
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pt single-atoms supported on nitrogen-doped carbon dots for highly efficient photocatalytic hydrogen generation†
Hui
Luo
 ab,
Ying
Liu
b,
Stoichko D.
Dimitrov
bc,
Ludmilla
Steier
d,
Shaohui
Guo
e,
Xuanhua
Li
e,
Jingyu
Feng
a,
Fei
Xie
ab,
Yuanxing
Fang
f,
Andrei
Sapelkin
b,
Xinchen
Wang
f and
Maria-Magdalena
Titirici
*a
ab,
Ying
Liu
b,
Stoichko D.
Dimitrov
bc,
Ludmilla
Steier
d,
Shaohui
Guo
e,
Xuanhua
Li
e,
Jingyu
Feng
a,
Fei
Xie
ab,
Yuanxing
Fang
f,
Andrei
Sapelkin
b,
Xinchen
Wang
f and
Maria-Magdalena
Titirici
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
bQueen Mary University of London, Mile End Road, London, E1 4NS, UK
cSPECIFIC IKC, College of Engineering, Swansea University, Swansea, SA2 7AX, UK
dDepartment of Materials, Imperial College London, White City Campus, London, W12 0BZ, UK
eNorthwestern Polytechnical University, Queen Mary University of London, Joint Research Institute of Advanced Materials and Structures, Xi'an, 710071, P. R. China
fState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350116, P. R. China
First published on 10th July 2020
Abstract
Single-atom catalysis has become the most active new frontier in energy conversion applications due to its remarkable catalytic activity and low material consumption. However, the issue of atom aggregation during the synthesis process or catalytic reaction must be overcome. In this work, we have developed a one-step photo-deposition process to fabricate Pt single-atom catalysts (SACs) on nitrogen doped carbon dots (NCDs). The Pt–NCDs were then hybridized with TiO2 to achieve high hydrogen generation activity and to understand the fundamentals at the Pt/NCD/TiO2 interface. The synergistic effect of Pt SAC and NCDs with maximized atomic efficiency of Pt and improved charge transfer capability provides a new strategy to rationally design a multi-scale photocatalyst structure to achieve high H2 evolution efficiency. The facile synthesis process also holds great potential for various applications such as electrocatalysis, heterogeneous catalysis and drug delivery, providing a promising way to reduce the high cost of noble metals.
Introduction
Hydrogen production with solar energy is a promising technology to address important renewable energy challenges. It requires efficient photocatalysts to convert solar energy into chemical fuels.1–3 Research into realizing the hydrogen economy has brought with it a greater demand for rare earth and noble metals. If the current consumption rate is maintained, it is predicted that 22 elements face depletion within the coming 50 years, including almost all Platinum Group Metals (PGMs) that are crucial catalyst components.4 Thus, increasing efforts have been made to downsize PGMs to form small but thermally stable active units, in order to produce more efficient metal catalysts with the additional benefit of reducing the costs.5,6The ultimate desired form of Pt is single atom, which offers advantages of both homogeneous and heterogeneous catalysis, building a bridge between the two. Ideally, in metal-based catalysis, all metal atoms should constitute single low-coordinated active sites. For such heterogeneous single-metal site catalysts the activity is superior compared to supported metal nanoparticles and bulk metal-containing materials,7,8 as the unsaturated surface atom exposed to reactants in solution can change dramatically the surface physical chemistry, resulting in high reactivity.9,10 However, when supported these species usually tend to aggregate during the synthesis processes, or in the course of catalytic reaction due to low barrier diffusion.11 High stability single atom catalysts are thus key to maximize the utilization and productivity of precious metals.
To design SACs, it is crucial to select a support which allows the creation of defined metallic sites on the surface and inhibits the aggregation of the metal atoms.8 The current approaches such as mass-selected soft-landing, metal leaching, atomic layer deposition involve high-cost synthesis processes and sometimes low-yield of active sites, which limits large-scale applications.12 In this regard, wet chemistry routes are more sustainable and straightforward. Previous studies have reported Pt single atoms anchored on carbon nitride,13 nitrogen doped graphene,8 mesoporous alumina14 and ceria.15 To the best of our knowledge, the utilisation of pristine carbon dots in synthesising Pt SACs have not yet been reported before.
Here we present a novel nitrogen doped carbon dot/Pt-SAC photocatalyst (denoted as Pt–NCD) for the hydrogen evolution reaction. The NCDs serve two functions in this system: (1) visible light absorption and (2) supporting material for highly stable Pt single atom catalysts with maximized atomic efficiency. Since the NCDs are soluble in aqueous solution, the Pt–NCDs are immobilised on a mesoporous TiO2 film, which improves catalyst recyclability, achieving a new, high-efficient and multi-scale structured photocatalytic system for H2 evolution reaction (HER). As a low cost, well-studied, most stable photocatalyst, TiO2 is an ideal system for investigating the interactions of Pt SACs/NCDs/support. The combination of efficient light harvesting, charge separation and highly active catalytic sites in the Pt-SACs/NCDs/TiO2 system leads to a synergistic effect in photocatalytic hydrogen evolution.
