
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen evolution from water using heteroatom substituted fluorene conjugated co-polymers†
Yang
Bai
 a,
Duncan J.
Woods
a,
Liam
Wilbraham
b,
Catherine M.
Aitchison
a,
Martijn A.
Zwijnenburg
*b,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*a
a,
Duncan J.
Woods
a,
Liam
Wilbraham
b,
Catherine M.
Aitchison
a,
Martijn A.
Zwijnenburg
*b,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*a
aDepartment of Chemistry and Material Innovation Factory, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: ssprick@liverpool.ac.uk; aicooper@liverpool.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
First published on 31st March 2020
Abstract
The photocatalytic performance of fluorene-type polymer photocatalysts for hydrogen production from water in the presence of a sacrificial hole scavenger is significantly improved by the incorporation of heteroatoms into the bridge-head. This improvement can be explained by a combination of factors, including changes in thermodynamic driving-force, particle size, dispersibility under photocatalytic conditions, and light absorption, all of which vary as a function of the heteroatom incorporated.
Introduction
The photocatalytic generation of hydrogen from water by water splitting using sunlight is an important area of research. Most photocatalysts studied are inorganic1–4 and the only organic material that has been studied very extensively is carbon nitride.5–8 Other π-conjugated organic materials, such as conjugated linear polymers,9–16 conjugated microporous polymers,17–21 covalent organic frameworks,22–26 and covalent triazine-based frameworks,27–30 have also begun to attract attention as organic photocatalysts due to their accessibility via low temperature routes9,31 and their synthetic tunability.17 It has been found that the incorporation of different but closely related monomer building blocks, such as dibenzo[b,d]thiophene and dibenzo[b,d]thiophene sulfone, can affect the photocatalytic performance dramatically.32,33 With this in mind, we considered the subtle change of the bridging atom in 9H-fluorene-based monomers to heteroatoms other than sulfur. Previously, the incorporation of different heteroatoms into the bridge-head of fluorene co-polymers was shown to affect the charge-transport in amorphous poly(triarylamine)s,34 stacking distances in the solid-state,35 and refractive indices.36Here, we explore the effect incorporating tetrels (silicon, germanium), pnictogens (nitrogen, phosphorus) and chalocogens (oxygen, sulfur) of periods 2 to 4 into the 9H-fluorene bridge-head position as alternative bridging