
Nitrogen-doped phosphorene for electrocatalytic ammonia synthesis†
Guangrui
Xu‡
ab,
Hao
Li‡
c,
Abdulaziz S. R.
Bati
 d,
Munkhjargal
Bat-Erdene
d,
Md J.
Nine
e,
Dusan
Losic
e,
Yu
Chen
*a,
Joseph G.
Shapter
*d,
Munkhbayar
Batmunkh
*df and
Tianyi
Ma
*b
d,
Munkhjargal
Bat-Erdene
d,
Md J.
Nine
e,
Dusan
Losic
e,
Yu
Chen
*a,
Joseph G.
Shapter
*d,
Munkhbayar
Batmunkh
*df and
Tianyi
Ma
*b
aKey Laboratory of Macromolecular Science of Shaanxi Province, School of Materials Science and Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: ndchenyu@gmail.com
bSchool of Environmental and Life Sciences, The University of Newcastle (UON), Callaghan, New South Wales 2308, Australia. E-mail: tianyi.ma@newcastle.edu.au
cDepartment of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: j.shapter@uq.edu.au
eSchool of Chemical Engineering and Advanced Materials, The University of Adelaide, Adelaide, South Australia 5005, Australia
fCentre for Clean Environment and Energy, Griffith University, Gold Coast, Queensland 4222, Australia. E-mail: m.batmunkh@griffith.edu.au
First published on 7th May 2020
Abstract
The rapid surface oxidation of phosphorene under ambient conditions is considered to be a serious issue for many applications, but is used here as a strategy to achieve efficient heteroatom doping. Highly crystalline nitrogen-doped phosphorene (N-phosphorene) is prepared using a combination of ball milling and microwave techniques. The prepared N-doped phosphorene nanosheets showed outstanding electrocatalytic performance as a new type of non-metallic catalyst for nitrogen (N2) to ammonia (NH3) conversion, with an NH3 yield rate and faradaic efficiency (FE) of up to 18.79 μg h−1 mgCAT−1 and 21.51%, respectively, at a low overpotential (0 V) versus the reversible hydrogen electrode (RHE). Density functional theory calculations revealed that the high nitrogen reduction reaction (NRR) FEs originate from the increased hydrophobicity at the N and O doped phosphorene surfaces, which in turn hinders the competing hydrogen evolution reaction (HER) in an alkaline environment and promotes the NRR. This work not only introduces an efficient strategy to chemically functionalize 2D phosphorene, but also opens a new avenue in using N-doped phosphorene nanosheets as a metal-free catalyst.
Introduction
Atomically thin two-dimensional (2D) layered nanomaterials have emerged as one key component to future materials development. Since 2014, black phosphorus (BP) – a layered 2D semiconductor – has attracted increasing attention due to its fascinating properties and unique structure.1 Phosphorene, mono- or few-layered BP obtained from bulk BP, exhibits a thickness-dependent tunable bandgap (0.3–2.0 eV) and excellent carrier mobility (∼1000 cm2 V−1 s−1) in addition to its high in-plane anisotropic properties,2–4 all of which make it attractive for a wide range of practical applications including electronic devices,5 energy storage,6 solar cells,7 and catalysis.8 Owing to its high carrier mobility, favorable electronic structure and large specific surface area, BP derivatives hold specific promise for both photocatalytic and electrocatalytic processes.9–11 Although excellent progress has been made using BP derivatives for the hydrogen and oxygen evolution reactions,10–15 practical demonstration of the use of 2D phosphorene for advanced catalysis for the nitrogen reduction reaction (NRR) and CO2 reduction reaction is very scarce.16 As such, developing efficient metal-free catalysts for these cutting-edge catalytic processes is of great interest.Few-layer phosphorene nanosheets tend to oxidize in the presence of ambient air and water, resulting in the rapid loss of their semiconducting properties.17,18 Although this high chemical reactivity of phosphorene with oxygen is typically considered undesirable for device applications, it may also become a broad utility for the chemistry of 2D phosphorene. Chemical functionalization has proven to be an effective strategy in modulating the chemical, electronic, electrical and structural properties of BP, while producing stable phosphorene nanosheets.19–21 In particular, elemental doping is known to be a reliable approach to modify the charge carrier concentration, thus modifying the material's properties. However, there has been limited effort in the elemental doping of 2D BP despite several theoretical studies suggesting great promise.22–24 Notably, a number of successful studies on doping of some metal elements (K, As, Se, and Te) into bulk BP crystals and/or mechanically exfoliated BP nanosheets have been reported,20,25–27 while non-metal element doping and solution processability of phosphorene are areas of great interest but have not progressed yet.
Herein, we present a facile and efficient strategy based on a combination of ball milling and microwave exfoliations to produce N-doped phosphorene (N-phosphorene) nanosheets and demonstrate their application as a novel metal-free electrocatalyst for electrochemical nitrogen-to-ammonia (N2 to NH3) synthesis under ambient conditions. Although the oxidation of BP is considered to be a significant issue, we elegantly used it to our advantage, providing abundant reactive sites for N-doping of phosphorene. Our N-phosphorene electrocatalyst exhibited excellent electrochemical performance with high NH3 yield rates and faradaic efficiencies (FEs) of 18.79 μg h−1 mgCAT−1 and 21.51% (determined using the indophenol blue method) and 16.67 μg h−1 mgCAT−1 and 19.08% (determined using an ammonia selective electrode), respectively, at a low overpotential (0 V) versus the reversible hydrogen electrode (RHE). The catalytic activity of functionalized phosphorene nanosheets has been systematically investigated using density functional theory (DFT) calculations.