Results and discussion
NCDs were prepared by a one-step solvothermal carbonization as we reported before.16,17 The detailed characterisation of NCDs can be found in our previous work.17 The N content in NCDs is approximately 6%, including pyridinic N, pyrrolic N and amine groups. With N-doping, the NCDs show better light absorption and longer charge carrier lifetime compared to undoped carbon dots, beneficial for photocatalytic process. In addition, the electron donating groups on the NCD surface, such as nitrogenated groups could act as active centre for chemically adsorbing metal atoms, making them potential candidate for supporting metal species.18 Pt–NCDs were prepared by a liquid-phase photoreduction reaction with NCDs and H2PtCl6, as illustrated in Fig. 1a and detailed in the ESI.† The visible light was chosen for a milder reduction process, because photo-reduction with UV light tends to induce Pt nanoparticle formation, unless spatial confinement has been applied.18,19 The Pt–NCDs were then uniformly decorated on nanocrystal cellulose templated mesoporous TiO2 film, followed by low-temperature annealing in H2, providing a mild synthetic route towards photoactive single-atom catalysts. The resulting triple phase composite is denoted Pt–NCD/TiO2. | ||
| Fig. 1 (a) Schematic illustration of the synthesis process of Pt–NCDs; TEM and atomic-resolution aberration-corrected HAADF-STEM images of Pt–NCDs (b and d) and Pt–NCDs/TiO2 (c and e); selected EDX mapping area on HAADF-STEM image (f) and the colour mix (g); (h–k) single element mapping. | ||
The morphology of Pt–NCDs and Pt–NCD/TiO2 was first investigated with transmission electron microscopy (TEM, Fig. 1b and c). No crystalline features assigned to Pt nanoparticles can be seen on the Pt–NCDs, indicating the Pt species did not form large particles. The Pt–NCDs are uniformly distributed on the TiO2 surface, which should provide good interaction for charge transfer. The size of Pt–NCDs determined from Fig. 1b is approximately 2–4 nm, and the TiO2 nanoparticles from Fig. 1c are ∼20 nm.
As a powerful tool for discerning individual heavy atoms,5,20 high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) was used to investigate the Pt distribution and configuration in Pt–NCDs and Pt–NCD/TiO2. For Pt–NCD/TiO2 (0.20 ± 0.003 wt% Pt loading determined by inductively coupled plasma mass spectroscopy (ICP-MS)), data in Fig. 1d and e clearly shows that the bright spots correspond to Pt atoms which are uniformly dispersed with no large nanoparticles observed. Energy-dispersive X-ray (EDX) mapping in Fig. 1f–k also shows single Pt atoms in the carbon-rich phase of the NCDs. It must be noted that when the deposition time increased to 10 min, the Pt loading increases to 0.25 ± 0.006 wt%, and the Pt atoms started to form nano clusters/particles, as shown in Fig. S1.† Therefore, we chose the Pt–NCDs with 5 min deposition time for further characterization.
Scanning electron microscopy (SEM) was employed to study the morphology of the TiO2 films. The SEM top-view image in Fig. S2a† displayed nanorod-shape arranged TiO2 nanoparticles, while the cross-section showed that the film is roughly 1 μm (Fig. S2b†), which was previously shown to have good charge carrier generation and collection ability.21 No Pt peaks can be found in the X-ray diffraction (XRD) pattern of Pt–NCDs/TiO2 shown in Fig. S3,† which might because of the low loading of Pt–NCDs below the detection limit and the dispersion of Pt without forming any crystalline nanoparticle supporting the single atom configuration.22,23
X-ray photoelectron spectroscopy (XPS) characterization was carried out to examine the change of the electronic structure of NCDs after the introduction of Pt atoms. As seen in Fig. S4,† the carbon bonds in NCDs and Pt–NCDs were similar. The C 1s peaks in Pt–NCDs shifted towards higher binding energy, indicating the electron density of some C atoms becomes lower in the presence of Pt atoms. The decrease in the electron density of the C atoms can be explained by considering the formation of Pt–C coordination bonds (as discussed further below).13,24 The Pt 4f spectrum for Pt–NCDs (Fig. 2a) shows two peaks, readily assigned to the Pt5/2 and Pt7/2 signals of a Pt2+ species (the Pt 4f5/2 feature was centred at 73.1 eV, typical for Pt2+).25 In this case, by possessing electron donating groups such as nitrogenated groups and carbon centered free radicals/defects, the carbon dots can act as reductive agents to reduce PtCl62− ions upon visible light irradiation. The acidic condition provided by chloroplatinic acid prevents further reduction into metallic Pt,26 providing an environment for single atom formation.