atoms. In total, 25 new conjugated polymers were prepared with these heterofluorenes as co-polymers with phenylene, dibenzo[b,d]thiophene sulfone, and spirobifluorene. All materials were tested as photocatalysts for hydrogen production from water in the presence of a sacrificial reagent and we found that this subtle change results in significant modulation of the catalytic activity for the materials.
Experimental
All polymers were synthesized using Pd(0)-catalyzed Suzuki–Miyaura polycondensation of diboronic acid ester-functionalized monomers with bromo-functionalized co-monomers (see ESI† for synthetic details).9,17 The phenylene co-polymers9 (p-XPh, Fig. 1), dibenzo[b,d]thiophene sulfone co-polymers11 (p-XS) and spirobifluorene conjugated microporous polymers21 (X-CMP3) were all insoluble in common organic solvents and in water. Chloroform-soluble polymers37 (sP-XS) were obtained by incorporating alkyl side-chains on the bridge-head atom. The materials were characterized by UV-vis spectroscopy (Fig. S24–S34†) and elementary analysis. Thermogravimetric analysis (TGA) indicated that all polymers were thermally stable in air up to 300 °C (Fig. S88–S90†), and PXRD patterns showed that all linear polymers were semi-crystalline, except for p-POPh and X-CMP3 materials, which were amorphous (Fig. S45–S47†). Solid-state CP/MAS 13C NMR spectroscopy was used to confirm the structures of a subset of the library (p-FS, p-TS, p-FuS and p-CzS, Fig. S20–S23†). As expected all materials show multiple broad resonances between ca. 160–100 ppm that can be assigned to aromatic carbon atoms. Furthermore, p-FS shows expected additional resonances at 47 and 26 ppm that can be ascribed to carbon atoms on the bridge-head position and methyl-groups, respectively.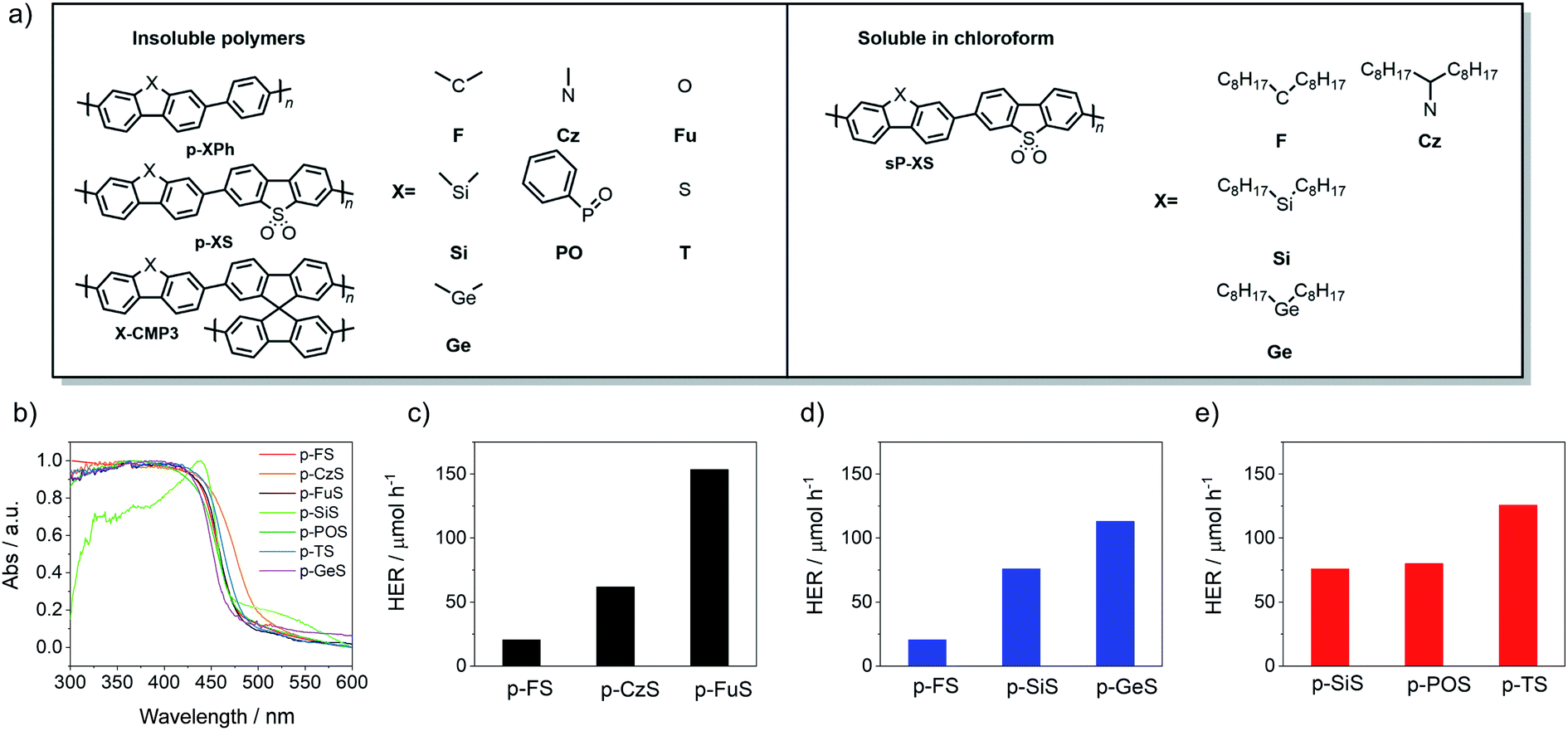 | ||
| Fig. 1 (a) Structures of the polymer photocatalysts in this study; (b) UV/vis spectra of the 7 dibenzo[b,d]thiophene sulfone-containing photocatalysts, as measured in the solid state; hydrogen evolution rates of (c) p-FS, p-CzS and p-FuS; (d) p-FS, p-SiS and p-GeS; (e) p-SiS, p-POS and p-TS under visible-light irradiation (λ > 420 nm, 300 W Xe light source). | ||
The potentials of the charge carriers in the different polymers were estimated using our standard approach38,39 based on ΔDFT calculation using the B3LYP40,41 density functional on a single oligomer chain of composition (AB)4 immersed in a dielectric continuum with εr 80.1 (water). Similarly, the optical gap values of the different oligomers were approximated by the energy of the lowest vertical singlet-to-singlet excitation as calculated using TD-B3LYP.
