W Xe light source) and rates of 10.8 μmol h−1 and 4.5 μmol h−1 were determined for H2 and O2 production for a Z-scheme consisting of P10/BiVO4 (4:50) with FeCl3 (Fig. 3c). Further evidence that overall water splitting was taking place was that (i) a colorimetric experiment confirmed the conversion of Fe3+ to Fe2+; (ii) the amount of O2 evolved was larger than the amount of Fe3+ in the FeCl3 solution (60 μmol); (iii) the total amount of H2 generated this experiment (125.2 μmol, Fig. S-24†) was larger than the amount of hydrogen in the P10 sample (56 μmol), thus ruling out the possibility that the H2 was produced from a self-corrosion process. Longer term stability was evaluated for 70 h under visible light irradiation (Fig. S-24†) followed by 5 hours full arc irradiation showing stabile water splitting over extended time. Post illumination analysis also showed no significant changes in the UV-vis, photoluminescence, FT-IR spectra, and PXRD patterns for the catalysts (Fig. S-31 to S-34†). We did observe that a small amount of H2 was produced by P10 in water only (i.e., in the absence of any intentionally added redox mediator, Fig. S-35†), possibly due to self-oxidation of the photocatalyst. However, when D2O was used as the proton source for P10 in the presence of FeCl2 D2 production was mostly observed (Fig. S-36†), which rules out that this self-oxidation is the source of the H2 in the Z-scheme experiments. Possibly the self-oxidation is also suppressed in the presence of electron mediator. We also note that the pH value remains virtually unchanged during the experiment, which shows that no Fe(OH)3 was formed.49 Decomposition of BiVO4 was not expected to occur at pH 2.4, but this been observed under more acidic conditions.48
The back reaction to form water from evolved H2 and O2 can be an issue for overall water splitting because noble metal co-catalysts can accelerate this.23,26 For the Z-scheme presented here, we did not observe a reduction in the amounts of H2 and O2 gas in the dark when the light source was switched off (Fig. S-23†), suggesting that no significant thermal back reaction was taking place.
We next examined the behaviour of the photocatalytic system by transient absorption (TA) spectroscopy to confirm the proposed mechanism of water splitting (Fig. 2). Initially, we studied P10 containing 0.33 wt% Pd in the absence of the redox mediator in water at pH 2.7 (Fig. 4a). Following excitation, a broad negative signal was observed at wavelengths below 740 nm, which has been assigned previously to stimulated emission by comparison to the photoluminescence spectrum of P10.28 From 740 nm to greater than 860 nm, a photoinduced absorption (PIA) was observed. Very similar behaviour was observed in a TA study of P10 in pure water, with the PIA assigned to singlet exciton formation. The decay kinetics of the PIA and the stimulated emission are complex, requiring greater than 4 exponential components to achieve a satisfactory fit likely due to the distribution of polymer structures present (Fig. S-37†). However, the time taken for the initially measured TA change to decay by 50% (t50%) for both the PIA and emission were similar (t50% ∼ 1.7 ps and 2.2 ps at 843 nm and 540 nm), respectively.