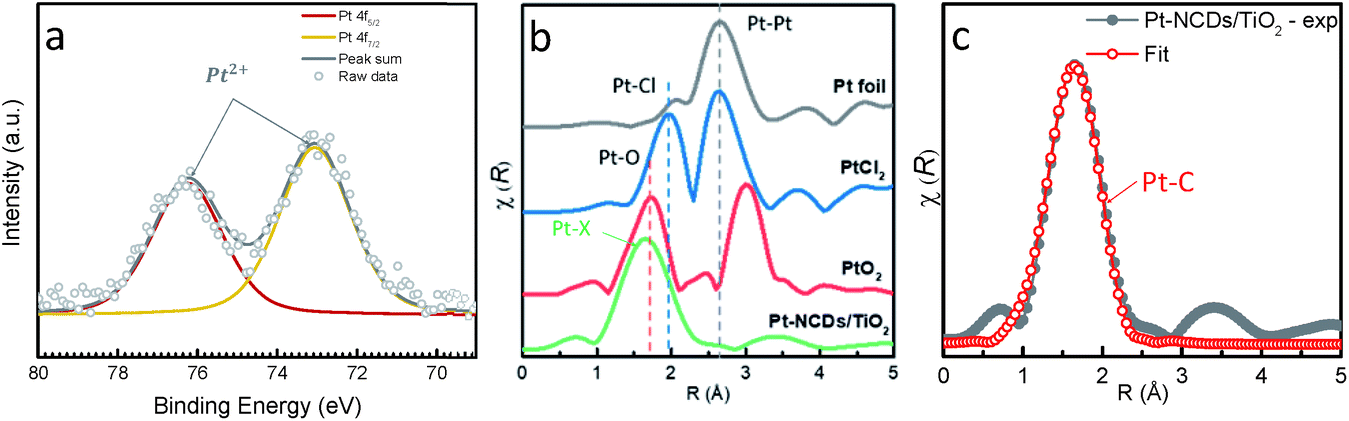 | ||
| Fig. 2 (a) High-resolution Pt 4f XPS spectra of Pt–NCDs_5 min; (b) the Fourier transform of EXAFS spectra derived from EXAFS, k-range is 3 to 12 Å−1, the X in Pt–X represents C or N; (c) FT-EXAFS curves between the experimental data and fit. | ||
To gain insights about the local environment of Pt on the Pt–NCDs/TiO2 sample and to confirm that Pt exists in an atomic form, X-ray absorption spectroscopy (XAS) measurements were performed at the Pt L3-edge before and after the treatment. The Fourier transforms (FTs) of normalized, k2 weighted extended X-ray absorption fine structure (EXAFS) k2χ(k) are displayed in Fig. 2b. The magnitude curve of FT has a strong peak in Pt–NCDs/TiO2 sample. Compared with the standard compounds PtO2, PtCl2 and Pt foil, no Pt–Pt bond peak in the region of 2–3.5 Å is observed in the Pt–NCDs/TiO2 sample, thus indicating solely the presence of isolated Pt atoms bound to the NCD support. This is in good agreement with STEM and EDX results in Fig. 1. FT of EXAFS signal of our sample gives one single peak at 1.6 Å, which could be attributed to the scattering pair of Pt and the coordinated light atoms. For comparison, the peak of PtO2 is present at 1.67 Å, which could be ascribed to the Pt–O first coordinated shell. Accordingly, the main peak of Pt–NCDs/TiO2 should belong to the Pt–N/C first coordination shell.8 Generally, EXAFS cannot distinguish between coordinated C and N atoms, because they give similar scattering parameters due to their neighbouring positions in the periodic table of elements.13 In order to better identify the coordination environment around the Pt atoms on the NCDs, a single scattering path for Pt–C or Pt–N (see Fig. S5†) was considered using least-squares refinement of the experimental data in the Artemis fitting process as shown in Fig. 2c and ESI Section 4, Fig. S5 and Table S1.† Detailed fitting process shows that the Pt atoms are more likely coordinated with C atoms than N atoms but the possibility of Pt–N coordination is not completely ruled out. The fitted Pt–C coordination number for the sample is 4.7, implying that each Pt atom coordinates with four or five carbon atoms on the support. Notably, the four-coordination mode is inherently preferred by zero or low valent Pt complexes. This is due to the highly functionalised surface providing a potential scaffold and strong coordination for firmly binding the SACs through spatial confinement,27 which partially explains the high stability of the Pt SACs in the system and makes NCDs a promising support for single-site catalysts.14
Based on the XPS and XAS results, here we summarise the formation mechanism of Pt SACs on NCD support. To describe it in detail, as the NCDs are physically mixed with H2PtCl6 solution, it can adsorb many PtCl62− ions from the solution. Firstly, NCDs have rich heteroatoms and functional groups such as amines and carbon centred free radicals/defects, which provide active sites for chemical adsorption of PtCl62− ions. Secondly, the highly functionalised surface provides a scaffold and strong coordination for firmly adsorbing the PtCl62− ions through spatial confinement. After adsorption, visible light is employed to irradiate the PtCl62− loaded NCDs. The absorbed visible light photons would excite the NCDs to generate electrons for reducing the PtCl62− ions, leading to the formation of Pt–C/N coordination. The same experiment with UV light irradiation resulted in large Pt nanoparticle formation, as shown in Fig. S6,† which proves the importance of visible light in single atom formation on photoactive NCDs. The concentration of H2PtCl6 (0.3 mM) is in excess, and the mixing of NCDs with PtCl62− is sufficient, as can be seen in Fig. 1d, that every NCD in the field are loaded with Pt atoms. However, further investigations are required to precisely control the metal loading. Unlike on NCDs, we found Pt nano cluster/particle formation on the surface of TiO2 (Fig. S7†) using same Pt-photoreduction reaction, proving the necessity of NCDs for Pt single atom formation, and also eliminate the possibility of forming Pt single sites by trapping effect from non-stoichiometric defects on TiO2 surface.
The above-mentioned Pt single site containing Pt–NCDs/TiO2 sample was then used as a visible light driven photocatalyst for HER. To test that we applied the Pt–NCDs on the mesoporous TiO2 film as an immobilized particulate system. As a proof-of-concept, the immobilization of a photocatalytic system onto a specific substrate is a way to scale up H2 production from the laboratory to the plant scale, which is beneficial in terms of recyclability and for effectively harvesting sunlight irradiation by tracking the movement of the sun.1 The NCD loading was determined to be 1.5 wt% from the mass loss using thermogravimetric analysis (Fig. S8†), while the Pt loading was confirmed to be 0.20 wt% by ICP-MS.
Compared to the pristine TiO2 film, the NCD/TiO2 and Pt–NCD/TiO2 films show extended visible light absorption with a tail up to 600 nm, as shown in Fig. 3a, indicating the good visible light response. However, it has to be noted that the as-prepared TiO2 sample also showed slight tail state light absorption beyond 420 nm, which means it can absorb visible light photons and generate electron–hole pairs, but within a very limited content. This fact could explain the observed small amount of hydrogen production from pristine TiO2 film in the following discussion.