Results and discussion
All of the materials in this series (Fig. 1) acted as photocatalysts for hydrogen evolution from water/triethylamine/methanol mixtures. Triethylamine (TEA) was used as the hole-scavenger,9,14 while methanol was added to aid mixing of the TEA with water.9,17 The measured hydrogen evolution rates (HERs) under visible light (λ > 420 nm, 300 W Xe light source) ranged from 3.2 μmol h−1 to 147.1 μmol h−1, with photocatalyst p-FuS being found to be the most active of the polymers in this study (Table 1). The hydrogen evolution rate for p-FuS is approximately 17% higher than our previously reported dibenzo[b,d]thiophene sulfone co-polymer p-TS (P1-64) under the same conditions (125.7 μmol h−1) (Table 1).32,33 Similarly, the phenylene co-polymer p-POPh (65.9 μmol h−1) is around twice as active as P7 (37.3 μmol h−1) (Table 1), which was among the most photocatalytically-active phenylene co-polymers reported previously.11,42 Comparing the different families of polymers reported here, the p-XS family is the most active, followed by the p-XPh family and the X-CMP3 networks, while the sp-XS family of soluble co-polymers are the least active, possibly due to poorer wetting through the incorporation of the solubilizing alkyl side-chains (Fig. S126†).37| Photocatalyst | Optical gapa [eV] | τ avg [ns] | Particle sizec [μm] | SABETd [m2 g−1] | Transmissione [%] | HERfλ > 420 nm [μmol h−1] | HERfλ > 295 nm [μmol h−1] | |
|---|---|---|---|---|---|---|---|---|
a Optical gap calculated from the absorption on-set.
b Estimated weighted average life-time of the excited state determined by time-correlated single-photon counting.
c Surface area mean diameter (Sauter mean diameter).
d Apparent BET surface area calculated from the N2 adsorption isotherm.
e Average transmittance of a polymer suspension in water/methanol/trimethylamine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1).
f Reaction conditions: 25 mg polymer was suspended in 25 mL water/methanol/triethylamine (1:1:1) solution, irradiated by 300 W Xe light source using suitable filters.
g Not determined. :1:1).
f Reaction conditions: 25 mg polymer was suspended in 25 mL water/methanol/triethylamine (1:1:1) solution, irradiated by 300 W Xe light source using suitable filters.
g Not determined.
