
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The nature of the methylamine–MAPbI3 complex: fundamentals of gas-induced perovskite liquefaction and crystallization†
Dmitry
Bogachuk
 ab,
Lukas
Wagner
ab,
Simone
Mastroianni
ac,
Michael
Daub
d,
Harald
Hillebrecht
cd and
Andreas
Hinsch
*a
ab,
Lukas
Wagner
ab,
Simone
Mastroianni
ac,
Michael
Daub
d,
Harald
Hillebrecht
cd and
Andreas
Hinsch
*a
aDepartment of Organic and Perovskite Photovoltaics, Fraunhofer Institute for Solar Energy Systems ISE, Heidenhofstr. 2, 79110, Freiburg, Germany
bINATECH, Albert-Ludwigs-Universität Freiburg, Emmy-Noether-Str. 2, 79110, Freiburg, Germany
cMaterials Research Center FMF, Albert-Ludwigs-Universität Freiburg, Stefan-Maier-Str. 21, 79104, Freiburg, Germany
dInstitute for Inorganic and Analytic Chemistry, Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104, Freiburg, Germany
First published on 28th April 2020
Abstract
The liquefaction and recrystallization of CH3NH3PbI3 perovskite with methylamine – MA0/CH3NH2 gas in solar cells have been intensively studied in the past years, yet the exact mechanisms remained hypothesized and unclear due to lack of experimental evidence. The investigation presented in this work connects the interatomic bonding in the methylamine–MAPbI3 complex during liquefaction and recrystallization with the crystal morphology and the lifetime of photo-generated carriers in planar and mesoporous layer structures. Raman shift analysis and XRD structural investigation have been performed, showing that MA0 gas forms a complex with MAPbI3via chemisorption of gas molecules to the perovskite crystal surface by replacing I− atoms in the PbI6 octahedra and via coordination of MA+. Through real-time photoluminescence, the changes in bonding during the liquid–solid transition are related to the radiative charge carrier recombination during the liquefaction and recrystallization. We found that the amount of MA0 gas reacting with perovskite determines the crystal morphology, pore-filling and, most importantly, the charge carrier lifetime.
Introduction
In the last decade, perovskite photovoltaics (PV) have been investigated by a tremendous number of scientists and researchers, reaching outstanding power conversion efficiencies (PCEs) which in the laboratory have already exceeded the record efficiencies of such mature technology as polycrystalline silicon, Cadmium Telluride (CdTe) and Copper Indium Gallium Selenide (CIGS).1Such rapid development was realized by continuous improvements of the molecular structure, film morphology and physical properties of perovskite crystals. One successful method to achieve this goal was proposed in 2015 by Zhou et al., where a peculiar phenomenon was described in which methylammonium lead iodide (MAPbI3) has be liquefied upon exposure to methylamine (CH3NH2/MA0) gas.2 This liquefaction process is reversible and upon removal of the MA0-gas, crystalline MAPbI3 is formed again. The chemical reaction has been suggested in literature as following:
| CH3NH3PbI3(s) + xCH3NH2(g) ⇆ CH3NH3PbI3·xCH3NH2(l) | (1) |
This process has often been used as a post-treatment step to improve film morphology and crystal quality.3–6 Enhancements of photovoltaic properties of solar cells using methylamine gas were repeatedly reported due to an improved film quality, charge carrier lifetime, perovskite film homogeneity and more favorable crystallinity,5,7–11 bridging to an efficiency of above 20%.12,13 On the technological side, Noel et al. have proposed to dissolve perovskite precursor in a mixed solvent of acetonitrile (ACN) and MA0 to produce a solution which can be used for manufacturing high-efficient perovskite solar cells with PCE of 18%.14 In this process the solvating capability of ACN was increased by methylamine bubbling, leading to formation of a clear perovskite solution. In 2018, we have developed a drop-casting method where a similar approach can be used for infiltration of the porous scaffold of carbon-based printable, low-cost, hole conductor-free perovskite solar cells (C-PSCs). The champion device had a Voc of over 1 V and a stabilized efficiency of 12.6% certified by Fraunhofer ISE CalLab.15 Consequentially, manufacturing the photo-absorber layer of a PV device using methylamine–perovskite interaction establishing itself as a promising, cost-effective approach, that can be implemented on a large scale module production.16–18
The question what stands behind the observed mechanism of perovskite liquefaction was already raised in previous reports. Zhou et al. supposed that the lone-electron pair in the nitrogen (N) atom of methylamine gas interacts with PbI6 octahedra, which leads to a liquefied state.2 It has also been noted that the “molten state” can be achieved by exposing the precursor salt PbI2 to methylamine or other amines which suggests that the reaction is driven by interaction between the alkylamines and Pb(II).7,8,19–21 The interaction between MAPbI3 and MA0 gas was also described as a moderate ligand coordination of the lead atoms changing the lead halide framework.8 Simultaneously, Rand et al. have reported that the proton transfer from PbI2 to alkylamines results in the formation of Pb–amide bonds, responsible for the perovskite liquefaction.20 Also the formation of colloids has been attributed to the presence of a xMA0–MAPbI3 complex which is strengthened by witnessing Tyndall effect in this complex.8,22 However, despite the substantial research in the field of MA0 gas treatment of CH3NH3PbI3 perovskites2–6,8–10,13,14,21–23 the exact mechanism of the methylamine–perovskite complex formation (which will be referred to as MPC further in this work) and the chemical bonding in the liquefied state remain unclear due to lack of experimental evidence. In particular the following underlying questions of this method remain pending: (1) how does the stoichiometry of the MPC formation reaction affects the crystal parameters, (2) what dictates the crystallization dynamics during up-take and removal of the MA0-gas and (3) what happens to the MAPbI3 lattice when it is exposed to MA0 gas molecules. We accentuate that the answers to those questions in this work contain vital importance not only for the fundamental chemistry scientists but also for numerous researchers in the perovskite community, which aim to reach high device efficiencies.
In this work, we were able to isolate single crystals and to determine the crystal structures of intermediates from the reaction of MA0 with MAPbI3. These empirical results clearly present how MA0 can coordinate Pb2+ and MA+ (CH3NH3) leading to the formation of crystalline materials, which are stable only in the presence of MA0-rich environment. We combine the knowledge of crystalline intermediates to elucidate how does methylamine interact with MAPbI3 perovskite, what are the possible reaction pathways and in particular how the properties of MPC affect the opto-electronic properties of the crystallized perovskite.
We demonstrate by Raman spectroscopy that, upon introduction of methylamine, the lead–halide framework changes through partial replacement of I− with MA0 during its coordination of Pb2+ and MA+. We also show the bonding environment in the molten state and how it correlates with the opto-electronic properties of the recrystallized films. Through real-time photoluminescence (PL) microscopic measurement during crystallization we reveal the underlying mechanisms happening inside a porous layer (ZrO2/graphite which are normally a part of C-PSCs) during the conversion of MPC to crystalline MAPbI3. In addition, we investigate the effect of methylamine/perovskite ratio in the solution on the crystallized absorber layer. Our extensive analysis sheds light onto the chemistry of the MPC, its crystallization kinetics and the mechanisms to control the fundamental properties of perovskite absorber, which define solar cell efficiencies.