Results and discussion
Synthesis and characterization of N-doped phosphorene
2D N-phosphorene sheets were prepared in N-methyl-2-pyrrolidone (NMP) solvent using a combination of ball milling and microwave-assisted exfoliation methods. The synthetic procedure of the N-phosphorene sheets is shown in Fig. 1a. It is well established that for chemical doping of 2D graphene and carbon materials, oxygen containing functional groups on the surface are responsible for the formation of a C–N bond (N-doping).28–30 Considering this, a bulk BP crystal was first ground in a mortar and pestle in air, creating abundant reactive sites on the surface to enhance its interaction with the N source. The ground BP crystal was then ball milled in NMP solvent with and without ammonia solution (NH4OH) as the N source to produce N-doped (N-phosphorene) and oxidized phosphorene (O-phosphorene), respectively.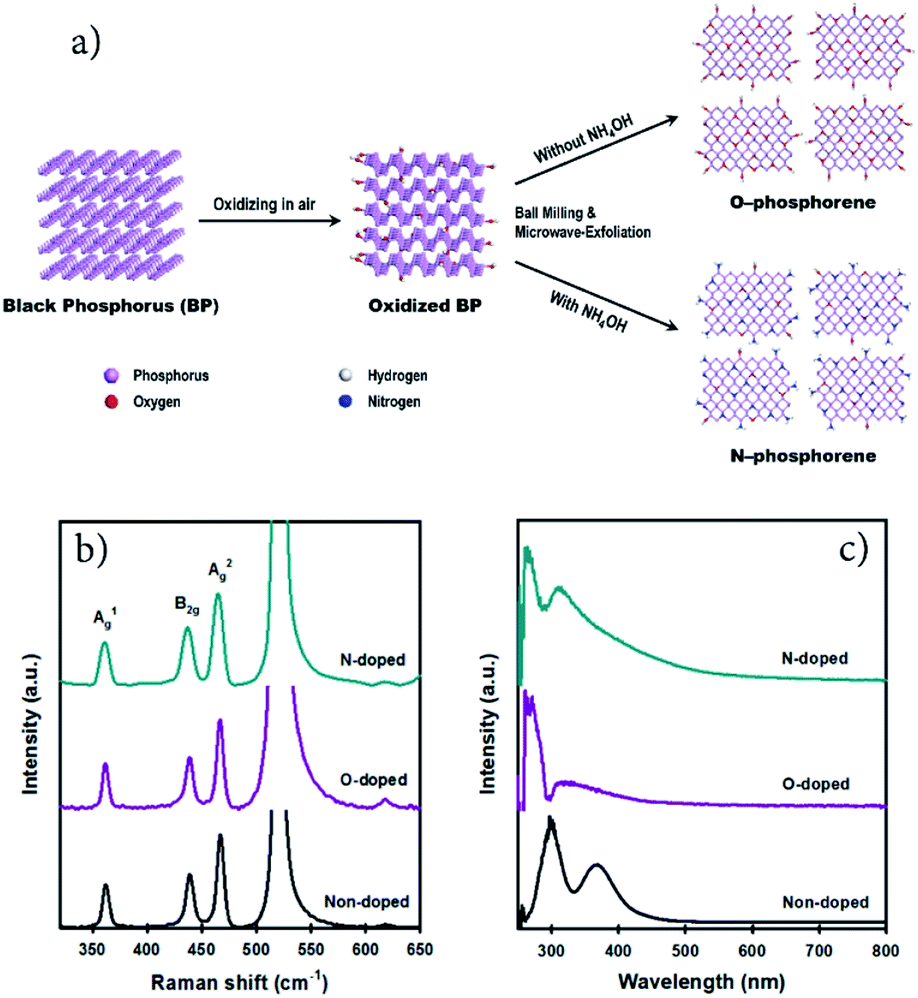 | ||
| Fig. 1 (a) Schematic illustration for the preparation of N-doped and O-doped phosphorene using a combination of ball milling and microwave-assisted LPE methods. (b) Raman (on a silicon (Si) substrate) and (c) UV-vis spectra of non-doped, O-doped and N-doped phosphorene. | ||
Ball milling is an effective approach for edge functionalization of 2D layered materials, generating abundant reactive species (radicals and ions) at the nanosheet edges.31–33 In the presence of the N source (NH4OH), the oxidized BP flakes can be N-doped and/or N-functionalized during the ball-milling process (Fig. 1a) with a slight preference at the flake edges if the O-phosphorene behaves as other 2D materials during ball-milling. Notably, ball milling BP flakes in the absence of NH4OH produced highly oxidized and/or oxygen doped phosphorene nanosheets (termed “O-phosphorene”). In addition, the van der Waals interaction between the flakes may be weakened during the ball milling, producing readily exfoliatable BP flakes. Then the ball milled BP flakes were further exfoliated in NMP using a microwave-assisted liquid-phase exfoliation (LPE) method to produce ultrathin (few-layer) phosphorene nanosheets. For comparison, we also prepared few-layer phosphorene nanosheets with controlled oxidation (termed “phosphorene”) using a microwave-exfoliation method (air oxidation and ball milling steps were not involved) using the procedure reported in our recent work.34
As depicted in Fig. S1a,† the height of the non-doped phosphorene nanosheet was measured to be ∼4.9 nm, which (considering that monolayer phosphorene is 0.85 nm thick)35,36 corresponds to about 5 atomic layers and is consistent with our previous measurements.34 The thickness of the O-phosphorene was ∼5.5 nm (6 layers) (Fig. S1b†), while the measured height for the N-phosphorene was ∼8.6 nm (10 layers) (Fig. S1c†). The thicker nanosheets of our N-phosphorene, as compared to the non-doped phosphorene, could be due to the presence of functional groups and doping elements increasing the measured AFM height,37 and/or acting as a barrier during the exfoliation. Another noticeable feature from the AFM images was the difference in the flake sizes of the samples. In particular, the lateral sizes of both O-doped and N-doped phosphorene sheets varied from 150 nm to 300 nm, while the non-doped phosphorene showed an average flake size of 600 nm. This significant difference in the lateral size of the non-doped and doped phosphorene nanosheets can be explained by the ball milling induced mechanochemical process. It is well established that ball milling 2D materials such as graphite results in a dramatic reduction in the flake size, but it generates reactive sites which can facilitate the doping and functionalization.31,32Fig. 1b shows the Raman spectra of our non-doped and doped phosphorene on a silicon (Si) substrate. The characteristic A1g, B2g, and A2g phonon modes centered at ∼362, ∼437, and ∼466 cm−1, respectively,17,38 were observed for all three samples, suggesting that the chemical doping/functionalization and ball milling treatments did not alter the structural integrity. Moreover, our Raman spectra are consistent with the signature Raman spectrum of partially oxidized few-layer black phosphorus.39
We further examined our non-doped and doped phosphorene dispersions using UV-vis spectroscopy (Fig. 1c). Two main absorption peaks can be observed for all samples and are consistent with previously reported theoretical results and the spectrum of mechanically exfoliated BP.40,41 In particular, the non-doped phosphorene dispersion exhibited peaks at around 300 nm and 370 nm, which are in excellent agreement with our previous study on microwave-exfoliated few-layer BP solution.34 Interestingly, despite the appearance of these two peaks, considerable shifts can be observed for the O- and N-doped phosphorene samples, as compared to the non-doped phosphorene (Fig. 1c). Recently, we used DFT calculations to determine the UV-vis absorbance of phosphorene,34 and observed the two characteristic peaks within the range 250–400 nm (Fig. S2†). More importantly, we theoretically found that the positions of the spectral peaks can shift depending on the BP flake size (blueshift as the flakes become smaller).34 Owing to the small flake sizes (confirmed by AFM), the peak positions of our doped phosphorene samples blueshifted to 260 nm and 310 nm, which qualitatively agree with the simulated UV-vis spectrum of the small phosphorene sheet with high accuracy (Fig. S2†). In addition, as shown in Fig. S3,† our N-phosphorene dispersion showed excellent stability over at least 20 days.