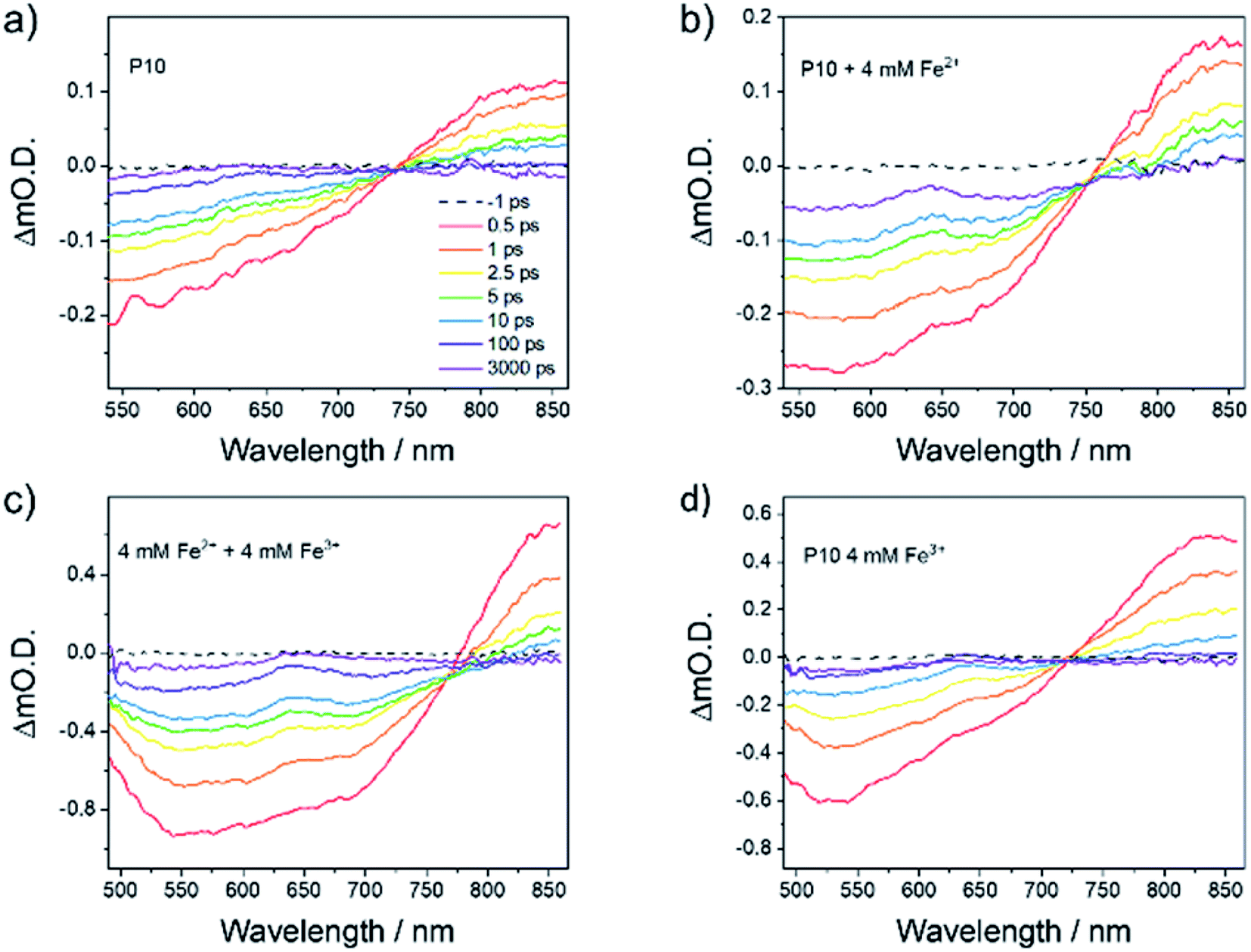 | ||
| Fig. 4 TA spectra of P10 suspension in (a) water at pH 2.7 (0.24 g L−1) and in the presence of (b) Fe2+ (4 mM), (c) Fe2+ and Fe3+ (both 4 mM) and (d) Fe3+ (4 mM). Spectra are recorded following 400 nm (150 nJ, 5 kHz) excitation. The presence of Fe2+ leads to the formation of a new long-lived TA band at 640 nm assigned to an electron polaron (P10−). | ||
Marked differences were observed in the TA spectra in the presence of 4 mM of Fe2+ (Fig. 4b). The features due to stimulated emission (<740 nm) and the initial singlet excitons (>740 nm) were still present, but a new band also grows in within 10 ps, centered at 640 nm. Similar features were assigned previously to the formation of the electron polaron with P10 in the presence of an amine electron donor.28 Here, we also assign this band to P10−, confirming the role of the Fe2+ species. In the presence of Fe2+, the rate of decay of the PIA at 843 nm (t50% ∼ 1.3 ps) and the bleach at 550 nm (t50% ∼ 2.1 ps) is similar to that measured in the absence of the electron donor (Fig. S-38†).