Experimental
Precursor solution preparation
The PbI2/MAI (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)–DMF complex (purchased from TCI, 99.99% trace metals basis) was heated at 100 °C under nitrogen atmosphere and thus a nearly pure MAPbI3 perovskite powder was obtained. The liquefied perovskite using MA0 gas (≥99%, purchased from Linde) of certain amount has been further diluted using acetonitrile with a ratio of 1 mg MAPbI3 to 1 μL of ACN (purchased from Carl Roth >99%).
:1)–DMF complex (purchased from TCI, 99.99% trace metals basis) was heated at 100 °C under nitrogen atmosphere and thus a nearly pure MAPbI3 perovskite powder was obtained. The liquefied perovskite using MA0 gas (≥99%, purchased from Linde) of certain amount has been further diluted using acetonitrile with a ratio of 1 mg MAPbI3 to 1 μL of ACN (purchased from Carl Roth >99%).
Sample preparation
The glass plates with fluorine-doped tin oxide on top were treated in an ultrasonic bath containing 3% deconex OP153 at 60 °C followed by rinsing with DI water. An ultrasonic treatment at 60 °C for 1 min in DI water was further operated to ensure removal of any detergent residual. Glass plates were then blown dry using nitrogen. ZrO2 (Zr-nanoxide paste obtained from Solaronix SA) of 2 μm thickness was screen printed on top of glass and sintered at 500 °C for 30 min, after which the graphite-based counter electrode of 12 μm thickness was screen printed, followed by sintering at 400 °C for 30 min. Samples filled with perovskite in ZrO2 and graphite layers were kept under MA0 environment for 30 minutes to facilitate a better pore impregnation, after which the samples were annealed at 80 °C for 30 minutes. Perovskite films were spin-coated on glass substrates, pre-treated with plasma and annealed at 80 °C for 30 minutes.For the complete perovskite solar cell devices (shown in ESI†) with carbon-based electrode, a compact TiO2 layer of 25–30 nm thickness was deposited by spray pyrolysis using a solution comprising 0.05 M of titanium diisopropoxy (bis)acetyl acetone (Sigma Aldrich) diluted with ethanol (≥99.5% from Carl Roth). Mesoporous layers of TiO2 (paste prepared via mixing Dyesol DSL-18NRT with terpineol in 1:0.75 weight ratio) and ZrO2 of 500 nm and 1 μm thicknesses respectively were screen printed and sintered at 500 °C. Afterwards the graphite-based layer (Elcocarb B/SP paste purchased from Solaronix SA) was screen printed and subsequently sintered as described above. The positive and negative electrodes were applied by ultrasonic soldering on the FTO. The applied silver paste on the electrodes completes the architecture of the cell. The device active area was 0.4 cm2.
Characterization
Raman spectra were measured on a Bruker Vertex 70 with a Bruker RAM II module Nd:YAG laser (λ = 1064 nm) in a range of 50–4000 cm−1. X-ray diffraction (XRD) patterns of films were collected with a Bruker D8 diffractometer (CuKα = 0.1540598 nm) equipped with a Lynxeye detector. The PL images were obtained by OLYMPUS BX50 stereomicroscope and sCMOS camera (“Zyla 5.5 sCMOS” by Andor) with a long-pass filter while the partial illumination of the sample was provided by a 623 nm red light-emitting diode (Thorlabs, SOLIS-623C). Due to the presence of long-pass we can exclude that the recorded signal originates from radiative recombination of free charge carriers and not from the reflectance of the excitation source. Time-resolved PL measurement were conducted using externally triggered CW laser (Coherent Orbis) of 660 nm wavelength with 100 mW power, with a fall and rise time (10% and 90%) below 2 ns. The laser was triggered to produce rectangular-shaped signals at 7 Hz frequency. The current-density and voltage curves of the devices were measured with a source meter (Keithley 2400) at a scan rate of 50 mV s−1 using a solar simulator providing a 100 mW cm−2 illumination intensity (of AM 1.5G).Result and discussion
Synthesis and characterization of intermediates MA0/MA+/PbI2
Following the conditions in the course of the liquefaction transformation process, single crystals of several intermediates were isolated and characterized. Passing a stream of MA0/CH3NH2 gas over powdered PbI2 yielded a highly viscous liquid from which pale-yellow platelets were crystallized. The crystal structure determination (Table S1†) revealed a neutral complex of Pb(II) with the composition (CH3NH2)4PbI2. A simplified description classifies (CH3NH2)4PbI2 as a neutral octahedral complex with two iodine anions in a cis-position (Fig. 1a, details Table S1†). CH3NH2 acts as a classical donor ligand to Pb2+.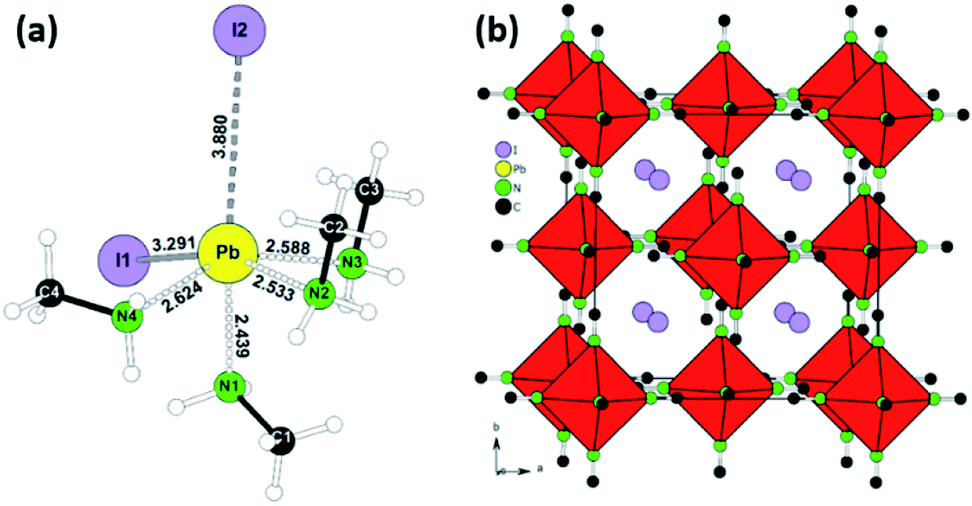 | ||
| Fig. 1 Visual representation of neutral complex PbI2(CH3NH2)4 with corresponding bonds lengths. (a) Crystal structure of [Pb(CH3NH2)6]I2 idealised as cubic K2PtCl6 type (b) as derived from the single crystal XRD measurement. | ||
Passing a strong stream of methylamine evaporated from the cold trap over MAPbI3 was readily liquefied and colorless crystals were precipitated from the MAPbI3xMA0. They were stable at room temperature if the sample was kept in a closed vessel with pure methylamine as equilibrium gas phase. For the measurement the crystals were directly transferred from the mother liquid into the cold N2-stream of the sc-XRD (at 100 K). Further examination revealed a multiply twinned crystal where the idealized structure is based on a face-centered cubic unit cell. The refinement (Table S1†) yielded a structure model close to the well-known K2PtCl6 type with octahedral [Pb(CH3NH2)6]2+ complexes and isolated I− anions (Fig. 1b, details Table S1†). This K2PtCl6-related structure is a very common motif for hexammine complexes of divalent transition metal halides [M(II)(NH3)6]X2 (M = V, Cr, Mn, Fe, Co, Ni; X = Cl, Br, I).24