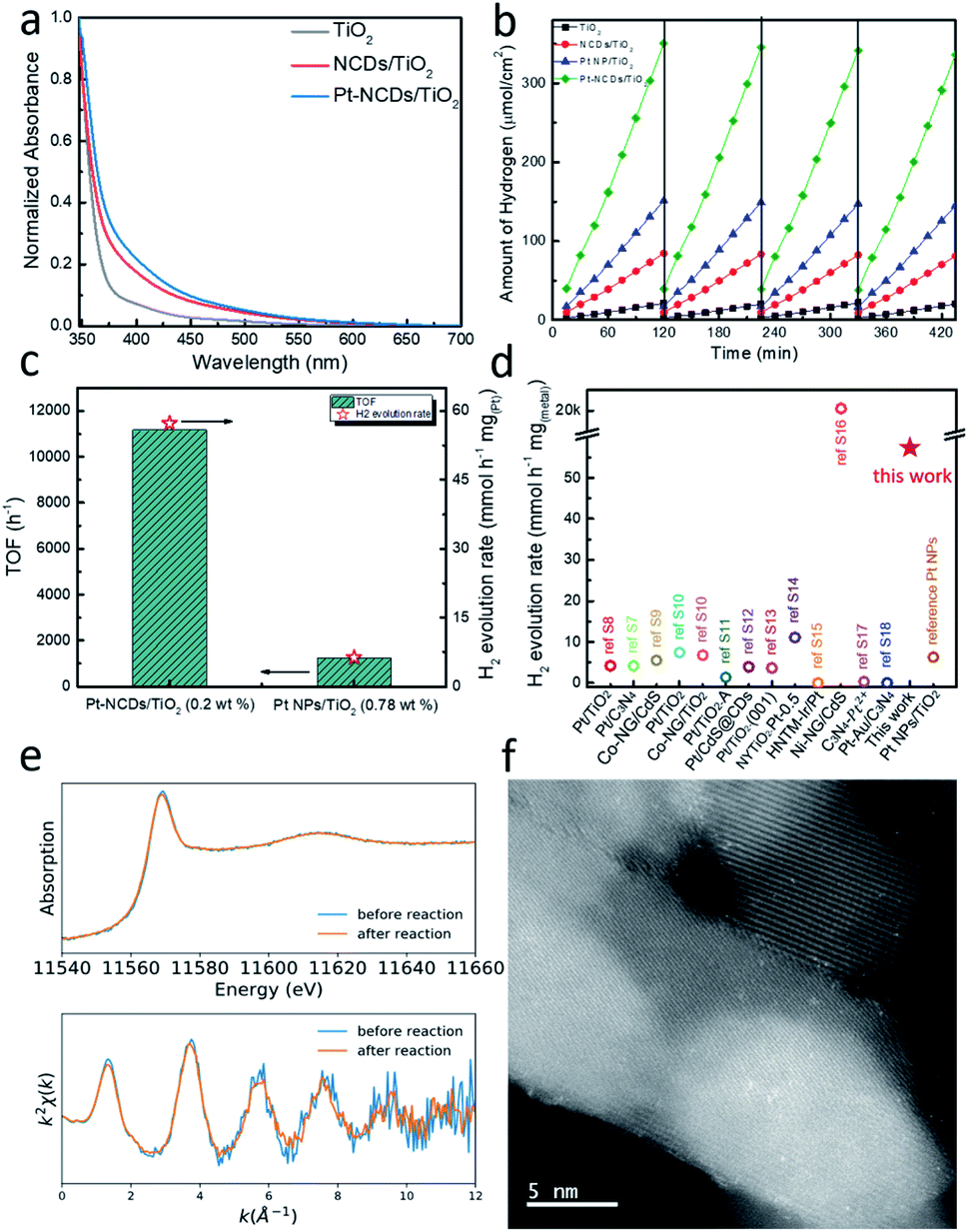 | ||
| Fig. 3 (a) UV-vis absorption spectra of different TiO2 films. (b) The HER performance of TiO2 films with different configurations (zero-bias, with hole scavenger); (c) the TOF and H2 evolution rate comparison of Pt–NCDs/TiO2 and Pt NPs/TiO2; (d) comparison of H2 generation rate of this work to literature; (e) XANES and EXAFS Chi(k) oscillations of Pt–NCDs/TiO2 before and after photocatalytic reaction; (f) HAADF-STEM image of the same sample as shown in Fig. 1e after reaction. | ||
To evaluate the effects, photocatalytic HER with different TiO2 films was measured using Na2S–Na2SO3 as the sacrificial agent at room temperature under visible light (λ > 420 nm). The reaction was carried out for four cycles, each of which lasted for 2 h.30 Photocatalytic H2 evolution with Pt–NCDs/TiO2 (0.20 wt% Pt loading) was measured by gas chromatography, and compared to NCDs/TiO2, nanoparticulate Pt/TiO2 and pristine TiO2 (Fig. 3b). TiO2 alone gives very limited activity for H2 evolution (10.5 μmol h−1 cm−2), owing to its large band gap and low visible light response (Fig. 3a). Loading with NCDs results in four times higher H2 yields, with an evolution rate of 42.2 μmol h−1 cm−2, due to the higher light utilisation by NCDs to produce light-induced electrons to drive HER. However, with single-atom Pt as co-catalyst, Pt–NCDs/TiO2 shows a H2 generation rate of 175.3 μmol h−1 cm−2, more than four times higher than that of NCDs/TiO2 and 17 times higher than pristine TiO2. This infers that apart from the activity enhanced by NCDs on TiO2, the Pt SACs plays a significant role on improving the photocatalytic efficiency. To evaluate the performance of the material, the catalyst layer was carefully scratched off from FTO glass and weighed (Fig. S9†). The average mass from 3 samples was 1.53 ± 0.21 mg. Under the present experimental conditions the H2 evolution rate and turnover frequency (TOF) with respect to the Pt content are calculated to be 57.3 mmol h−1 mgPt−1 and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 171 h−1, respectively (Fig. 3c).13 As summarized in Fig. 3d and Table S2,† this photocatalytic H2 production activity is one of the highest records for metal-based cocatalysts. After 4 cycles, the H2 evolution rate for each sample was only reduced by around 4%, showing the excellent stability of the composites. The stable H2 evolution rate of Pt–NCDs/TiO2 in the test reflects the stability of single-atom Pt in the framework of NCDs loaded on the TiO2 surface. For comparison, a catalyst comprising of Pt nanoparticles (Pt NPs) decorated on TiO2 film was also tested. Even though the Pt loading is higher (0.78 ± 0.011 wt% by ICP-MS), the H2 generation rate was only 7.0 mmol h−1 mgPt−1 and TOF 1359 h−1, suggesting a lower atomic efficiency. These results demonstrate that the maximised atomic efficiency of single-atom co-catalyst remarkably boosts the photocatalytic performance of TiO2 and NCDs/TiO2.13 The overall performance of Pt–NCDs/TiO2 is higher than the sum of Pt NPs/TiO2 and NCDs/TiO2, which might due to the efficient charge exchange processes in Pt SACs/NCDs/TiO2 system as well as to the presence of more effective active sites, achieving synergistic effect for photocatalytic reactions.7