X-ray photoelectron spectroscopy (XPS) is an important tool to characterize the chemical structure of nanostructured functional materials. In this work, we used XPS to explore the oxidation and functionalization/doping states of our phosphorene samples. Fig. 2a–c show the high-resolution (HR) P 2p XPS spectra of our non-doped, O-doped and N-doped phosphorene nanosheets. All phosphorene samples show the P 2p3/2 and P 2p1/2 doublet at around 129.9 and 130.8 eV, respectively, which can be assigned to the signals of P–P bonds.19 In addition, broad peaks at around 134.4 eV, corresponding to P–O bonds (PxOy),10 can be observed for all samples. However, significant differences in the intensity of these peaks associated with PxOy can be used to evaluate the degree of oxidation. As expected, only slight oxidation was observed for the non-doped phosphorene nanosheets (Fig. 2a) as no ball milling or air oxidation steps were involved in the sample preparation. Moreover, all experimental steps except the microwave-exfoliation were carried out in a N2-filled glovebox to control the oxidation. As shown in Fig. 2b, the O-phosphorene exhibited an intense PxOy peak centered at 134.4 eV, confirming the strong oxygen doping and/or functionalization of BP. Due to its tendency to oxidize in ambient air, the as-produced phosphorene nanosheets were heavily oxidized during the air oxidation and ball milling in the absence of any doping agent. Interestingly, when the doping agent (NH4OH) was used during the ball milling, the peak at higher binding energy (131.2–136.0 eV) broadened, in addition to the existence of two signals from P–P bonds (Fig. 2c). The peak can be deconvoluted into three main spin–orbit split doublets with a splitting of 0.86 eV. While the P 2p3/2 peaks centered at around 129.8 eV and 134.3 eV can be assigned to P–P and PxOy, the extra peak at ∼133 eV is attributed to the P–N bonds.21,33,42 More importantly, the N-phosphorene sample shows lower PxOy peak intensity as compared to the O-phosphorene, suggesting that some of the oxygen containing groups were replaced by nitrogen functional groups during ball milling. Ball milling is an effective method not only for functionalizing graphene nanosheets;31,32 it was also found to be a powerful strategy to form stable phosphorus–carbon (P–C) bonds in a BP and graphite composite.43 The successful N-doping was further corroborated by the XPS survey scan (Fig. S4†) and HR N 1s spectrum (Fig. 2d) of the N-phosphorene sample.
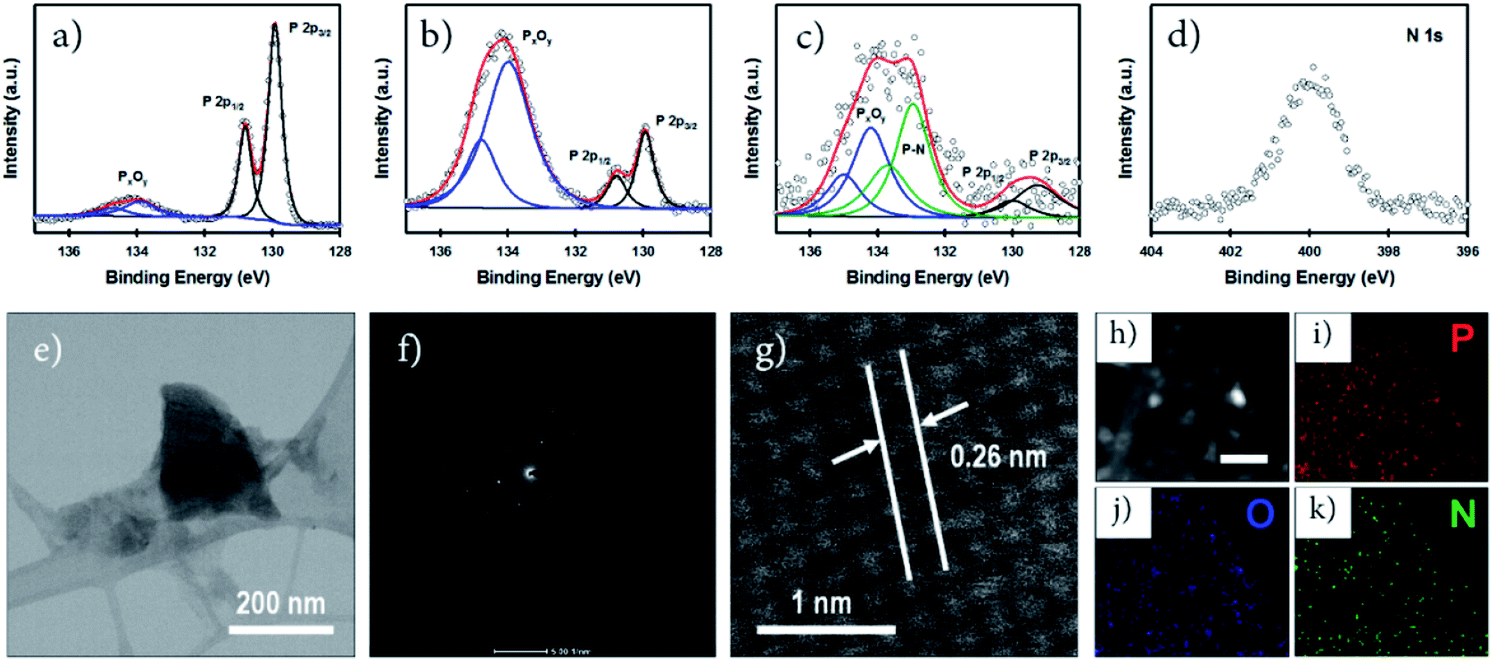 | ||
| Fig. 2 High resolution P 2p XPS spectra of (a) non-doped, (b) O-doped, and (c) N-doped phosphorene nanosheets. (d) N 1s XPS spectrum, (e) TEM image, (f) SAED pattern and (g) HRTEM image of the N-phosphorene. (h) High-angle annular dark-field-scanning transmission electron microscope (HAADF-STEM) image of an N-phosphorene nanosheet (scale bar: 50 nm), and (i–k) the corresponding EDX elemental mapping images. | ||
We further characterized our N-phosphorene sample using transmission electron microscopy (TEM). The TEM image presented in Fig. 2e shows that the flake size of our N-phosphorene nanosheets is around 200 nm, which is in agreement with the lateral sizes measured from the AFM images (Fig. S1c†) and UV-vis absorption spectrum of a small phosphorene nanosheet (Fig. S2†). As depicted in the inset of Fig. 2f, the selected area electron diffraction (SAED) pattern of our N-phosphorene confirms the high-quality single-crystal structure of BP, and is also consistent with a previous report of exfoliated BP.44 The measured lattice spacing (0.26 nm) of our N-phosphorene is in excellent agreement with previous literature (Fig. 2g).9,11 As shown in Fig. S5,† ultrasmall atomic size dots (white) can be observed in the HRTEM images of the N-phosphorene nanosheet, particularly at the edge (as expected),32 which could be due to the significant functionalization and doping. Furthermore, the successful N-doping and the presence of each element (P, O and N) in our N-phosphorene sample was confirmed by energy-dispersive X-ray spectroscopy (EDX) elemental mapping (Fig. 2h–k).