In some cases, small colorless needles were formed as by-product which can't be recovered as MAPbI3 in the course of the re-crystallization process. The small needles were characterized by sc-XRD as Pb(OH)I.25,26 Additionally, pale-yellow platelets were separated from the bulk material, which were characterized as (CH3NH3)5(CH3NH2)2Pb2I9. This compound contains [Pb2I9]5− units consisting of two face-sharing octahedra and MA0/MA+ dimers [CH3NH3–NH2CH3]+ bridged by a hydrogen bond (Fig. S1, details Table S1†). The formation is explained by moisture according the following reaction:
| 4CH3NH3PbI3 + 4CH3NH2 + 2H2O → (CH3NH3)5(CH3NH2)2Pb2I9 + 2Pb(OH)I + CH3NH3I | (2) |
Composition of MA0–MAPbI3 complexes in solution of liquefied MAPbI3 and evolution/precipitation of perovskite
From the above-mentioned findings, it is strongly suggested that the liquefaction of MAPbI3 in the presence of MA0 is driven by its coordination of Pb2+ and MA+. To gain new insights into the interaction between MA0 and CH3NH3PbI3, an MPC solution (diluted in ACN) with high excess of MA0 was produced in a closed vial. The Raman spectrum was then measured as the vial was opened in ambient conditions at room temperature and MA0 gradually left the liquid over a course of 125 minutes throughout which the pale-yellow liquid gradually turned to saturated clear yellow and, consequently with additional annealing, turned dark, resembling crystalline MAPbI3. In the liquified state (Fig. 2, purple) at high MA0 pressure, the most intense peak is at 78 cm−1, followed by a peak at 94 cm−1. Upon opening of the vial, an intermediate state is introduced which has a dominant peak at 104 cm−1 and a smaller peak at 82 cm−1. Finally, upon additional annealing the bands at 94 cm−1 and 82 cm−1 arise. Furthermore, the bands at 155 cm−1 and 290 cm−1 are only present in the MPC and both of them disappear during crystallization, while the new bands at 120 cm−1 and 242 cm−1 occur.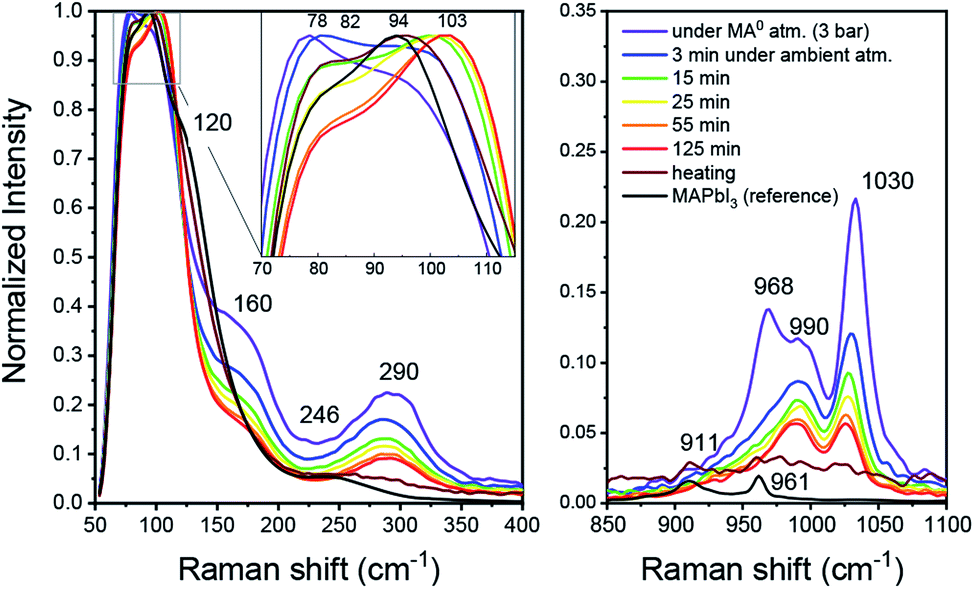 | ||
| Fig. 2 Raman spectra of a liquified MAPbI3xMA0 complex (MPC) during liquid–solid conversion as the MA0 leaves the complex. Note the shifts in the 70–115 cm−1 region (inset) corresponding to Pb–I symmetric and asymmetric stretching vibrations, suggesting that the PbI6 octahedra undergo changes. | ||
In the context of MAPbI3 Raman shift spectrum, the region between 70 and 120 cm−1 is usually attributed to Pb–I framework interactions, such as symmetric and asymmetric Pb–I bond stretching.28–31 As soon as the vial with the MPC is opened, MA0 molecules start to leave and both peaks shift, indicating a structural reorganization of the Pb–I framework (i.e. a change in Pb–I valence vibrations) during the transition between the liquid and solid phases. The Raman shifts in wavenumber region of 150–3000 cm−1 is commonly attributed to numerous vibrational modes of organic cations such CH3 bending, C–N stretching, N–H stretching and others29,32 (complete Raman shift spectra are presented in Fig. S2 in the ESI†). Notably, we highlight the presence of a band at 290 cm−1 existing only in the liquid state under high pressure, which we affiliate with a rotational mode of MA+. In a recent report from Park et al. a similar band at 288 cm−1 was found to be responsible for a rotational mode of MA+ cation, which is normally inactive, but can be seen here either due to the fact that in the MPC the local symmetry of PbI3 framework is lower or because MA+ form MAI–MA0x during the MPC formation as will be shown later.30
As seen from Fig. 2, the band at 290 cm−1 gradually reduces during the crystallization, while the band at 120 cm−1 rises, which can be attributed to MA+ libration mode.29 In addition, the bands at 1030 cm−1 and 990 cm−1 which nearly ideally correspond to the CH3–NH3 rocking and C–N stretching vibrations respectively for a free MA+ in vacuum, vanish during crystallization.32 Simultaneously, the bands assigned to the same vibrational modes in MAPbI3 appear at 911 cm−1 and 961 cm−1 for CH3–NH3 rocking and C–N stretching, respectively. Such band shifts suggest the reduction of free MA+ cations and the increase of the Pb–I framework symmetry due to insertion of MA+ yields recrystallized MAPbI3. In the presence of humidity, H2O and CH3NH2 are reacting in an acid base reaction to produce MA+ cations and OH− anions. In this case degassing results in the formation of crystalline (CH3NH3)3(CH3NH3–NH2CH3)2Pb2I9 and colorless rod-shaped crystals of Pb(OH)I. (CH3NH3)3(CH3NH3–NH2CH3)2Pb2I9 is build up by isolated [Pb2I9]5− dimers of face sharing PbI6-octahedra (Fig. 3). Charge balance is achieved by MA+ as well as the dimeric (MA–MA0)+ cations. Further degassing yields also MAPbI3, but Pb(OH)I stays as a by-product, which emphasizes that humidity should be avoided during the recrystallization of MAPbI3 with MA0. Thus, the MAPbI3 liquefaction may be driven by two effects, MA0 bonding to Pb2+ atom via lone electron pair as well as by the formation of MA0–MA+ dimers. Very recently (this manuscript had already been submitted at that time), another publication on the liquefaction of MAPbI3 using methylamine gas was published.27 In accordance with our own work, they also found the existence of methylammonium–methylamine dimers. The dimers can be regarded as large cations with low charge density, reducing the lattice energy of the corresponding salt, which yields a situation comparable to ionic liquids.