171 h−1, respectively (Fig. 3c).13 As summarized in Fig. 3d and Table S2,† this photocatalytic H2 production activity is one of the highest records for metal-based cocatalysts. After 4 cycles, the H2 evolution rate for each sample was only reduced by around 4%, showing the excellent stability of the composites. The stable H2 evolution rate of Pt–NCDs/TiO2 in the test reflects the stability of single-atom Pt in the framework of NCDs loaded on the TiO2 surface. For comparison, a catalyst comprising of Pt nanoparticles (Pt NPs) decorated on TiO2 film was also tested. Even though the Pt loading is higher (0.78 ± 0.011 wt% by ICP-MS), the H2 generation rate was only 7.0 mmol h−1 mgPt−1 and TOF 1359 h−1, suggesting a lower atomic efficiency. These results demonstrate that the maximised atomic efficiency of single-atom co-catalyst remarkably boosts the photocatalytic performance of TiO2 and NCDs/TiO2.13 The overall performance of Pt–NCDs/TiO2 is higher than the sum of Pt NPs/TiO2 and NCDs/TiO2, which might due to the efficient charge exchange processes in Pt SACs/NCDs/TiO2 system as well as to the presence of more effective active sites, achieving synergistic effect for photocatalytic reactions.7
The stability of the samples was further confirmed by EXAFS and HAADF-STEM. Fig. 3e shows the comparison of XANES and EXAFS Chi(k) oscillation curves before and after the photocatalytic reaction. The spectra remain identical, indicating that no chemical environmental changes happened to the Pt atoms during the reaction. The HAADF-STEM images in Fig. 3f reveal that the Pt species are still in the form of isolated single atoms with no aggregation being observed. Combined with the stable H2 generation rate during the photocatalytic process, these results demonstrate that the Pt single atom catalysts maintain structural integrity and hence an excellent performance.
The mechanism behind SACs' photocatalytic activity has been mainly studied using theoretical DFT calculation, which attributed the enhanced photocatalytic hydrogen production performance to the maximized catalyst atomic efficiency,7,8,20 and the effectively lowered Gibbs free energy barriers for hydrogen adsorption. Guo and co-workers calculated the capability of Pt single atoms on the TiO2 surface to trap photoelectrons and found that the photoelectrons were extracted from the subsurface to the surface around the Pt sites, which can lower the generated charge migration barrier, facilitating the proton adsorption and reduction process. The Pt single atoms form unoccupied electronic states within the bandgap, which are primarily due to the hybridization of Pt and Ti5c orbitals and can be effective electron acceptors.10 The formation of intermediate trap states was also demonstrated by Xie et al., who revealed an accelerated charge transfer by Pt single atoms coordinated with carbon nitride via femtosecond transition absorption spectroscopy.13 On the other hand, the Pt atoms can also reduce the adsorption energy for hydrogen at 0 K, which is considered to determine the reaction energetics of the elementary steps of HER. The H* adsorption energy calculated by Yang and co-workers is near to the optimum adsorption strength, which is rationalized by the facilitated removal of H* into H2 through either the Tafel or Heyrovsky mechanism.31 The Gibbs free energy profiles of the HER comparison between anatase TiO2, Pt nanoparticle and Pt single atom reveals that the TiO2 has a poor photocatalytic activity owing to the weak hydrogen adsorption ability, and that metallic Pt has a too strong hydrogen adsorption leading to a limited H2 formation. Pt single atoms show a ‘in-between’ scenario with optimum adsorption position and should therefore be more suitable for HER.32 The same finding has also been described by Qiu et al. with atomic layers of Pt anchored on Mo2C nanorods.33 Those works have successfully demonstrated the favourable role of Pt single atoms in photocatalytic applications. In this work the mechanism of Pt SACs on enhancing the photocatalytic HER performance is investigated with experimental approaches.