Electroreduction of N2 to NH3
The NRR electrocatalytic activity of all catalysts was evaluated in an N2-saturated 0.1 M KOH solution. Linear sweep voltammograms (LSVs) of N-phosphorene were recorded in 0.1 M KOH solution saturated with Ar or N2 (Fig. 3a). It can be seen that the measured current density in N2-saturated solution was higher than that of the Ar-saturated solution when using N-phosphorene as the cathodic catalyst, indicating the effective catalysis of the NRR using N-phosphorene. The NRR occurred at a potential of +0.05 V, which was close to the equilibrium thermodynamic potential (+0.533 V vs. RHE) for the NRR under the same conditions. The low onset potential of N-phosphorene for the NRR was similar to that of Ru@ZrO2/NC (ruthenium single atom in ZrO2/N-doped porous carbon) which showed a FE of up to 21% at −0.11 V.45 With N2 gas bubbling into the cathodic compartment, the chronoamperometry curves of electrolysis at different potentials were measured under ambient conditions using N-phosphorene, O-phosphorene and phosphorene as the catalysts (Fig. S6a–c†). | ||
| Fig. 3 (a) LSV curves of N-phosphorene in Ar- and N2-saturated 0.1 M KOH measured with a scan rate of 5 mV s−1. (b) Yield and FE of N-phosphorene, O-phosphorene and phosphorene determined using two different methods (ammonia selective electrode method and indophenol blue method). (c) NH3 yield rates and (d) FEs of the three catalysts at different potentials ranging from +0.05 V to −0.5 V determined using the ammonia selective electrode (solid line) and the indophenol blue method (dashed line), and (e) the corresponding current densities. (f) EIS spectra at an applied potential of 0 V vs. RHE of the three electrocatalysts, and their corresponding equivalent-circuit diagram. | ||
The production of NH3 was detected and quantified using two different methods, namely an ammonia selective electrode method46,47 (Fig. S7†) and the indophenol blue method48,49 (Fig. S8†). The possible by-product N2H4 was detected using the Watt and Chrisp method (Fig. S9†).48–50 As shown in Fig. 3b, N-phosphorene exhibited an excellent yield rate of 16.67 μg h−1 mgCAT−1 at 0 V determined using an ammonia selective electrode, which was more than three times and five times higher than that of O-phosphorene and phosphorene, respectively. The FE of NH3 generation catalyzed by N-phosphorene was measured to be about 19.08% at 0 V, which was significantly higher than that of the O-phosphorene (4.86%) and phosphorene (3.19%). The prominent catalytic activity of N-phosphorene could be attributed to the exposure of enhanced active sites of the nanosheets as well as the electronic structure obtained through N-doping. We also used the indophenol blue method to quantify the produced NH3. UV-vis absorption spectra of the electrolytes colored with the indophenol indicator in the dark for 3 h are plotted in Fig. S6d–f.† The results show the highest absorbance intensity when electrolyzed at 0 V vs. RHE. Although the yield rates of N-phosphorene, O-phosphorene and phosphorene determined by the indophenol blue method were slightly higher than those determined using the ammonia selective electrode method, the same trends were observed for both methods. In particular, the yield rate for our N-phosphorene measured using the indophenol blue method was 18.79 μg h−1 mgCAT−1 and its corresponding FE was 21.51% at a low overpotential (0 V) (see Fig. 3b). These values are comparable to or even higher than those of some metal catalysts reported so far. Table S1† compares the NRR performance of our N-phosphorene and the state-of-the-art electrocatalysts reported in the literature. Notably, no by-product N2H4 was detected by the Watt and Chrisp method (Fig. S10†), suggesting good selectivity of N-phosphorene for the NRR.
To confirm the origin of the NH3 produced, several control experiments were performed. No NH3 was detected in Ar-saturated electrolyte using N-phosphorene on a copper foam (CF) (Fig. S11†), suggesting that the raw material for the NH3 comes from the feed gas injected in the electrolyte. Also, no NH3 was detected in N2-saturated electrolyte with a pure CF electrode (Fig. S12†). These results indicate that NH3 was produced using only the N2 gas injected into the electrolyte catalyzed by the electrocatalysts.
To explore the differences among the three catalysts in detail, the NH3 yield rates, FEs and current densities of N-phosphorene, O-phosphorene and phosphorene at different potentials ranging from +0.05 V to −0.5 V are illustrated in Fig. 3c–e, respectively. The NH3 yield rates were measured to be around 15 μg h−1 mgCAT−1 at all potentials, but the FEs gradually reduced as the potential shifted from +0.05 to −0.5 V. This is due to the fact that the hydrogen evolution reaction (HER) process has gradually dominated the reaction as the potentials become more negative. Compared to the O-phosphorene and phosphorene, the NH3 yield rates and FEs of N-phosphorene were much higher at all applied potentials while the measured current densities were roughly similar, especially at low applied potentials (Fig. 3e), confirming the better activity of N-phosphorene for the NRR. Particularly, at potentials below −0.4 V, the current densities measured for all our electrocatalysts were very similar, but the N-phosphorene exhibited significantly higher FEs than the other two catalysts, verifying the significant role of N-phosphorene in suppressing the HER during the NRR at low overpotentials.
In order to gain better insights into the intrinsic activity for the NRR, electrochemical impedance spectroscopy (EIS) was used to measure the charge transfer resistance (Rct) of the electrodes. As shown in Fig. 3f, the Rct of N-phosphorene (24.2 Ω) is much lower than that of O-phosphorene (34.1 Ω) and phosphorene (56.3 Ω), suggesting a favorable charge transfer mechanism for the N-doped phosphorene system due to the presence of the heteroatoms.