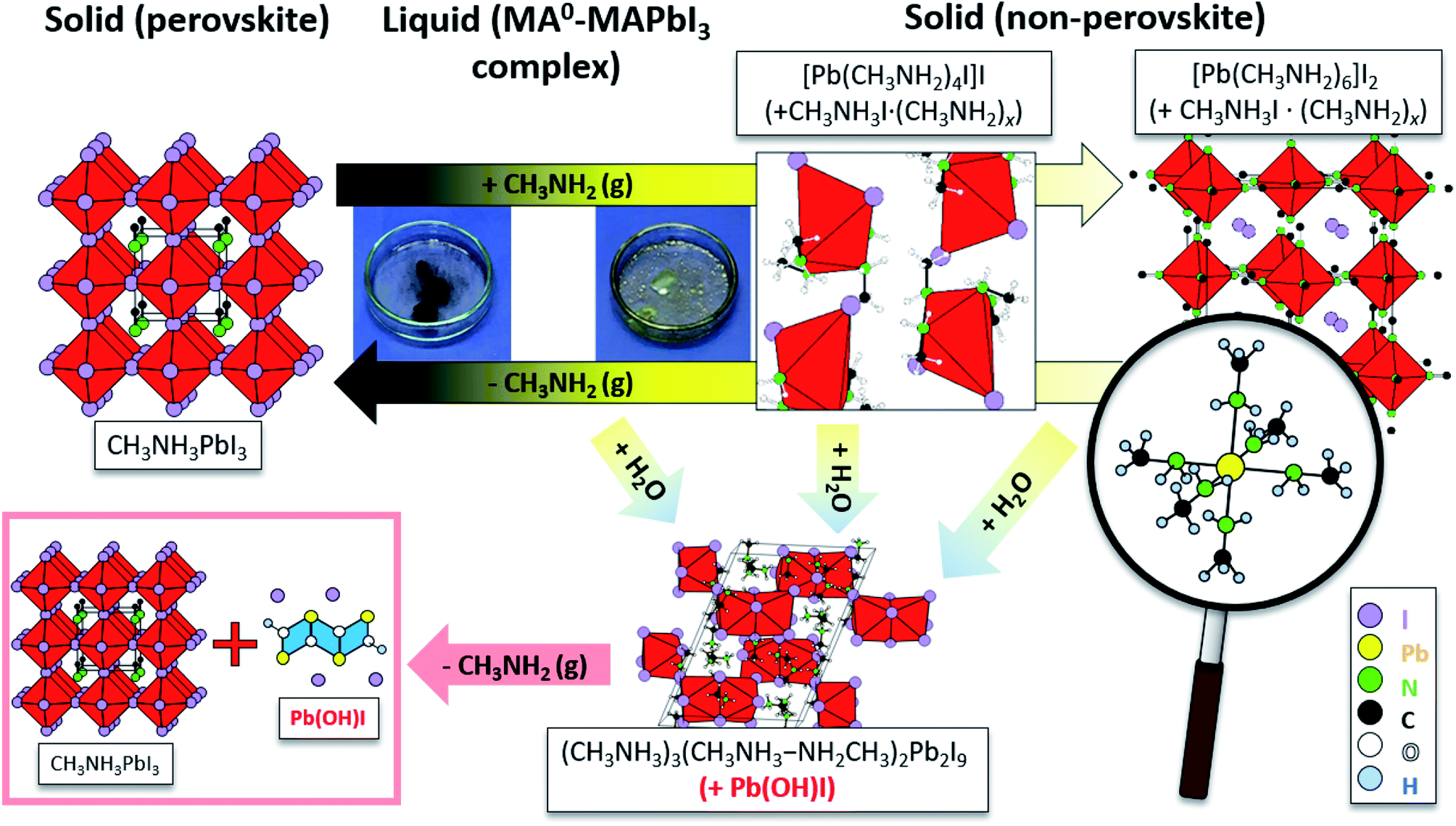 | ||
| Fig. 3 Structural changes during the liquefaction of MAPbI3 upon the introduction of MA0 and formation of the neutral complex [PbMA04I]I (+MAI·MA0x) in which Pb2+ is coordinated by MA0 molecules. The MA0 amount increase into the complex leads to the increase of coordinated MA0 and to the formation of crystalline PbMA06I2 (+MAI·MA0x). Note that in the presence of humidity a crystalline (CH3NH3)3(CH3NH3–NH2CH3)2Pb2I9 and Pb(OH)I are produced. Upon further degassing Pb(OH)I stays as an unwanted by-product. These measurements are derived from detailed XRD pattern analysis and visualized via embedded software. | ||
Real-time crystallization dynamics observed by photoluminescence
We investigated the crystallization dynamics using real-time photoluminescence microscopy. Such characterization tool allows us to link the emission of photons from radiative recombination with the surface chemistry of the crystallizing layer to understand the behavior of nucleation and absorber layer formation inside the mesoporous scaffold.33 A layer of graphite (12 μm) on top of thick ZrO2 layer (2 μm) on a glass substrate was filled with perovskite solution (MPC with ACN) and subsequently annealed at 70 °C to crystallize the perovskite. Then, the substrate was placed in a transparent gas chamber (as depicted in Fig. S4†), to which MA0 gas is later supplied, inducing the perovskite liquefaction. After reaching the liquefied state, a small outlet was opened in the box allowing MA0 gas to leave and upon rise in temperature induced by a transparent hotplate, the methylamine molecules start to evaporate even more rapidly inducing a crystallization within the mesoporous layers. A more detailed description of the setup and the procedure is shown in Fig. S4 and S5.† We ascribe several processes to the different stages observed during the liquid–solid conversion of the perovskite crystals. The real-time PL measurement starts with solid perovskite without MA0 (denoted as stage (i) in Fig. 4). Upon introduction of MA0 to the system, in a first instance the PL sharply rises, reaching a PL intensity 4 times higher than initially and subsequently drops to PL counts close to zero (stage (ii)) since the perovskite now is in a liquified form (stage (iii)). When the MA0 is removed again, the nucleation takes place, and a rise in the PL signal (stage (iv)) is observed, followed by a slow decay (stage (v)) during which the annealing takes place (stage (vi)). After that in the final stages ((vii) and (viii)) the PL signal slightly rises and stabilizes at a value close to the initial one. The real-time PL microscopy measurement is provided in detail in Video S1 in ESI.†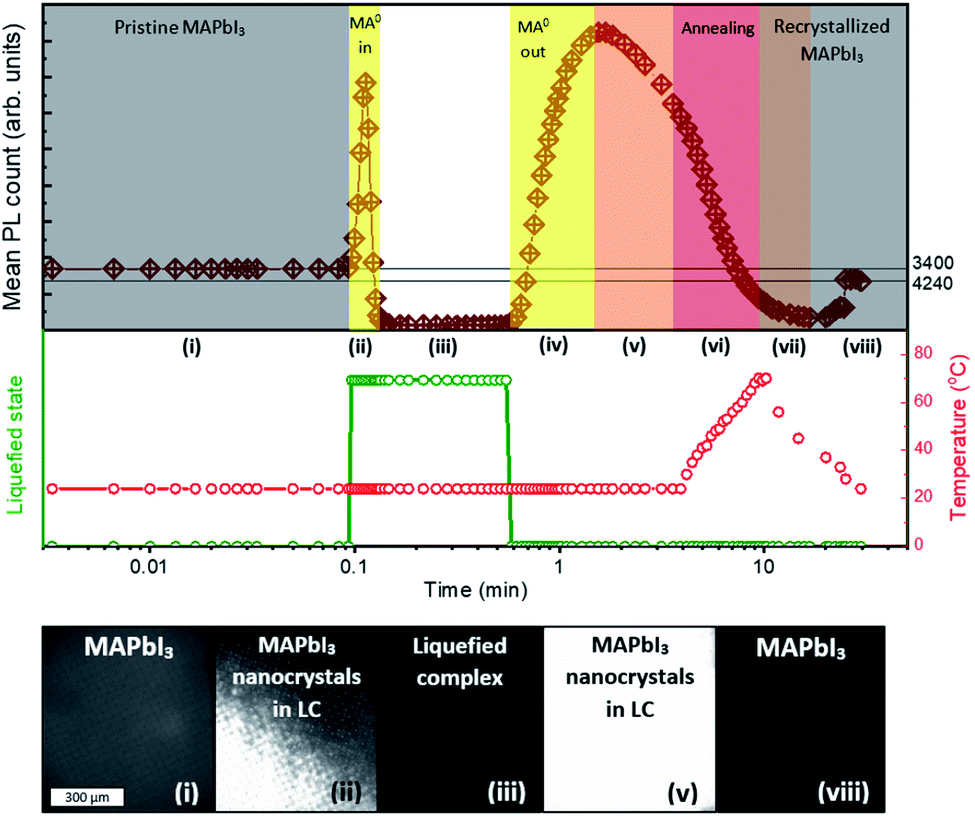 | ||
| Fig. 4 Investigation of liquid–solid evolution of MPC in mesoporous layers. The photoluminescence is monitored in real-time on the glass/ZrO2/graphite device. When the MA0 is supplied through an inlet, the layer turns pale-yellow, causing a drop in the PL signal due to the collapse of the ABX3 structure. Once, the inlet is opened, the MA0 leaves the MPC, reforming perovskite and causing a rise in PL. When the sample is heated up to ∼70 °C, it induces a more rapid leave of MA0 and the subsequent crystallization. The full PL microscopy measurement can be found in Video S1 in ESI.† | ||