It is notable that the 640 nm band is very long-lived (>3.3 ns; the longest timescale we can study here). By contrast, in water alone, minimal TA signals remain after this timescale and, if present at all, the 640 nm band is much weaker in intensity (Fig. 4a). When both Fe2+ (4 mM) and Fe3+ (4 mM) are in the P10 suspension, we see similar behaviour to when Fe2+ alone is present, with the efficient formation of the electron polaron still occurring (Fig. 4c), persisting to timescales beyond the maximum that can be studied here (Fig. S-39†) with no notable loss in lifetime. It is therefore apparent that despite the presence of Fe3+, which might be expected to act as an electron scavenger, long-lived P10− species can still be formed, which is known to be a requirement since H2 evolution is thought to occur on the micro- to millisecond timescale.28
A significantly decreased intensity of the 640 nm PIA is observed using Fe3+ (Fig. 4d). The small absorption at 640 nm could be due to the presence of a not fully charge separated state, with spectral features similar to that of the electron polaron. Alternatively, it may indicate that a small population of the P10 electron polaron can be formed, may be due to the build-up of Fe2+ following the excitation of the sample for prolonged periods.
Attempts to study BiVO4 by TA here were unsuccessful due to the colloidal instability of the suspensions. However, electron scavenging by Fe3+/2+ following the photoexcitation of BiVO4 has been studied previously by TA spectroscopy. There,50 electron scavenging with Fe3+ occurred within a few microseconds of BiVO4 excitation, with the photogenerated holes on BiVO4 being then retained for >100 μs, indicating that both back electron transfer to Fe2+ and the transfer of the hole into water occur on a slower timescale. Therefore, in light of our new experiments and the literature results28,50 discussed above, we are able to propose timescales for the individual steps in the Z-scheme system (Fig. 5).
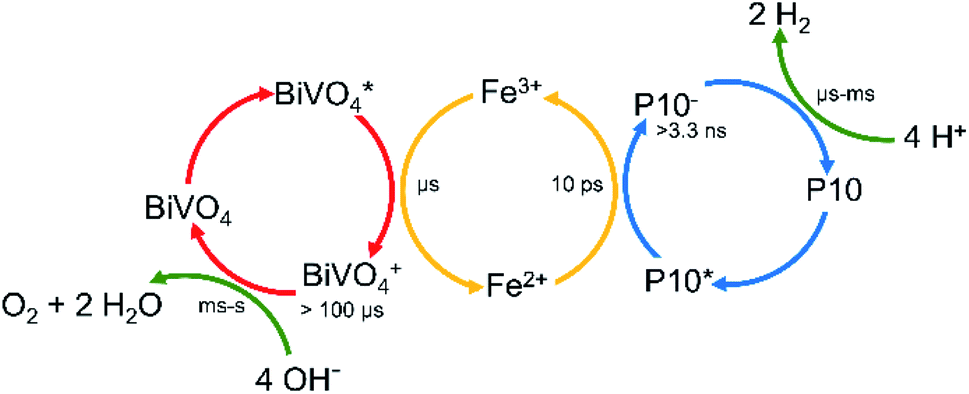 | ||
| Fig. 5 Time scales of the individual processes taking place in the Z-scheme. | ||
Conclusions
In summary, conjugated polymer photocatalysts can be coupled with inorganic photocatalysts to produce a Z-scheme that performs overall water splitting under visible light irradiation. In this first example, the organic polymer photocatalyst is less dense than its inorganic counterpart and therefore much smaller amounts of the organic catalyst are needed (4:50 w/w P10:BiVO4). While the overall solar-to-hydrogen efficiency of this first system is very low (0.0014%), this proof-of-concept study opens the door for other linear polymer–inorganic Z-schemes in the future. It is possible, for example, that solid-state organic–inorganic Z-schemes using solution processable polymers10,51–54 might give better performance without the need for a soluble redox shuttle.