|
||||||||
| Phenylene co-polymers | p-FPh | 2.79 | 2.50 | 2.94 | 4 | 22.5 | 3.4 (±0.1) | 17.7 (±0.1) |
| p-CzPh | 2.72 | 0.54 | 2.53 | 14 | 76.3 | 3.2 (±0.1) | 14.2 (±0.2) | |
| p-FuPh | 2.77 | 0.18 | 2.75 | 35 | 2.0 | 48.1 (±0.6) | 81.5 (±4.6) | |
| p-SiPh | 2.89 | 0.39 | 7.76 | 55 | 0.7 | 5.0 (±1.2) | 38.6 (±3.5) | |
| p-POPh | 2.74 | 0.54 | 2.82 | 27 | 58.7 | 65.9 (±2.5) | 91.0 (±6.9) | |
| p-TPh | 2.79 | 0.26 | 2.1 | 29 | 2.3 | 10.8 (±0.1) | 41.5 (±0.3) | |
| p-GePh | 2.85 | 0.19 | 2.69 | 6 | 48.6 | 13.6 (±2.9) | 36.2 (±2.5) | |
| Dibenzo[b,d]thiophene sulfone polymers | p-FS | 2.59 | 0.46 | 8.28 | 114 | 0.2 | 20.7 (±1.0) | 38.6 (±1.9) |
| p-CzS | 2.44 | 4.51 | 3.46 | 3 | 34.8 | 61.9 (±1.1) | 73.9 (±4.0) | |
| p-FuS | 2.57 | 2.54 | 0.87 | 67 | 0.4 | 147.1 (±6.3) | 209.3 (±12.5) | |
| p-SiS | 2.66 | 1.07 | 3.53 | 5 | 54.1 | 72.3 (±5.6) | 85.8 (±3.9) | |
| p-POS | 2.58 | 0.52 | 1.76 | 9 | 42.3 | 80.2 (±3.6) | 81.1 (±8.9) | |
| p-TS | 2.56 | 2.26 | 2.63 | 127 | 5.1 | 125.7 (±5.3) | 256.9 (±14.4) | |
| p-GeS | 2.63 | 0.60 | 2.3 | 30 | 0.3 | 113.0 (±3.2) | 248.7 (±14) | |
| Spirobifluorene polymers | F-CMP3 | 2.77 | 0.35 | 6.31 | 596 | 0.4 | 13.5 (±0.8) | 63.6 (±0.5) |
| Cz-CMP3 | 2.72 | 0.17 | 4.81 | 422 | 4.0 | 11.0 (±0.8) | 17.6 (±0.6) | |
| Fu-CMP3 | 2.80 | 0.22 | 10.7 | 513 | 3.6 | 15.0 (±0.6) | 46.9 (±3.1) | |
| Si-CMP3 | 2.81 | 0.47 | 5.66 | 566 | 74.1 | 3.0 (±0.2) | 28.0 (±1.5) | |
| PO-CMP3 | 2.64 | 0.28 | 3.4 | 153 | 0.3 | 60.3 (±2.4) | 65.2 (±2.8) | |
| T-CMP3 | 2.78 | 0.22 | 5.22 | 508 | 13.9 | 12.2 (±0.6) | 41.6 (±3.1) | |
| Ge-CMP3 | 2.70 | 0.36 | 8.81 | 653 | 5.4 | 4.7 (±0.4) | 15.3 (±0.5) | |
| Soluble polymers | sP-FS | 2.80 | 0.48 | 9.86 | —g | 75.3 | 17.1 (±0.9) | n.d.g |
| sP-CzS | 2.76 | 0.21 | 3.05 | —g | 34.6 | 16.2 (±2.5) | n.d.g | |
| sP-SiS | 2.79 | 0.75 | 24.3 | —g | 54.1 | 48.4 (±8.2) | n.d.g | |
| sP-GeS | 2.90 | 0.60 | 179 | —g | 79.1 | 0.02 (±0.0) | n.d.g | |
We next tried to correlate the observed hydrogen evolution rates with a range of different measured and predicted polymer properties: we focused in this comparison on co-polymers in the p-XPh and p-XS families because they allow for the most like-for-like comparison. In previous work,30,32 we found that the key properties to consider are the optical gap, the wavelength below which the polymer starts absorbing light (Fig. 1b),17 the thermodynamic potentials of charge-carriers in the polymer (Fig. 2),12 and the dispersibility of the polymer in the reaction medium.11 We therefore started by considering the same factors here, all of which were observed to vary with the heteroatom and/or co-monomer chosen and hence potentially to be tunable in this way.