We manifest that the MA0 gas forms the MPC with MAPbI3via chemisorption of gas molecules to the perovskite crystal lattice by replacing I− atoms, thus coordinating Pb2+ as well as MA+. Therefore, when MA0 is introduced to a perovskite bulk, it diffuses through the grain boundaries and is adsorbed by the crystal surface resulting in the formation of separated nano-crystals. This phenomenon is accompanied by the sharp rise in PL (as seen in stage (ii) Fig. 4), which arises from the reduced number of available bulk recombination centers per perovskite crystal. In literature, high PL in nano-sized perovskite crystals and quantum dots has been observed too.34–36 Then the PL signal drops to nearly zero and the absorber layer color becomes transparent-yellow, highlighting that the ABX3 crystal structure is collapsed leading to the inability of the perovskite to have a periodic wave function. Upon the removal of MA0 from the MPC during the crystallization process, the (PbIxMA06−x)2−x octahedra first start to form into clusters, as the number of free MA+ decreases (seen from the decrease of Raman shift band at 290 cm−1), and the number of MA+ embedded into the ABX3 structure increases (seen from the increase of band at 120 cm−1). We note that (PbIxMA06−x)2−x are charged species where the total charge of a cluster is determined by the number of residing I− atoms. These clusters act then as nuclei in the beginning of the crystallization process. The supposed process is sketched in Fig. 5.
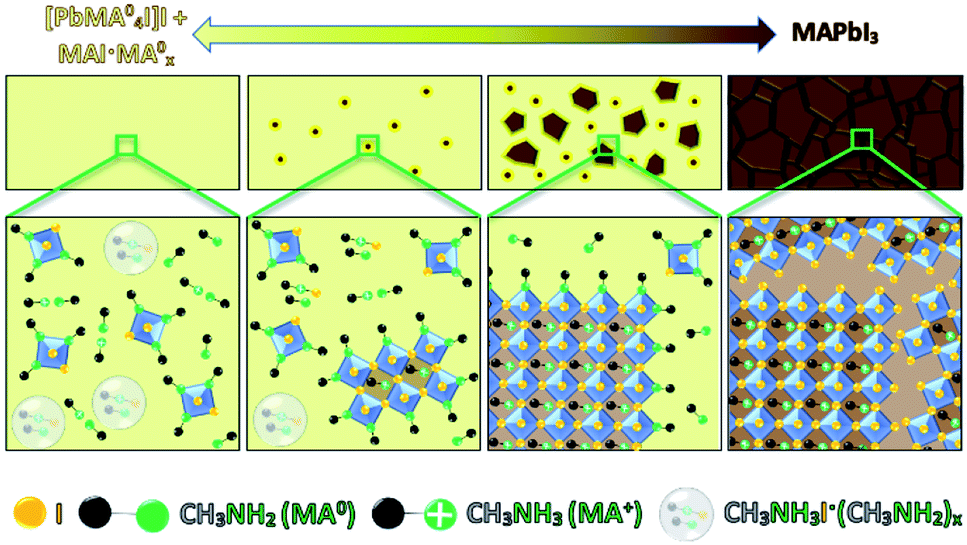 | ||
| Fig. 5 Schematic illustration of MAPbI3 perovskite recrystallization during desorption of MA0 from the methylamine–perovskite complex or crystalline [PbMA04I]I + MAI·MA0x. The double-sided arrow highlights that the reaction and mechanisms are reversible. The upper row depicts the crystal morphology change, while the bottom row illustrates the interaction of PbI6 octahedra with MA0 and the by-products in the solution such as MAI–MA0x and MA+·MA0. | ||
Revealingly, the adsorption and desorption of MA0 both tend to increase the PL signal, since the non-radiative recombination mechanisms in bulk are suppressed. Between stages (iv) and (v), when the PL signal reaches its maximum (t = 1.55 min) the bulk recombination starts to dominate because of crystal growth whereas surface passivation by MA0 starts to fade away, decreasing the overall PL. During the recrystallization for ∼20 min, stabilization is reached as is indicated by constant slightly higher PL. During the stabilization, improvement of perovskite quality through the change of grain structure and removal of residual MA0 caused by annealing, has been reported before.14 A similar overall trend, where the PL starts to rise, then drops and rises slightly again during annealing until stabilization has been observed also before for the 2-step solution processing, where PbI2 instead of MA0 passivates the grain boundaries14,37 until its complete conversion to the CH3NH3PbI3, after which the PL decreases.33,38
Influence of MA0 concentration in the MPC solution on the fully-crystallized photoabsorbers
Accurate estimation of the reaction products during liquefaction is challenging, therefore we quantitatively compare the effect of various given amounts of MA0 in the solution by reacting it with the known quantity of CH3NH3PbI3.We prepared three different solutions using x (ratio of moles of MA0 per moles of perovskite) of 19.2, 14.4 and 11.5. The estimation of x is shown in the ESI.† The lowest value (x = 11.5) already allows the complete melting of the perovskite, whilst x > 20 lead to formation of white precipitates in the solution.