The durability of the N-phosphorene was evaluated by performing six consecutive electrolysis cycles at a constant potential of 0 V (Fig. 4a). The NH3 yield rate and FE decreased slightly after six cycles of chronoamperometric runs, indicating the good stability of N-phosphorene during continuous NRR electrocatalysis. In addition, the long-term electrolytic test revealed that the N-phosphorene has excellent stability over time, maintaining stable current density over 15 h (Fig. 4b), which further confirmed the good stability of N-phosphorene for the NRR electrocatalysis. The inherent properties of the N-phosphorene should be explored after the catalysis as phosphorene nanosheets are known to be unstable in water and in oxygen-enriched environments.16 Our N-phosphorene nanosheets were characterized after six cycles of continuous NRR electrocatalysis to determine whether the structure and composition have changed during the NRR. The existence of P, O and N in the elemental mapping of N-phosphorene nanosheets after six cycles of continuous catalysis suggests excellent stability. As depicted in Fig. S13,† the increased signal for N in the catalytic material can be explained by the adsorption of N2 during the catalysis, which is not surprising and was expected. Furthermore, the HR P 2p XPS spectrum, shown in Fig. S14,† indicates that the surface composition of N-phosphorene barely changed during the electrolysis process.
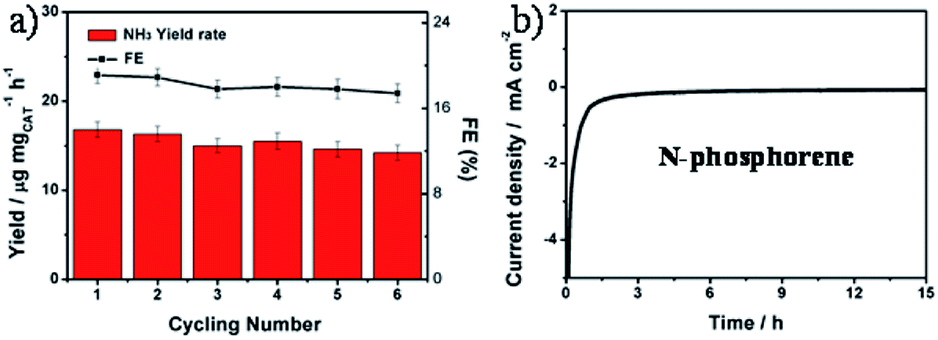 | ||
| Fig. 4 (a) Cycling stability results of N-phosphorene at 0 V vs. RHE. (b) Long-term durability test with chronoamperometry of N-phosphorene toward the electrocatalytic NRR in a N2-saturated 0.1 M KOH solution for 15 h at 0 V vs. RHE. | ||
As our catalyst contains N, it is important to consider the origin of the N in the NH3 produced. As stated earlier, when Ar was bubbled through the electrolyte instead of N2, no ammonia was observed (Fig. S11†). Additionally using the XPS survey spectra, an estimate of the amount of N present in the catalyst can be obtained. XPS is done on oxidized Si wafers so there are for example multiple contributions to the O 1s peak (catalyst and substrate). We have used very conservative assumptions for the contributions to various peaks to maximize the N content in the catalyst. Using these assumptions, the amount of N present in the catalyst is between 8 and 17 μg. The amount of N produced during the durability test (in Fig. 4a) was calculated to be 22.9 μg considering that the total mass of NH3 produced after six consecutive electrolysis cycles is 27.8 μg. Clearly all this N could not have come from the catalyst and in fact given the very small change in NH3 produced per hour over six cycles (6 hours), it is clear that the catalyst must make a very minor or even no contribution to the production of NH3.
Theoretical investigation
To understand the electrocatalytic results found in the experiments, DFT calculations were performed to study (i) the thermodynamic positions of N and O in a phosphorene structure, (ii) the hydrophobicity of phosphorene surfaces, and (iii) the free energy pathways of the NRR at different possible sites. Firstly, the thermodynamic doping positions of N and O were evaluated by using a multilayer phosphorene model, with the N/O doped at the upper site of the first layer as the energy reference (Fig. 5). It was found that both N and O tend to stay at the topmost surface of phosphorene. The formation energies of the N- and O-doped phosphorene were also calculated (Fig. S16†). The formation of the O-doped structure has negative energy, while the formation of the N-doped structure is endothermic. This helps to explain why there is a high amount of O in the N-phosphorene as found from our XPS (Fig. 2c) and EDX (Fig. 2h–k) results. Nevertheless, due to the kinetically controlled synthetic method and the relatively abundant N and O after synthesis, we expect that the N and O atoms are doped in all of the layers, while the topmost layer tends to possess higher ratios of N and O, as compared to other layers.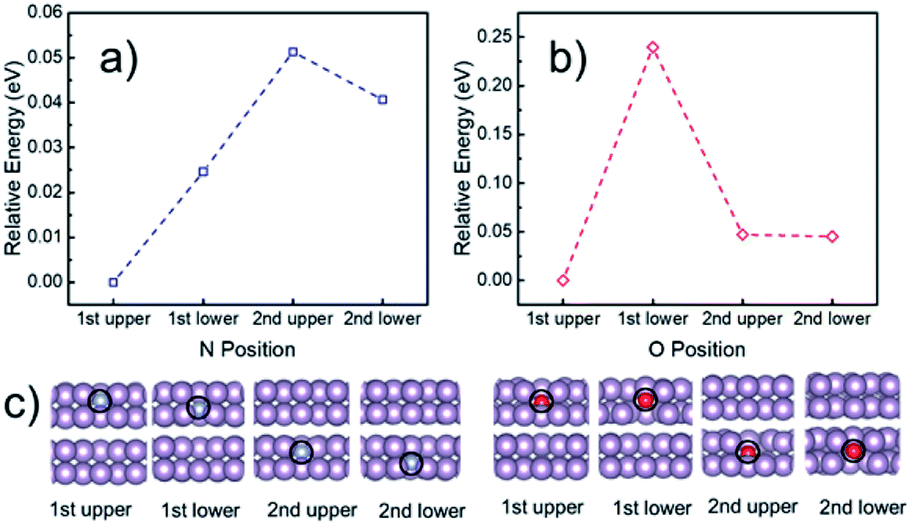 | ||
| Fig. 5 Calculated relative energy of the phosphorene structure doped with (a) N and (b) O at different positions, with the optimized structures of the first two layers shown in (c). The N and O doped at the upper layer of the topmost (i.e., first) phosphorene surface were used as the energy references for (a) and (b), respectively. The complete optimized structures can be found in Fig. S15.† Purple, blue, and red spheres represent P, N, and O, respectively. | ||
Next, DFT calculations were performed to evaluate the surface hydrophobicity by calculating the adsorption free energy of water at different sites (Fig. 6a), with the reason that water adsorption and dissociation are the most important steps that determine the HER activity in an alkaline solution.51 The pure, doped, and defected surfaces of a layered phosphorene were considered for the calculations, based on our experimental characterization (Fig. 2) and the DFT results (Fig. 5). Since the catalytic reactions are usually site-specific,52 we evaluated six typical sites at the surface of the phosphorene structure: P, P–N (P-site), P–N (N-site), P–O (P-site), P–O (O-site), and P-vacancy (Fig. 6a and b) for water and the NRR-related adsorbates. Interestingly, it is found that although a pure phosphorene surface is relatively hydrophobic already (which is slightly exothermic), the doping of N or O can further increase the hydrophobicity by having an endothermic water adsorption free energy at all P, N, and O sites. All the optimized adsorption geometries indicate that unlike those metallic surfaces,53 water does not form a stabilized binding geometry at all these evaluated phosphorene surfaces. This hydrophobic nature was also discovered in other p-block elements such as sulfur, by combined experimental and DFT methods.54,55 The hydrophobicity found at all the doped phosphorene surfaces indicates that the HER process is relatively hindered at these surfaces, which in turn improves the FEs of the NRR. We expect that there is a similar chance for the protonation of the surface or N2 under various hydrophobicities. However, similar to previous studies on electrocatalytic oxygen reduction,56 it has been broadly discussed that surface hydrophobicity significantly influences the prefactor of kinetics of the reactants in electrochemistry. Therefore, a surface with higher hydrophobicity is expected to enlarge the prefactor of the kinetics of N2 adsorption and protonation, making it easier for N2 and protonated N2 molecules to penetrate the water layer which leads to a more facile NRR process.