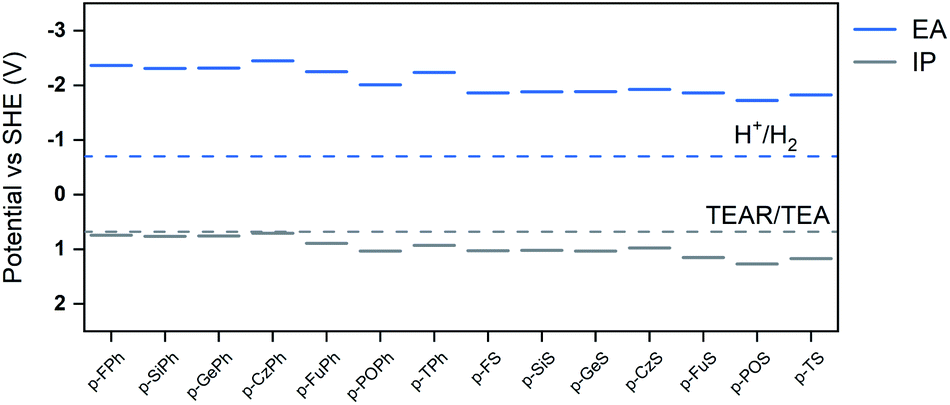 | ||
| Fig. 2 Ionization potentials and electron affinities of the different photocatalysts as predicted using DFT (B3LYP), as well as the potentials of the different solution reactions at pH 11.5, the likely pH of a TEA solution (TEAR is the deprotonated TEA radical N(Et)2CHCH3). The potential for the overall oxidation of TEA to diethylamine (DEA) and acetaldehyde (AcO) is not shown because it lies at a similar value to the H+/H2 potential; the potential for the oxidation of TEAR to DEA and AcO is not shown since it is more negative than −3 V. | ||
All polymers studied can absorb visible light because they all have experimental optical gaps that are smaller than 2.95 eV (λ > 420 nm). There is a moderate variation of ∼0.2 eV in the optical gap within the different families of polymers driven directly by heteroatom substitution. The variation in the combined set of p-XPh and p-XS families is larger due to the combined effect of heteroatom substitution and different co-monomers. Most notably, the optical gap of the p-XS polymers is red-shifted relative to the p-XPh polymers, probably because of the more planar structure of the former.9 The polymers with the highest hydrogen evolution rates are those with the smallest experimental optical gaps (Fig. 3a) and the envelope that encloses all the points shows that the hydrogen evolution rate clearly increases with decreasing optical gap. Fig. S131,† which plots the hydrogen evolution rate versus the TD-DFT predicted optical gap, shows exactly the same trend.
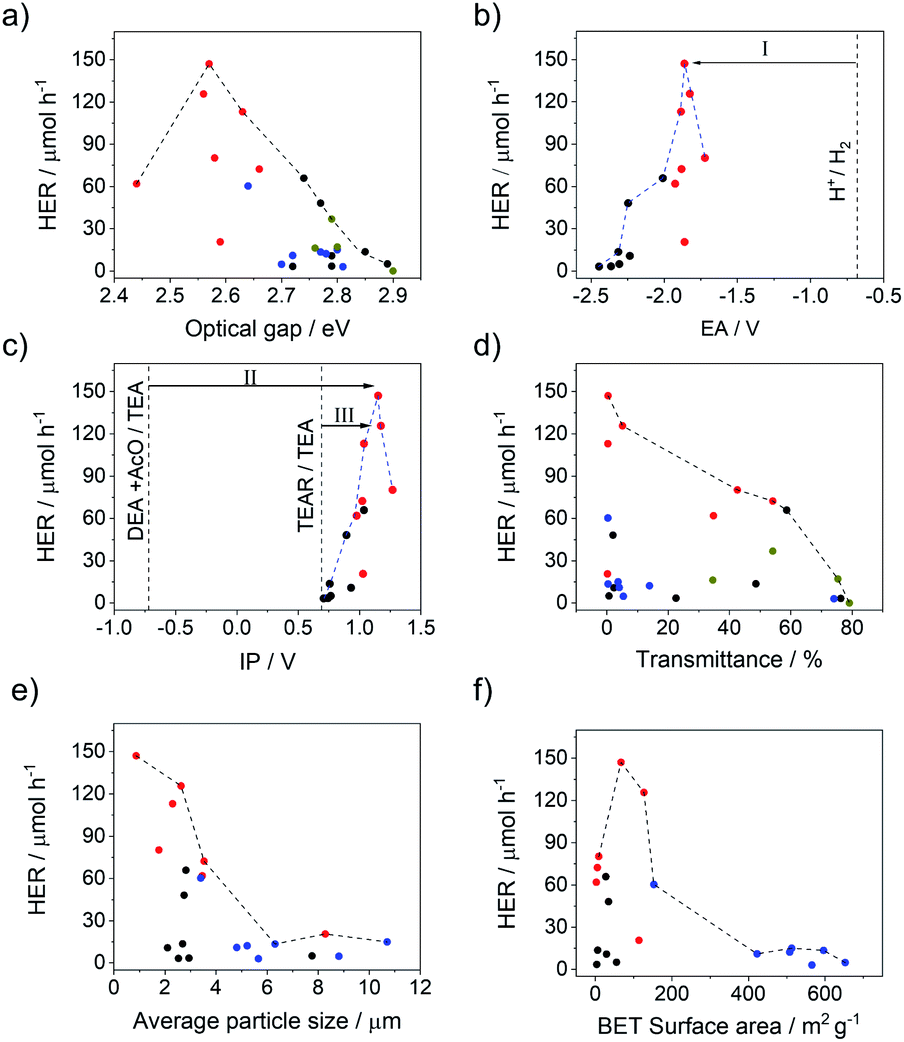 | ||
| Fig. 3 Photocatalytic hydrogen evolution rates (HER) of the phenylene co-polymers (black), dibenzo[b,d]thiophene sulfone co-polymers (red), conjugated microporous polymers (blue), and soluble polymers (dark yellow) in TEA/MeOH/H2O mixture under a visible light plotted versus (a) optical gaps of photocatalysts (see Fig. S131† for the equivalent plot using predicted rather than measured