Perovskite films were deposited on glass substrates by spin-coating the MPC (diluted with ACN) of different ratios x of MA0 in it. The obtained XRD patterns of each sample are shown in Fig. 6d, where the (110) and (220) diffraction peaks increase with higher concentration of MA0 in the complex, while most of other reflections decrease. Such trend highlights how the concentration of methylamine can directly affect the preferential orientation of the crystal lattice. Looking at the most pronounced diffraction signal of (110) plane, an increase in full-width at half maximum (FWHM) from 0.095° over 0.119° to 0.165° can be observed as the amount of MA0 in the MPC rises. The change in FWHM indicates that by varying the MA0 ratio in the MPC, the perovskite grain size can be tuned (i.e. large grain sizes are grown using high amounts of MA0), which can also clearly be seen in SEM top-view images in Fig. 6a–c and crystal area distributions (Fig. S7†), obtained by analysing these images via ImageJ.
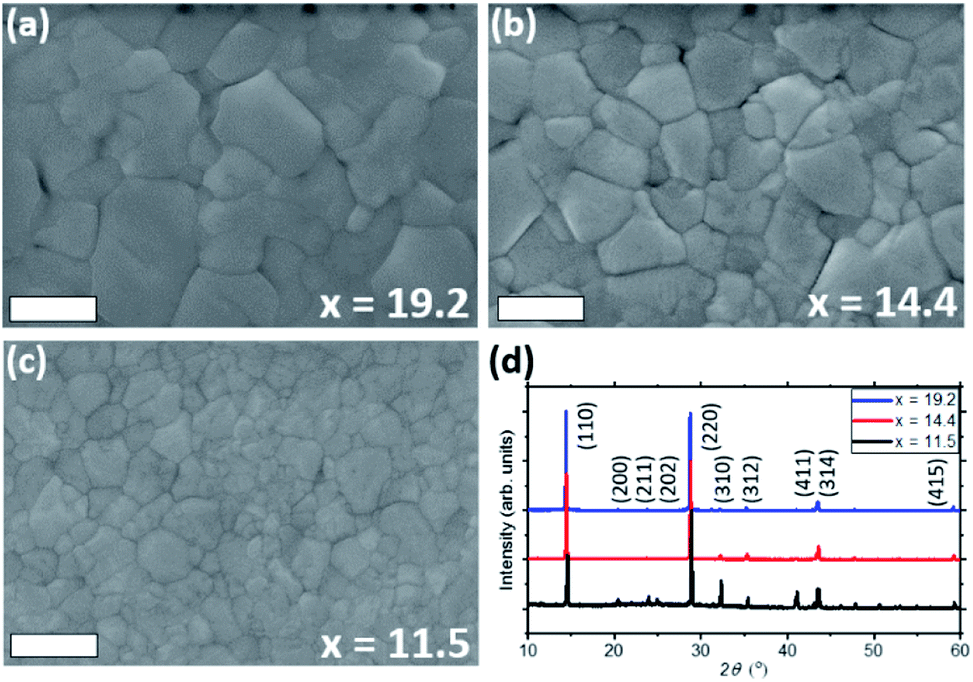 | ||
| Fig. 6 SEM images of MAPbI3 crystallized from various amounts of MA0 in the liquefied xMA0·MAPbI3 complex (a–c), where the crystal size increases with higher MA0 concentration, which can also be seen from an decrease in FWHM of (110) and (220) diffraction peaks at highly concentrated (with MA0) samples. The presented XRD patterns of three samples show how strongly MA0 coordinates the recrystallization of MAPbI3 along (110) and (220) planes (d). Scale bar – 300 nm. | ||
It has been numerously reported that the change of stoichiometry in the precursor perovskite solution leads to different crystallization dynamics, which can lead to variation in such fundamental material properties as crystal size, charge carrier transport and contact at the interface between perovskite and charge extracting layer.39–42 A similar dependency on the solution composition is seen in the MPC, where, as a result of the MA0/perovskite ratio, crystals with different domain size have been grown which coincides with the observations by Chen et al.3 The differences in FWHM in the XRD measurements correlate with the observations by SEM. We attribute such behavior to a stronger chemical coordination of Pb2+ and MA+ with the higher MA0 concentration, which causes a more preferential orientation of the crystal lattice along 〈hk0〉 planes such as (110) and (220),8,43 resulting in the growth of larger crystals. It is worth mentioning that the commonly observed peak at 2θ = 12.7° which is usually observed when PbI2 is formed28,37,44,45 was absent in all the samples. However, this residual PbI2 could be screened by the formation PbI2-alkylamide impurities, resulting in the reduced carrier lifetime of the crystallized absorber. The peaks at 2θ = 9.84,° 19.74° and 29.79° which are attributed to MAI reflections28,46 are also not present, concluding that upon crystallization the MPC forms a pure, stoichiometric MAPbI3 photoabsorber from an additive-free “perovskite melt”.
The finding that the morphology of the final crystal layers changes as a function of the x (MA0/perovskite) ratio suggests that the crystallization kinetics could also be different within the mesoporous layers of the C-PSCs. While perovskite crystals using MPC can even reach sizes of >1 μm on planar substrate,43 the crystal growth inside the porous layers of C-PSCs is clearly different, as the majority of pores have diameters of less than 20 nm. Therefore, crystallization is confined by the pore size, which means that the interface plays a big role in the nucleation process as it provides multiple nucleation sites.47 To gain a better understanding of the effect of ratio x in the solution on the crystallized absorber layer in porous isolating ZrO2 scaffold, we use steady-state and time-resolved photoluminescence (TRPL) intensity measurements. A series of perovskite-filled mesoporous layers with different x variations were characterized. As seen from Fig. S8,† the steady-state PL intensity image of layers crystallized from a low-concentrated solution show strong PL signal. With higher concentration of MA0 the average number of PL counts decreases, while the overall homogeneity of the PL signal increases as a result of better infiltration in the mesoporous layers. We quantify the homogeneity (h) of the PL image by comparing the histogram of the PL intensity shown in Fig. S9.† Due to a higher concentration of MA0 in the solution, the liquified complex becomes less viscous enabling an easier infiltration through the pores. Zhou et al. suggested that the excess of MA0 in the MPC “trims” the clusters, leading to a decrease in cluster size and lower viscosity.48
A TRPL was employed to derive the carrier lifetime (τ). Generally, as it has been demonstrated in numerous works, the PL-decay is affiliated with the transient behavior of radiative and non-radiative recombination of charge carriers in a semiconductor.9,14,49–51 In the absence of a quencher (as in this case), the longer the PL-decay, the longer the excited diffusing carriers are present in the perovskite, and therefore the longer carrier lifetime and diffusion length, both of which are vital characteristics of any PV device.