To reveal the interaction between the TiO2 support and Pt–NCDs, electron energy-loss spectroscopy (EELS) was employed to give qualitative evidence of the electronic state changes in the TiO2 matrix. As can be seen in Fig. 4a, the detected difference of low-loss EELS reflects a decreased free electron density within the TiO2 support matrix after decorating it with Pt–NCDs.34 This can be attributed to the electron transfer from TiO2 to Pt–NCDs, resulting in an average electron enrichment around the Pt atoms.35 Correspondingly, in Fig. 4b the detected shift of the Ti-L2,3 edge towards a higher energy, and the slightly increased t2g–eg crystal-splitting of Ti-L2,3 after the loading of Pt–NCDs, which changed from 2.00 eV to 2.07 eV in L3 and 1.75 eV to 1.97 eV in L2, further confirms the less effective charges on Ti atoms due to the electron transfer to Pt–NCDs.34,36
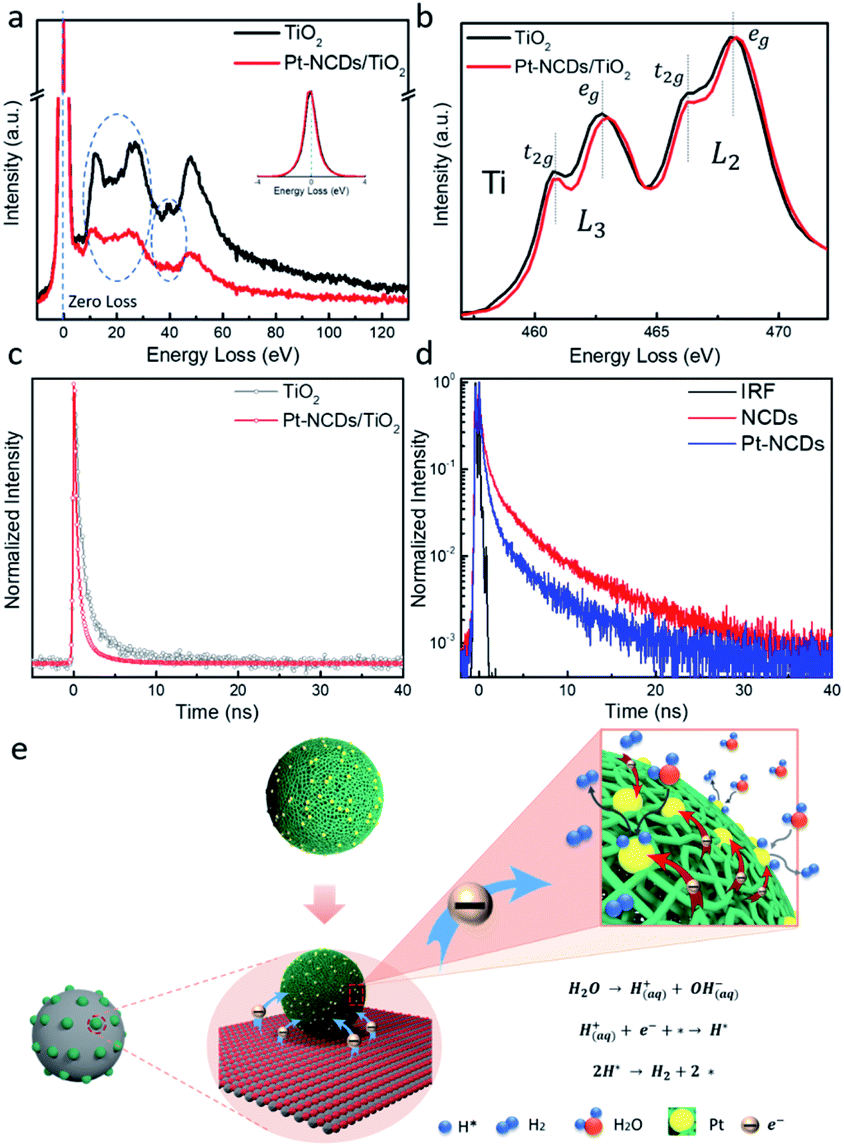 | ||
| Fig. 4 Ti-L2,3 low-loss (a) and core-loss (b) EELS spectra of pristine TiO2 and Pt–NCDs decorated TiO2. Fluorescence decay curves recorded using TCSPC equipped with 405 nm excitation laser diode and detected at 520 nm of (c) TiO2 and Pt–NCDs/TiO2 samples and (d) NCDs and Pt–NCDs films in air (black curve corresponds to the TCSPC's instrument response function); (e) the schematic multi-scale design of Pt–NCDs/TiO2 photocatalyst. | ||
Nanosecond spectroscopic measurements were employed to directly monitor the charge transfer process within the Pt–NCDs/TiO2 system, using time-correlated single photon counting (TCSPC). The photoluminescence (PL) decay measurements on pristine TiO2 and Pt–NCDs/TiO2 have been recorded, as displayed in Fig. 4c. The decay kinetics were fitted with a bi-exponential function, and the sample containing Pt–NCDs showed a shorter PL lifetime (1.26 ns) compared to 3.14 ns of TiO2. The decay time of TiO2 has two components: 5.52 ns (ca. 13.7%) and 0.962 ns (ca. 86.3%). After loading with Pt–NCDs, the fast decay component decreased sharply: 2.23 ns (ca. 14.4%) and 0.45 ns (85.6%). The reduced lifetime suggests an ultrafast electron transfer from TiO2 to the Pt–NCDs, possibly introduced by the electron accepting ability of NCDs and the high electron affinity of Pt single atoms,10,13,37,38 consistent with the findings from EELS. Facilitated charge transfer within the system results in a more sufficient charge separation that can effectively improve the photocatalytic activity.39
What is more interesting, as shown in Fig. S10,† is the observed fluorescence decays of Pt–NCDs aqueous solution compared with those of neat NCDs. The direct comparison between the curves suggests that no meaningful quenching effects on the fluorescence decays of NCDs by the surface-deposited Pt single atoms occurs. However, when drop casting the NCDs and Pt–NCDs on glass substrates and measure in air, the films showed a distinct difference, by which the Pt–NCDs film sample decaying faster than that of NCDs (Fig. 4d). The corresponding observed fluorescence quantum yield was reduced dramatically, from 9.7% for the NCDs film to 5.8% for the Pt–NCDs film, suggesting a charge transfer from NCDs to the surface loaded Pt single atoms, which act as active sites for efficient H2 evolution reactions under visible light. Such effects on fluorescence intensities and decays can be understood in terms of static quenching, meaning that the quenchers are near-neighbours to the emissive species. In this case, the near-neighbour configuration must be absolute with the Pt atoms (the quencher) right at the emissive sites on the carbon dots surface, thus the observation of static fluorescence quenching.40 It has been reported that N-doping can affect the PL properties of carbon dots by strong orbital interactions with the adjacent atoms, suggesting that N dopants are close to emissive sites as well.41–44 Considering the fitted EXAFS results which have shown that the Pt is likely bonded with C or N or both, here we infer that the N atoms may help stabilize the Pt atoms by an atom trapping effect.29,45,46 A DFT study reported previously has shown that doping the graphene lattice with N can enhance the Pt–C support interaction energy, effectively stabilizing the metal single atoms, attributing to the creation of preferred binding sites for metal atoms,28 and improve the photocatalytic activity with efficiently facilitated charge transfer.