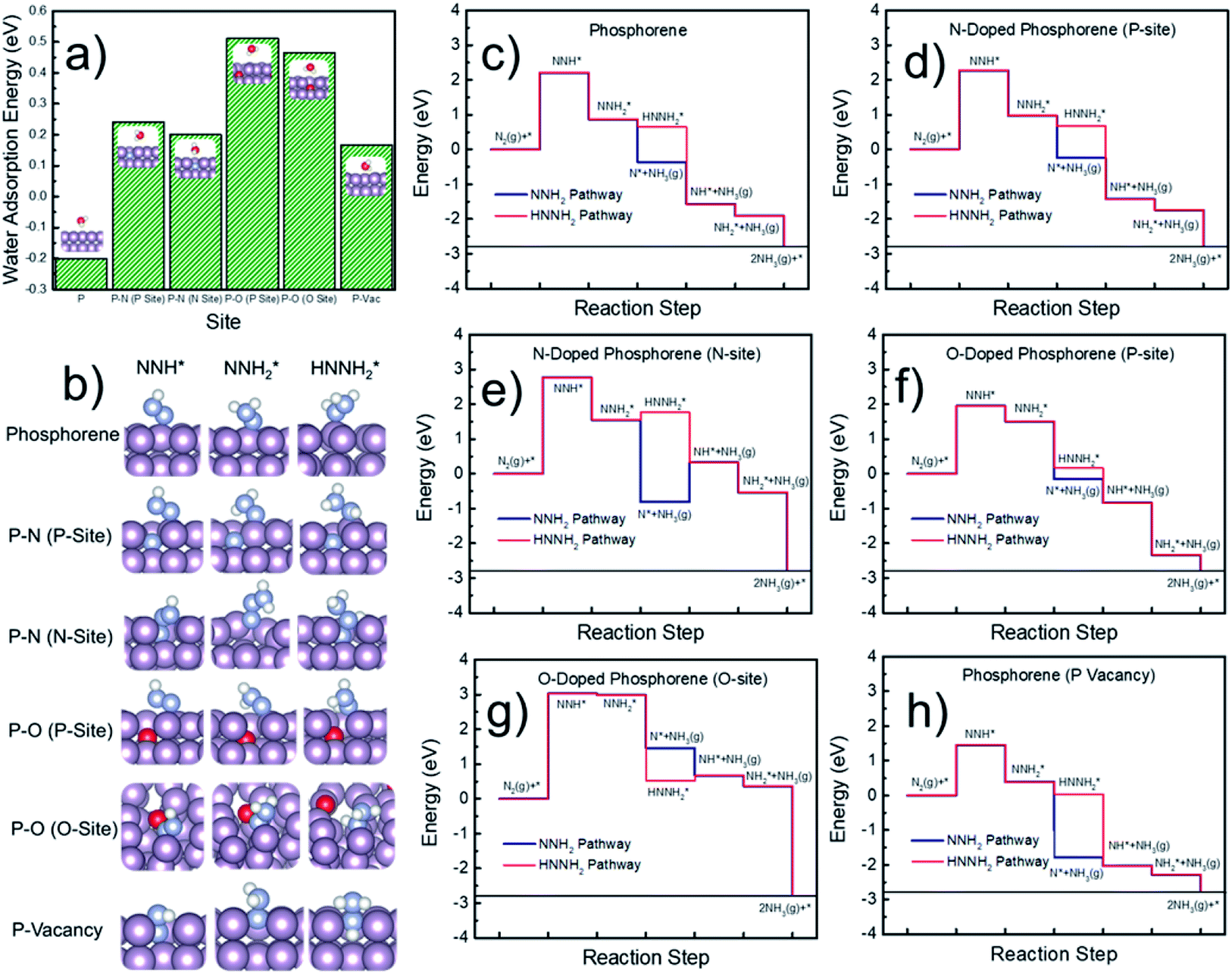 | ||
Fig. 6 (a) Calculated adsorption free energy of water at six typical sites on the phosphorene surfaces. Insets show the optimized water adsorption geometries. (b) Optimized structures of NNH*,  , and , and 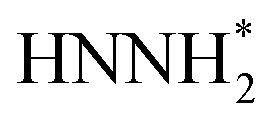 species adsorbed at the six typical sites. (c–h) Calculated free energy pathways of the NRR at six typical sites on the phosphorene surfaces. Purple, blue, white, and red spheres represent P, N, H, and O, respectively. species adsorbed at the six typical sites. (c–h) Calculated free energy pathways of the NRR at six typical sites on the phosphorene surfaces. Purple, blue, white, and red spheres represent P, N, H, and O, respectively. | ||
Finally, the free energy pathways of the NRR were calculated at those six studied sites using a computational hydrogen electrode method (Fig. 6c–h).57 Because the direct activation of N–N bonds at the protonated N2-species was found to be difficult on the phosphorene surfaces and the surfaces do not adsorb hydrazine (H2NNH2), only two mechanisms were found to be available at the phosphorene surfaces: the pathways, respectively, through the dissociation of NNH2 and HNNH2. For each pathway, the formation of NH3 gas is considered as the following elementary step after the formation of NNH2 or HNNH2, because the surface does not have a stabilized NH3 adsorption geometry. It can be seen that the potential-determining step (PDS) is the direct formation of NNH* at all the reactive sites. Except for the O-site, all the other sites show more favorable energetics for the NNH2 pathway. The P-vacancy site shows the lowest PDS (Fig. 6h). However, due to the high formation energy of a P-vacancy (1.03 eV), it is expected that this site is not abundant in the system. The P-site at the O-doped phosphorene surface shows the second lowest PDS, suggesting that the electronic effects from the oxygen nearby could improve the NRR activity. Although the P-site in the O-doped phosphorene model has lower potential-determining free energy (Fig. 6f), the O-site in this system has a very high free energy for the formation of NNH* (Fig. 6g), which is significantly higher than those of all other evaluated sites. Therefore, we conclude that doping with O makes P-sites more active for the NRR, and it also dilutes the ratio of active sites due to the relatively inert O-sites. In contrast, doping with N does not lead to a significant up-shift of the reaction free energy as compared to that of the pure phosphorene, and meanwhile, its improved hydrophobicity leads to a more facile NRR process due to the expected larger prefactor in NRR kinetics (Fig. 6a).