We quantify the charge carrier lifetime by fitting the PL-decay to a bi-exponential function, accounting for Schockley–Read–Hall recombination processes at surfaces and in the bulk. The TRPL results in Fig. S10† show that the decay is much slower (longer carrier lifetime) in the photoabsorbers fabricated with MPC using low ratio of MA0/MAPbI3 in it (273.65 ns and 140.67 ns for x = 8.6 and 11.1, respectively). Contrarily, for the higher ratio of MA0 the PL-decay is significantly faster: 20.9 ns for x = 14.8 and 22.56 ns for x = 18.2. As seen from Table 1, large amounts of MA0 (x > 12) yield an absorber with lower carrier lifetime having lower averaged PL, indicating that despite strong advantages from crystallographic perspective (as seen from XRD patterns) such ratios of MA0 are not desired due to poor opto-electronic properties of the layers. Conings et al. have also found that the recrystallized layer after the MA0 treatment helps the infiltration through the pores, but, on the other side, leading to an inferior electronic quality.10 Since no quenching layers are involved, the observed reduction in carrier lifetime is likely related to a more efficient bulk/surface non-radiative recombination processes. This means that there are either more of faster recombination channels, most likely in the form of trap-assisted recombination. Since only the stoichiometry of the MPC changes without introduction of any other additive, the trap capture cross-section (i.e. quenching efficiency of each trap) should remain unchained. We therefore suggest that at high x, high number of trap energy states inside the bandgap outweigh the advantages of smoother film and better infiltration and start to play major role in defining the electronic properties of the absorber. Such a dependency of τ on x emphasizes the importance of a careful control of xMA0 (in reaction (1)) which should be within a certain processing window when producing solutions for perovskite PV devices.
| xMA0 | x = 8.6 | x = 11.1 | x = 14.8 | x = 18.2 |
| PL (cts) | 6.6 × 103 | 4 × 103 | 3.8 × 103 | 3.8 × 103 |
| Homogeneity | 2.6 × 10−4 | 3.3 × 10−4 | 7.4 × 10−4 | 8 × 10−4 |
| τ (ns) | 273.65 | 140.67 | 20.9 | 22.56 |
Conclusions
In this work, the nature of methylamine–MAPbI3 complex formation and its re-crystallization during the degassing in perovskite solar cells is presented. We show through XRD single crystal measurements, that MA0 simultaneously forms neutral complexes with both Pb2+ and MAI leading to the collapse of the perovskite crystal structure. In the excess (≫ 1 atm) of MA0 this reaction induces the formation and precipitation of non-perovskite solid crystals of [Pb(CH3NH2)4I]I and [Pb(CH3NH2)6]I2. We highlight that the presence of H2O leads to the irreversible formation of Pb(OH)I, which act as impurities in the photoabsorbing layer. Therefore, the formation of pure methylamine–perovskite complex needs to be carried out under normal atmospheric pressure of the MA0, and in the absence of water.During the degassing of MA0 from the complex, the iodide bonds with Pb2+, forming PbI6 octahedra, while MA+ cations get embedded in the A-site of the original pseudo-cubic ABX3 structure. Real-time photoluminescence (PL) microscopy as a measure of the crystal quality visualizes the interaction of MA0 gas with perovskite crystals. We postulate that the liquefaction starts from diffusion of MA0 gas molecules through grain boundaries, leading to a break-up of bulk crystals and therefore reducing bulk recombination. This process is then followed by a complete collapse of the perovskite structure, as the PL signal approaches zero. When the MA0 gas is desorbed afterwards in inert atmosphere, in accordance with the PL recovery a reversible process takes place, where the isolated non-bulk crystals of MAPbI3 start to re-emerge from a liquefied state till the crystal coalescence is complete.
Furthermore, we find that the amount of MA0 in the complex plays a vital role in the control of crystal size and trap-assisted charge carrier lifetime. In planar films with high MA0 concentration, the MA0–MAPbI3 complex leads to growth of larger crystals, while the low concentration leads to smaller crystals. In perovskite-filled mesoporous layers time-resolved PL decay shows that high amount of MA0 in the complex causes an increase in trap-assisted monomolecular charge carrier recombination suggesting the formation of numerous trap energy states.
With this work we show that the understanding of liquefaction processes and crystallization kinetics is paramount for the further development of perovskite photovoltaics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially funded within the projects APOLO (Grant Agreement No. 763989) as a part of European Union's HORIZON 2020 Research and Innovation Program, PROPER financed from the German Ministry of Education and Research under funding number 01DR19007 and UNIQUE supported under umbrella of SOLAR-ERA.NET_cofund by ANR, PtJ, MIUR, MINECO-AEI and SWEA, within the EU's HORIZON 2020 Research and Innovation Program (cofund ERA-NET Action No. 691664). D. B. and L. W. acknowledge the scholarship support of the German Federal Environmental Foundation (DBU). The authors thank Jutta Zielonka for conducting the SEM measurements.Notes and references
- NREL, Best Research-Cell Efficiency Chart, 2019 Search PubMed.
- Z. Zhou, Z. Wang, Y. Zhou, S. Pang, D. Wang, H. Xu, Z. Liu, N. P. Padture and G. Cui, Angew. Chem., Int. Ed., 2015, 54, 9705–9709 CrossRef CAS PubMed.
- Y. Jiang, L. Tu, H. Li, S. Li, S.-E. Yang and Y. Chen, Crystals, 2018, 8(1), 44, DOI:10.3390/cryst8010044.
- T. Zhao, S. T. Williams, C.-C. Chueh, D. W. deQuilettes, P.-W. Liang, D. S. Ginger and A. K.-Y. Jen, RSC Adv., 2016, 6, 27475–27484 RSC.
- L. Hong, Y. Hu, A. Mei, Y. Sheng, P. Jiang, C. Tian, Y. Rong and H. Han, Adv. Funct. Mater., 2017, 27, 1703060 CrossRef.
- Y. Jiang, E. J. Juarez-Perez, Q. Ge, S. Wang, M. R. Leyden, L. K. Ono, S. R. Raga, J. Hu and Y. Qi, Mater. Horiz., 2016, 3, 548–555 RSC.
- C. Li, S. Pang, H. Xu and G. Cui, Sol. RRL, 2017, 1, 1700076 CrossRef.