Our experimental findings help gain further insights on the stabilisation of Pt SAC active sites and explain the enhanced photoactivity which can be assigned to the photo-induced charge separation.
Based on the findings from UV-vis absorption, EELS and TCSPC, here we propose the following mechanism of Pt–NCDs enhancing the photocatalytic HER activity of TiO2. As shown in Fig. 4e, the photocatalyst can be described as a multi-scale design. The micrometer thick electrode consisting of nanometer sized TiO2 support provides large surface area to anchor the Pt–NCDs, and the slight tail state light absorption contributes to the generation of some electron–hole pairs upon visible light irradiation. The nano-meter sized NCDs can enhance the light response of the TiO2 support, as well as generate electrons themselves under visible light irradiation. The N species in the NCDs help stabilizing Pt single atoms and contribute to longer photo-generated carrier lifetime by suppressing the interlayer non-radiative energy dissipation. EELS and TCSPC results revealed the electron transfer from TiO2 to the Pt–NCDs, leading to an overall electron-enriched environment around Pt–NCDs. The harvested electrons then transfer to the stabilized atomic scale Pt SACs, which act as active sites to effectively reduce water into hydrogen. The maximised atom efficiency, optimal H* adsorption energy and low energy barriers of Pt SACs also lead to a higher H2 production rate and turnover frequency. In summary, with NCDs supported Pt SACs used as co-catalyst in photocatalytic HER, a synergistic effect of higher light utilisation, better charge transportation, maximised atomic efficiency and lower kinetic energy barrier could be achieved, representing a promising approach for designing highly active photocatalytic co-catalysts.
Conclusions
In conclusion, we have used a facile one-step photo-deposition method to synthesise Pt single-atom-catalysts on nitrogen doped carbon dots which then anchored on the surface of a mesoporous TiO2 model system. HR-STEM combined with EXAFS have proven the existence of single Pt atoms, which remained stable after photocatalytic HER reactions. The electron donating ability of NCDs contributes to the reduction of PtCl62− in acidic conditions, while the highly functionalised surface provides a potential scaffold and a strong coordination for firmly trapping the SACs through spatial confinement. The presence of N doping can further stabilise the isolated single atoms during thermal treatment and photocatalytic reactions. EELS and TCSPC were employed to study the mechanism of the enhanced HER performance, which have shown an improved charge transfer process between TiO2, NCDs and Pt SACs, leading to the sufficient charge separation. The NCDs harvest visible light and create an electron enriched environment for the anchored Pt SACs, and the Pt SACs with maximized atomic efficiency act as active sites and contribute to the better HER performance. The synergistic effect of single-atom co-catalyst and carbon dots provides a new strategy to rationally design a multi-scale photocatalyst structures to achieve high H2 evolution efficiency. The facile process of producing single atom catalyst with nitrogen-doped carbon dots also holds great potential in various other applications such as electrocatalysis, heterogeneous catalysis and drug delivery, providing a promising way to reduce the high cost of noble metals in many fields and pave a new avenue for the development of highly efficient materials.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
HL thanks the Chinese government for the award of CSC scholarships. SD is grateful for financial support by Edinburgh Instruments. This work is part-funded by the European Regional Development Fund through the Welsh Government. We thank the Diamond Light Source for access and support in use of the electron Physical Science Imaging Centre (Instrument E01 proposal MG22447 and E02 proposal EM20116) that contributed to the results presented here. AS thanks the Diamond BAG access scheme and help from Giannantonio Cibin at B18 beamline. XW thanks the National Natural Science Foundation of China (21861130353).Notes and references
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 4, 387–399 CrossRef.