values); (b) predicted EA of photocatalysts, with driving force for proton reduction shown as arrow I; (c) predicted IP of photocatalysts, with driving force for the overall oxidation of TEA shown as arrow II and driving force for the one-hole oxidation of TEA shown as arrow III; (d) light transmission of the photocatalysts dispersed in TEA/MeOH/H2O; (e) particle size of the photocatalyst in TEA/MeOH/H2O; (f) BET surface area. The dotted lines on graphs (a) to (f) are envelopes that trace the maximum HER observed across each property range. | ||
All polymers were estimated to have a very negative electron affinity (EA) and a significantly positive ionization potential (IP). As results the polymers should have ample driving force for proton reduction (Fig. 3b), significant driving force for the overall TEA oxidation to diethylamine and acetaldehyde and a negligible to moderate driving force for the first one-hole oxidation step of TEA (Fig. 3c). Just as the case of the optical gap, the choice of heteroatom and co-monomer have a clear effect on the IP and EA values but the largest variation in IP and EA is obtained when both are varied.
As can be seen from Fig. 3c, when considering the envelope that encloses all polymers, the hydrogen evolution rate increases, in general, when the polymer's predicted IP becomes more positive and the driving force for the one-hole and overall oxidation of hence TEA increases. By contrast, as can be seen from Fig. 3b, the hydrogen evolution rate decreases when the polymer's predicted EA becomes more negative and the driving force for proton reduction increases. This, while apparently counter-intuitive, suggests that the rate of TEA oxidation and hence the driving force for TEA oxidation controls the hydrogen evolution rate of the polymers. The apparent trend in hydrogen evolution with EA is then simply due to the fact that the IP and EA of the polymers are correlated and polymers with the most positive IP values also tend to have the least negative EA values.