Although the formation of NNH* has relatively high energy, the energetics of this step is electronically tunable and expected to be significantly lowered under applied potentials. It should be noted that these are only the thermodynamic trends for the site-specific NRR activity; we believe that a more complicated model with an explicit solvation model can help to precisely evaluate the free energy of the PDS which can be directly comparable to the experimental overpotentials. However, due to the tremendous computational cost, we only evaluated the trends in the NRR activity in this study. Meanwhile, since P, N, and C are typical inert p-block elements, we expect that the reaction energy of the PDS is intrinsically higher than that of other transition metal catalysts for the NRR, due to the lack of active electronic structures. Therefore, the hydrophobicity may be the key factor that leads to the relatively high FE in our experimental observations. It can be observed from Fig. 6a that the calculated P–O structures have higher hydrophobicity than the P–N structures. We note that according to our XPS (Fig. 2c) and EDX results (Fig. 2h–k), our N-doped phosphorene has an almost equal amount of P–O and P–N in the system. Taking this into account, our N-phosphorene system should also have strong hydrophobicity due to the co-existence of both O and N. With the significant hydrophobic properties after doping with N and O (Fig. 6a), the NRR is predicted to show a significant FE, in qualitative agreement with our experimental studies.
Conclusions
We have developed a facile and efficient strategy for preparing N-doped phosphorene nanosheets using oxidized BP to our advantage, providing abundant reactive sites for the doping. The newly designed N-phosphorene shows an impressive catalytic activity and stability for the NRR process, exhibiting an excellent NH3 yield rate (18.79 μg h−1 mgCAT−1) and FE (21.51%) at a low potential under ambient conditions. DFT calculations indicate that the thermodynamically favorable doping of N and O at the phosphorene surface leads to a significant increase of hydrophobicity, which improves NRR efficiency. We believe that the development of heteroatom doped phosphorene opens a promising avenue for metal-free catalyst synthesis, which deserves further research in terms of energy conversion.Author contributions
T. M., M. B., Y. C., and J. G. S. conceived and designed the research work. G. X. and H. L. conducted the major experimental work (catalysis) and theoretical calculations, respectively. A. S. R. B., M. B. E., M. J. N., and D. L. made significant contributions in materials preparation, characterization and data analysis. Y. C. and T. M. provided all facilities for catalysis experiments, while J. G. S. and M. B. provided the necessary instruments and facilities for materials synthesis, functionalization and characterization. Y. C., J. G. S., M. B. and T. M. supervised all the research phases. G. X. and M. B. wrote the first draft of the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the Australian Research Council (ARC) through the Discovery Early Career Researcher Award (DE150101306), Linkage Project (LP160100927), Discovery project (DP160101301) and ARC Research Hub for Graphene Enabled Industry Transformation funding (IH150100003), Faculty of Science Strategic Investment Funding 2019 of University of Newcastle, CSIRO Energy, Liaoning Revitalization Talents Program – Pan Deng Scholars (XLYC1802005), Liaoning BaiQianWan Talents Program, the National Science Fund of Liaoning Province for Excellent Young Scholars, Science and Technology Innovative Talents Support Program of Shenyang (RC180166), Fundamental Research Funds for the Central Universities (GK201901002 and 2017TS049), the 111 Project (B14041), and China Scholarship Council. We acknowledge the use of the Queensland node of the Australian National Fabrication Facility (ANFF-Q) and Centre for Microscopy and Microanalysis (CMM) at the University of Queensland. The authors thank Dr Ashley D. Slattery of Adelaide Microscopy at the University of Adelaide for his help with Titan TEM analysis.Notes and references
- M. Batmunkh, M. Bat-Erdene and J. G. Shapter, Adv. Mater., 2016, 28, 8586–8617 CrossRef CAS PubMed.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372 CrossRef CAS PubMed.
- M. Buscema, D. J. Groenendijk, S. I. Blanter, G. A. Steele, H. S. J. van der Zant and A. Castellanos-Gomez, Nano Lett., 2014, 14, 3347–3352 CrossRef CAS PubMed.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980 CrossRef CAS PubMed.
- M. Batmunkh, M. Bat-Erdene and J. G. Shapter, Adv. Energy Mater., 2018, 8, 1701832 CrossRef.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 2001 CrossRef PubMed.
- M. Batmunkh, A. Shrestha, M. Bat-Erdene, M. J. Nine, C. J. Shearer, C. T. Gibson, A. D. Slattery, S. A. Tawfik, M. J. Ford, S. Dai, S. Qiao and J. G. Shapter, Angew. Chem., Int. Ed., 2018, 57, 2644–2647 CrossRef CAS PubMed.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- X. Zhu, T. Zhang, Z. Sun, H. Chen, J. Guan, X. Chen, H. Ji, P. Du and S. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, R. Hua, Q. Lei and Y. Tian, Nat. Commun., 2018, 9, 1397 CrossRef PubMed.
- M. Batmunkh, M. Myekhlai, A. S. R. Bati, S. Sahlos, A. D. Slattery, T. M. Benedetti, V. R. Gonçales, C. T. Gibson, J. J. Gooding, R. D. Tilley and J. G. Shapter, J. Mater. Chem. A, 2019, 7, 12974–12978 RSC.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS PubMed.
- X. Ren, J. Zhou, X. Qi, Y. Liu, Z. Huang, Z. Li, Y. Ge, S. C. Dhanabalan, J. S. Ponraj, S. Wang, J. Zhong and H. Zhang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- L. Zhang, L.-X. Ding, G.-F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-L'Heureux, N. Y. W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826 CrossRef CAS PubMed.