- T. Takata and K. Domen, ACS Energy Lett., 2019, 4, 542–549 CrossRef CAS.
- A. J. Hunt, A. S. Matharu, A. H. King and J. H. Clark, Green Chem., 2015, 17, 1949–1950 RSC.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104–114 CrossRef CAS.
- J. Tian, Y. Leng, Z. Zhao, Y. Xia, Y. Sang, P. Hao, J. Zhan, M. Li and H. Liu, Nano Energy, 2015, 11, 419–427 CrossRef CAS.
- X. Cui, K. Junge, X. Dai, C. Kreyenschulte, M. M. Pohl, S. Wohlrab, F. Shi, A. Brückner and M. Beller, ACS Cent. Sci., 2017, 3, 580–585 CrossRef CAS PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- F. Han, Z. Zhou, X. Zhang, Z. Huang, M. Li and L. Guo, J. Phys. Chem. C, 2018, 122, 2546–2553 CrossRef CAS.
- S. Tosoni and G. Pacchioni, Surf. Sci., 2017, 664, 87–94 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- F. Dvořák, M. F. Camellone, A. Tovt, N. D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- D.-W. Zhang, N. Papaioannou, N. M. David, H. Luo, H. Gao, L. C. Tanase, T. Degousée, P. Samorì, A. Sapelkin, O. Fenwick, M.-M. Titirici and S. Krause, Mater. Horiz., 2018, 5, 423–428 RSC.
- H. Luo, S. D. Dimitrov, M. Daboczi, J.-S. Kim, Q. Guo, Y. Fang, M.-A. Stoeckel, P. Samorì, O. Fenwick, A. B. Jorge Sobrido, X. Wang and M.-M. Titirici, ACS Appl. Nano Mater., 2020, 3, 3371–3381 CrossRef CAS.
- T. Li, J. Liu, Y. Song and F. Wang, ACS Catal., 2018, 8, 8450–8458 CrossRef CAS.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li, M. Lei, C. Zhang, J. Irawan, L. M. Liu and H. Wu, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Z. Chen, H. N. Dinh and E. Miller, Photoelectrochemical water splitting: standards, experimental methods, and protocols, Springer, 2013 Search PubMed.
- J. Jin, C. Wang, X. N. Ren, S. Z. Huang, M. Wu, L. H. Chen, T. Hasan, B. J. Wang, Y. Li and B. L. Su, Nano Energy, 2017, 38, 118–126 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- K. Kamiya, R. Kamai, K. Hashimoto and S. Nakanishi, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- C. Dong, C. Lian, S. Hu, Z. Deng, J. Gong, M. Li, H. Liu, M. Xing and J. Zhang, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- Y. Wang, Y. Wang and R. Xu, J. Phys. Chem. C, 2013, 117, 783–790 CrossRef CAS.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, L. Jiang, Z. Wu, D. J. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- S. Guo, X. Li, X. Ren, L. Yang, J. Zhu and B. Wei, Adv. Funct. Mater., 2018, 28, 1–13 Search PubMed.
- J. Xing, H. B. Jiang, J. F. Chen, Y. H. Li, L. Wu, S. Yang, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 15258–15264 RSC.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem.–Eur. J., 2014, 20, 2138–2144 CrossRef CAS PubMed.
- Y. Qiu, Z. Wen, C. Jiang, X. Wu, R. Si, J. Bao, Q. Zhang, L. Gu, J. Tang and X. Guo, Small, 2019, 15, 1–9 Search PubMed.
- Y. Xu, C. Zhang, L. Zhang, X. Zhang, H. Yao and J. Shi, Energy Environ. Sci., 2016, 9, 2410–2417 RSC.
- P. Pichat, J.-M. Herrmann, J. Disdier, M.-N. Mozzanega and H. Courbon, in Catalysis on the Energy Scene, ed. S. Kaliaguine and C. Mahay, Elsevier, 1984, vol. 19, pp. 319–326 Search PubMed.
- C. M. Wang, Z. G. Yang, S. Thevuthasan, J. Liu, D. R. Baer, D. Choi, D. H. Wang, J. G. Zhang, L. V. Saraf and Z. M. Nie, Appl. Phys. Lett., 2009, 94, 2–5 Search PubMed.
- R. Shi, Z. Li, H. Yu, L. Shang, C. Zhou, G. I. N. Waterhouse, L. Z. Wu and T. Zhang, ChemSusChem, 2017, 10, 4650–4656 CrossRef CAS PubMed.
- H. Yu, Y. Zhao, C. Zhou, L. Shang, Y. Peng, Y. Cao, L. Z. Wu, C. H. Tung and T. Zhang, J. Mater. Chem. A, 2014, 2, 3344–3351 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- J. Xu, S. Sahu, L. Cao, C. E. Bunker, P. Ge, L. Yamin, K. a. S. Fernando, P. Wang, E. a. Guliants, M. J. Meziani, H. Qian and Y.-P. Sun, Langmuir, 2012, 28, 16141–16147 CrossRef CAS PubMed.
- D. Qu, M. Zheng, J. Li, Z. Xie and Z. Sun, Light: Sci. Appl., 2015, 4, e364 CrossRef CAS.
- M. Sudolská and M. Otyepka, Appl. Mater. Today, 2017, 7, 190–200 CrossRef.
- S. S. K. Das, Y. Liu, S. Yeom, D. Y. D. Kim and C. C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and characterization data & supporting figures. See DOI: 10.1039/d0ta04431h |
| This journal is © The Royal Society of Chemistry 2020 |