We also measured optical transmittance of the solutions, which is a measure of how well the polymer particles disperse in the reaction mixture. We found, as for other systems,30,32 that the most active materials are also the most dispersible in the water/methanol/TEA mixture used for photocatalysis experiments (transmittance ∼ 0%; Fig. 3d). There is a clear downward trend in the hydrogen evolution rate with increasing transmittance and, hence, decreasing dispersibility. A plot of the hydrogen evolution rate against the average particle-size, as measured by static light scattering (SLS) (Fig. 3e), suggests that this might be an important factor in the dispersibility, since the most dispersible polymers also tend to have the smaller average particles (Fig. S76†). Scanning electron microscopy (SEM) was conducted for all dibenzo[b,d]thiophene sulfone polymers (Fig. S77–S83†). These show that most materials consist of small particles of similar sizes that are fused together into micron-sized particles, with the exception of p-FuS (Fig. S79†) which consists of a mixture of submicron and micron-sized particles. These observations that agreed well with the SLS measurements. Again, the heteroatom and co-monomer choice have a clear effect on both the measured optical transmittance and particle-size.
The wettability of the dibenzo[b,d]thiophene sulfone co-polymers, another potential contributor to the dispersibility of the polymers, was studied by contact angle measurements with water (Fig. S84 and S85†). The contact angles against water for these polymers ranged from 52.8° to 85.0°, with p-POS being the most hydrophilic polymer, and p-GeS being the most hydrophobic polymer, demonstrating a clear effect of the heteroatom choice on the polymers' wettability. In contrast to the particle size, however, the contact angles do not correlate well with the dispersibility (Fig. S86†) or hydrogen evolution rate (Fig. S87†), suggesting that small particle-size trumps wettability for these materials, though this might be different if the same polymers could be prepared with different particle size distributions.
In contrast to the case of the external surface area, increasing the internal surface area seems to have a negative effect on the catalytic performance of these materials (Fig. 3f). This may be due to the generally poor wettability of these materials and a lack of water penetration into these rather non-polar CMPs. Also, the low physical density of these amorphous microporous polymers might reduce charge transport to a degree that offsets any mass transport benefits associated with high surface areas. Furthermore, it is also relevant in this context that the conjugated microporous polymers (X-CMP3) are not formally conjugated due to the linker used, unlike the linear p-XS, p-XPh and sp-XS materials.
Residual palladium within these materials acts as the co-catalyst as shown in previous studies.18,21 All insoluble polymers were found to have residual amounts of Pd from the synthesis (ranging from 0.25 wt% to 1.72 wt%), while soluble polymers contained less residual Pd (ranging from 0.052 wt% to 0.17 wt%). Therefore, a Pd content control experiment was performed for the soluble polymer with the highest photocatalysis activity, sP-SiS (Fig. S118†). A loading of 1 wt% of Pd for sP-SiS gave a higher hydrogen evolution rate compared with the rate of sP-SiS without Pd loading, but the dependency on Pd concentration was relatively small, with hydrogen evolution rates varying between 48.4 μmol h−1 (without additional co-catalyst) and 59.3 μmol h−1 (loaded with 1 wt% Pd).
We also considered some other properties, such as the lifetime of the excited-state of the polymer in the reaction mixture as estimated using time-correlated single photon counting (TCSPC, Fig. S57–S70†) and found that average weighted life-times ranged from 0.17 to 4.51 ns. We found no overall correlation, but all materials the best performing materials (HER > 70 μmol h−1) have estimated life-times longer than 0.5 ns (see Fig. S71†).
As observed in our previous work, the hydrogen evolution rate appears to correlate with a combination of contributing factors. A polymer that has an ‘optimal’ value for only one property—for example, low transmittance/high dispersibility—is not necessarily catalytically active. For example, p-FS disperses very well in the reaction mixture, but it also has one of the least positive IP values within the p-XS family, and an optical gap value that is larger than more active catalysts such as p-CzS – hence, its photocatalytic activity is low. Moreover, we find that heteroatom substitution is a useful strategy of modulating these factors that contribute to the hydrogen evolution rate and hence optimise the polymers' photocatalytic activity.