- M. Bat-Erdene, M. Batmunkh, S. A. Tawfik, M. Fronzi, M. J. Ford, C. J. Shearer, L. Yu, M. Dadkhah, J. R. Gascooke, C. T. Gibson and J. G. Shapter, Adv. Funct. Mater., 2017, 27, 1704488 CrossRef.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597 CrossRef CAS PubMed.
- C. Han, Z. Hu, L. C. Gomes, Y. Bao, A. Carvalho, S. J. R. Tan, B. Lei, D. Xiang, J. Wu, D. Qi, L. Wang, F. Huo, W. Huang, K. P. Loh and W. Chen, Nano Lett., 2017, 17, 4122–4129 CrossRef CAS PubMed.
- Y. Liu, P. Gao, T. Zhang, X. Zhu, M. Zhang, M. Chen, P. Du, G.-W. Wang, H. Ji, J. Yang and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 1479–1483 CrossRef CAS PubMed.
- A. Hashmi and J. Hong, J. Phys. Chem. C, 2015, 119, 9198–9204 CrossRef CAS.
- N. Yang, L. Li, J. Li and Z. Wei, Phys. Chem. Chem. Phys., 2019, 21, 4899–4906 RSC.
- X. Xuan, Z. Zhang and W. Guo, Nanoscale, 2018, 10, 7898–7904 RSC.
- B. Liu, M. Köpf, A. N. Abbas, X. Wang, Q. Guo, Y. Jia, F. Xia, R. Weihrich, F. Bachhuber, F. Pielnhofer, H. Wang, R. Dhall, S. B. Cronin, M. Ge, X. Fang, T. Nilges and C. Zhou, Adv. Mater., 2015, 27, 4423–4429 CrossRef CAS PubMed.
- Y. Xu, J. Yuan, L. Fei, X. Wang, Q. Bao, Y. Wang, K. Zhang and Y. Zhang, Small, 2016, 12, 5000–5007 CrossRef CAS PubMed.
- B. Yang, B. Wan, Q. Zhou, Y. Wang, W. Hu, W. Lv, Q. Chen, Z. Zeng, F. Wen, J. Xiang, S. Yuan, J. Wang, B. Zhang, W. Wang, J. Zhang, B. Xu, Z. Zhao, Y. Tian and Z. Liu, Adv. Mater., 2016, 28, 9408–9415 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- M. Batmunkh, C. J. Shearer, M. J. Biggs and J. G. Shapter, J. Mater. Chem. A, 2016, 4, 2605–2616 RSC.
- I.-Y. Jeon, H.-J. Choi, S.-M. Jung, J.-M. Seo, M.-J. Kim, L. Dai and J.-B. Baek, J. Am. Chem. Soc., 2013, 135, 1386–1393 CrossRef CAS PubMed.
- J. Xu, I.-Y. Jeon, J.-M. Seo, S. Dou, L. Dai and J.-B. Baek, Adv. Mater., 2014, 26, 7317–7323 CrossRef CAS PubMed.
- L. Shao, H. Sun, L. Miao, X. Chen, M. Han, J. Sun, S. Liu, L. Li, F. Cheng and J. Chen, J. Mater. Chem. A, 2018, 6, 2494–2499 RSC.
- M. Bat-Erdene, M. Batmunkh, C. J. Shearer, S. A. Tawfik, M. J. Ford, L. Yu, A. J. Sibley, A. D. Slattery, J. S. Quinton, C. T. Gibson and J. G. Shapter, Small Methods, 2017, 1, 1700260 CrossRef.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- M. Batmunkh, K. Vimalanathan, C. Wu, A. S. R. Bati, L. Yu, S. A. Tawfik, M. J. Ford, T. J. Macdonald, C. L. Raston, S. Priya, C. T. Gibson and J. G. Shapter, Small Methods, 2019, 3, 1800521 CrossRef.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- W. Lu, H. Nan, J. Hong, Y. Chen, C. Zhu, Z. Liang, X. Ma, Z. Ni, C. Jin and Z. Zhang, Nano Res., 2014, 7, 853–859 CrossRef CAS.
- B. Tian, B. Tian, B. Smith, M. C. Scott, Q. Lei, R. Hua, Y. Tian and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4345–4350 CrossRef CAS PubMed.
- B. Sa, Y.-L. Li, J. Qi, R. Ahuja and Z. Sun, J. Phys. Chem. C, 2014, 118, 26560–26568 CrossRef CAS.
- T. Ahmed, S. Balendhran, M. N. Karim, E. L. H. Mayes, M. R. Field, R. Ramanathan, M. Singh, V. Bansal, S. Sriram, M. Bhaskaran and S. Walia, npj 2D Mater. Appl., 2017, 1, 18 CrossRef.
- J. Wu, X. Zheng, C. Jin, J. Tian and R. Yang, Carbon, 2015, 92, 327–338 CrossRef CAS.
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- J. Kang, J. D. Wood, S. A. Wells, J.-H. Lee, X. Liu, K.-S. Chen and M. C. Hersam, ACS Nano, 2015, 9, 3596–3604 CrossRef CAS PubMed.
- H. Tao, C. Choi, L.-X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang, A. W. Robertson, S. Hong, Y. Jung, S. Liu and Z. Sun, Chem, 2019, 5, 204–214 CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- G.-R. Xu, M. Batmunkh, S. Donne, H. Jin, J.-X. Jiang, Y. Chen and T. Ma, J. Mater. Chem. A, 2019, 7, 25433–25440 RSC.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- H.-M. Liu, S.-H. Han, Y. Zhao, Y.-Y. Zhu, X.-L. Tian, J.-H. Zeng, J.-X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- M. Bat-Erdene, G. Xu, M. Batmunkh, A. S. R. Bati, J. J. White, M. J. Nine, D. Losic, Y. Chen, Y. Wang, T. Ma and J. G. Shapter, J. Mater. Chem. A, 2020, 8, 4735–4739 RSC.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
- B. Yu, H. Li, J. White, S. Donne, J. Yi, S. Xi, Y. Fu, G. Henkelman, H. Yu, Z. Chen and T. Ma, Adv. Funct. Mater., 2020, 30, 1905665 CrossRef CAS.
- J. Xu, A. Avellan, H. Li, X. Liu, V. Noël, Z. Lou, Y. Wang, R. Kaegi, G. Henkelman and G. V. Lowry, Adv. Mater., 2020, 32, 1906910 CrossRef CAS PubMed.
- Z. Cao, H. Li, X. Xu and J. Xu, Chem. Eng. J., 2020, 394, 124876 CrossRef CAS.
- V. Tripkovic and T. Vegge, J. Phys. Chem. C, 2017, 121, 26785–26793 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta03237a |
| ‡ These two authors made an equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2020 |