Experimental procedures
Synthesis of the hydrogen evolution photocatalyst (HEP) and the oxygen evolution photocatalyst (OEP)
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1, EP/P034497/1 and EP/S017623/1. Y. B. thanks the China Scholarship Council for a Ph.D. studentship and The Great Britain Sasakawa Foundation (No. 5611) for financial support. We also thank JSPS KAKENHI Grant Numbers 17H06433 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)” and 17H0127 for financial support. Dr Qian Wang is acknowledged for useful discussions. Rob Clowes is acknowledged for help with water sorption measurements and Kenta Watanabe for ESR measurements.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120(2), 919–985 CrossRef CAS PubMed.
- Y. Fang, Y. Xu, X. Li, Y. Ma and X. Wang, Angew. Chem., 2018, 130, 9897–9901 CrossRef.
- G. Peng, J. Qin, M. Volokh and M. Shalom, ACS Appl. Mater. Interfaces, 2019, 11, 29139–29146 CrossRef CAS PubMed.
- J. Xia, N. Karjule, L. Abisdris, M. Volokh and M. Shalom, Chem. Mater., 2020, 32, 5845–5853 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- C. L. Chang, W. C. Lin, C. Y. Jia, L. Y. Ting, J. Jayakumar, M. H. Elsayed, Y. Q. Yang, Y. H. Chan, W. S. Wang, C. Y. Lu, P. Y. Chen and H. H. Chou, Appl. Catal., B, 2020, 268, 118436 CrossRef.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- P.-J. J. Tseng, C.-L. L. Chang, Y.-H. H. Chan, L.-Y. Y. Ting, P.-Y. Y. Chen, C.-H. H. Liao, M.-L. L. Tsai and H.-H. H. Chou, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 1907296, 1–52 Search PubMed.
- A. F. M. El-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 641, 1–10 Search PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- L. Y. Ting, J. Jayakumar, C. L. Chang, W. C. Lin, M. H. Elsayed and H. H. Chou, J. Mater. Chem. A, 2019, 7, 22924–22929 RSC.
- J. Jayakumar and H. Chou, ChemCatChem, 2020, 12, 689–704 CrossRef CAS.
- Y. Bai, Z. Hu, J. X. Jiang and F. Huang, Chem.–Asian J., 2020, 15, 1780–1790 CrossRef CAS PubMed.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol., C, 2011, 11, 179–209 CrossRef.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- Y. Qi, Y. Zhao, Y. Gao, D. Li, Z. Li, F. Zhang and C. Li, Joule, 2018, 2, 2393–2402 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 21, 7102–7106 CrossRef PubMed.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- Z.-A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS PubMed.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS PubMed.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- G. Zhao, X. Huang, F. Fina, G. Zhang and J. T. S. Irvine, Catal. Sci. Technol., 2015, 5, 3416–3422 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Z. Pan, Y. Zheng, F. Guo, P. Niu and X. Wang, ChemSusChem, 2017, 10, 87–90 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416–2417 RSC.
- E. A. Kozlova, T. P. Korobkina and A. V. Vorontsov, Int. J. Hydrogen Energy, 2009, 34, 138–146 CrossRef CAS.
- K. Sayama, R. Abe, H. Arakawa and H. Sugihara, Catal. Commun., 2006, 7, 96–99 CrossRef CAS.
- T. M. Suzuki, A. Iwase, H. Tanaka, S. Sato, A. Kudo and T. Morikawa, J. Mater. Chem. A, 2015, 3, 13283–13290 RSC.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, P.-A. Glans, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, Chem. Mater., 2014, 26, 5365–5373 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133–137 CrossRef CAS.
- A. Kudo, MRS Bull., 2011, 36, 32–38 CrossRef CAS.
- N. Aiga, Q. Jia, K. Watanabe, A. Kudo, T. Sugimoto and Y. Matsumoto, J. Phys. Chem. C, 2013, 117, 9881–9886 CrossRef CAS.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, D. S. Ginger, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. Alves Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, crystallographic data, hydrogen and oxygen evolution setup and measurement data, UV-vis spectra, gas sorption data, optical transmittance data, scanning electron microscope, transmission electron microscope, time correlated single photon counting, and static light scattering. See DOI: 10.1039/d0ta04754f |
| This journal is © The Royal Society of Chemistry 2020 |