To evaluate the stability of these new photocatalysts, we studied the dibenzo[b,d]thiophene sulfone-dibenzo[b,d]furan co-polymer (p-FuS) with repeat catalytic runs under visible irradiation (300 W Xe light source, λ > 420 nm) over a total of 30 hours, with intermittent degassing and replacement of the water/MeOH/TEA mixture after 19 hours. The HER was reduced by about 10% after 10 hours, but the material was still active when irradiation was continued for a total of 30 hours (Fig. S119†). The material did not show significant changes in its UV/vis, photoluminescence, FT-IR spectra or its PXRD pattern after 30 hours of irradiation compared to the as made material (Fig. S123–S125†). Furthermore, control experiments show that as expected no hydrogen is produced in the dark (Fig. S120†) and isotope labelling experiments are clear evidence that the origin of the gas evolution products is indeed from proton reduction (Fig. S121†).
Conclusions
Substitution of the carbon atom in the bridge-head of the fluorene-type co-polymers by a heteroatom leads to some of the highest-performing polymer photocatalysts that we have found so far with the dibenzo[b,d]thiophene co-polymer with dibenzofuran having a hydrogen evolution rate of 147 μmol h−1 under visible illumination. The modulation of the activity of heteroatom substituted polymers compared to their fluorene analogs is observed to be the result of how the presence of the heteroatom influences material properties, such as optical gap, ionisation potential, electron affinity, dispersibility and particle-size, properties we have previously demonstrated explains the majority of the variation in the hydrogen evolution rates of polymers.Comparing the different families of co-polymers studied, the dibenzo[b,d]thiophene sulfone co-polymers are the most active, followed closely by the phenyl co-polymers and the spirobifluorene CMPs. The family of soluble co-polymers were found to be the least active, however, offer the advantage of solution processability. The latter is likely beneficial when fabricating advanced devices, such as high-surface area coatings and heterojunctions, for which an intimate contact between both phases is required.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under grant EP/N004884/1. YB thanks the CSC for a scholarship. We acknowledge the National Mass Spectrometry Facility at Swansea University for mass spectrometry measurements.Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- T. Jafari, E. Moharreri, A. Amin, R. Miao, W. Song and S. Suib, Molecules, 2016, 21, 900 CrossRef PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120(2), 919–985 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- X. Zong, X. Miao, S. Hua, L. An, X. Gao, W. Jiang, D. Qu, Z. Zhou, X. Liu and Z. Sun, Appl. Catal., B, 2017, 211, 98–105 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- L. Li, W. Y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903–6909 CrossRef CAS.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- F. Haase, T. Banerjee, G. Savasci, C. Ochsenfeld, B. V. Lotsch, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Faraday Discuss., 2017, 162, 165–169 Search PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J. S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- C. B. Meier, R. Clowes, E. Berardo, K. E. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- Z.-A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS PubMed.
- R. S. Sprick, M. Hoyos, J. J. Morrison, I. M. Grace, C. Lambert, O. Navarro and M. L. Turner, J. Mater. Chem. C, 2013, 1, 3327–3336 RSC.
- F. Caffy, N. Delbosc, P. Chávez, P. Lévêque, J. Faure-Vincent, J.-P. Travers, D. Djurado, J. Pécaut, B. Grévin, N. Lemaitre, N. Leclerc and R. Demadrille, Polym. Chem., 2016, 7, 4160–4175 RSC.
- V. H. K. Fell, A. Mikosch, A.-K. Steppert, W. Ogieglo, E. Senol, D. Canneson, M. Bayer, F. Schoenebeck, A. Greilich and A. J. C. Kuehne, Macromolecules, 2017, 50, 2338–2343 CrossRef CAS.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, J. Phys. Chem. C, 2017, 121, 1498–1506 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta02599b |
| This journal is © The Royal Society of Chemistry 2020 |