- M. Long, T. Zhang, H. Zhu, G. Li, F. Wang, W. Guo, Y. Chai, W. Chen, Q. Li and K. S. Wong, et al. , Nano Energy, 2017, 33, 485–496 CrossRef CAS.
- A. J. Ramadan, N. K. Noel, S. Fearn, N. Young, M. Walker, L. A. Rochford and H. Snaith, Chem. Mater., 2018, 30, 7737–7743 CrossRef CAS.
- B. Conings, S. A. Bretschneider, A. Babayigit, N. Gauquelin, I. Cardinaletti, J. Manca, J. Verbeeck, H. J. Snaith and H.-G. Boyen, ACS Appl. Mater. Interfaces, 2017, 9, 8092–8099 CrossRef CAS PubMed.
- J.-a. Yang, T. Qin, L. Xie, K. Liao, T. Li and F. Hao, J. Mater. Chem. C, 2019, 7, 10724–10742 RSC.
- Y. Zhang, Z. Zhou, F. Ji, Z. Li, G. Cui, P. Gao, E. Oveisi, M. K. Nazeeruddin and S. Pang, Adv. Mater., 2018, 30, e1707143 CrossRef PubMed.
- Y. Zhang, G. Grancini, Z. Fei, E. Shirzadi, X. Liu, E. Oveisi, F. F. Tirani, R. Scopelliti, Y. Feng and M. K. Nazeeruddin, et al. , Nano Energy, 2019, 58, 105–111 CrossRef CAS.
- N. K. Noel, S. N. Habisreutinger, B. Wenger, M. T. Klug, M. T. Hörantner, M. B. Johnston, R. J. Nicholas, D. T. Moore and H. J. Snaith, Energy Environ. Sci., 2017, 10, 145–152 RSC.
- L. Wagner, S. Chacko, G. Mathiazhagan, S. Mastroianni and A. Hinsch, ACS Energy Lett., 2018, 3, 1122–1127 CrossRef CAS.
- M. Wong-Stringer, T. J. Routledge, T. McArdle, C. J. Wood, O. S. Game, J. A. Smith, J. E. Bishop, N. Vaenas, D. M. Coles and A. R. Buckley, et al. , Energy Environ. Sci., 2019, 12, 1928–1937 RSC.
- Q. Liu, Y. Zhao, Y. Ma, X. Sun, W. Ge, Z. Fang, H. Bai, Q. Tian, B. Fan and T. Zhang, J. Mater. Chem. A, 2019, 7, 18275–18284 RSC.
- D. Burkitt, R. Swartwout, J. McGettrick, P. Greenwood, D. Beynon, R. Brenes, V. Bulović and T. Watson, RSC Adv., 2019, 9, 37415–37423 RSC.
- R. F. Warren and W. Y. Liang, J. Phys.: Condens. Matter, 1993, 5, 6407–6418 CrossRef CAS.
- R. A. Kerner, T. H. Schloemer, P. Schulz, J. J. Berry, J. Schwartz, A. Sellinger and B. P. Rand, J. Mater. Chem. C, 2019, 7, 5251–5259 RSC.
- H. Chen, F. Ye, W. Tang, J. He, M. Yin, Y. Wang, F. Xie, E. Bi, X. Yang and M. Grätzel, et al. , Nature, 2017, 550, 92–95 CrossRef CAS PubMed.
- C. Wu, H. Li, Y. Yan, B. Chi, J. Pu, J. Li, M. Sanghadasa and S. Priya, Nano Energy, 2017, 36, 295–302 CrossRef CAS.
- S. R. Raga, Y. Jiang, L. K. Ono and Y. Qi, Energy Technol., 2017, 5, 1750–1761 CrossRef CAS.
- R. Essmann, G. Kreiner, A. Niemann, D. Rechenbach, A. Schmieding, T. Sichla, U. Zachwieja and H. Z. Jacobs, Z. Anorg. Allg. Chem., 1996, 622, 1161–1166 CrossRef CAS.
- R. Nasanen, SUOMEN KEMISTILEHTI, 1966, 39(4), 105 Search PubMed.
- O. I. Siidra, D. Y. Zenko, A. N. Suknotova and S. V. Krivovichev, Mineral. Mag., 2013, 77, 3239–3248 CrossRef CAS.
- X. Huang, R. Chen, G. Deng, F. Han, P. Ruan, F. Cheng, J. Yin, B. Wu and N. Zheng, J. Am. Chem. Soc., 2020, 142, 6149–6157 CrossRef CAS PubMed.
- S. M. Jain, B. Philippe, E. M. J. Johansson, B.-w. Park, H. Rensmo, T. Edvinsson and G. Boschloo, J. Mater. Chem. A, 2016, 4, 2630–2642 RSC.
- C. Quarti, G. Grancini, E. Mosconi, P. Bruno, J. M. Ball, M. M. Lee, H. J. Snaith, A. Petrozza and F. D. Angelis, J. Phys. Chem. Lett., 2014, 5, 279–284 CrossRef CAS PubMed.
- D.-N. Jeong, D.-K. Lee, S. Seo, S. Y. Lim, Y. Zhang, H. Shin, H. Cheong and N.-G. Park, ACS Energy Lett., 2019, 4, 1189–1195 CrossRef CAS.
- M. Park, N. Kornienko, S. E. Reyes-Lillo, M. Lai, J. B. Neaton, P. Yang and R. A. Mathies, Nano Lett., 2017, 17, 4151–4157 CrossRef CAS PubMed.
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky and A. Pucci, et al. , J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS PubMed.
- L. Wagner, L. E. Mundt, G. Mathiazhagan, M. Mundus, M. C. Schubert, S. Mastroianni, U. Würfel, A. Hinsch and S. W. Glunz, Sci. Rep., 2017, 7, 14899 CrossRef PubMed.
- S. Bonabi Naghadeh, B. Luo, G. Abdelmageed, Y.-C. Pu, C. Zhang and J. Z. Zhang, J. Phys. Chem. C, 2018, 122, 15799–15818 CrossRef CAS.
- Y.-F. Li, J. Feng and H.-B. Sun, Nanoscale, 2019, 11, 19119–19139 RSC.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, CsPbX3, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- L. Wang, C. McCleese, A. Kovalsky, Y. Zhao and C. Burda, J. Am. Chem. Soc., 2014, 136, 12205–12208 CrossRef CAS PubMed.
- M. Anaya, J. F. Galisteo-López, M. E. Calvo, C. López and H. Míguez, J. Phys. Chem. C, 2016, 120, 3071–3076 CrossRef CAS PubMed.
- V. D'Innocenzo, A. R. Srimath Kandada, M. d. Bastiani, M. Gandini and A. Petrozza, J. Am. Chem. Soc., 2014, 136, 17730–17733 CrossRef PubMed.
- S. I. Seok, M. Grätzel and N.-G. Park, Small, 2018, 14, e1704177 CrossRef PubMed.
- J.-H. Im, I.-H. Jang, N. Pellet, M. Grätzel and N.-G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- S. Mastroianni, F. D. Heinz, J.-H. Im, W. Veurman, M. Padilla, M. C. Schubert, U. Würfel, M. Grätzel, N.-G. Park and A. Hinsch, Nanoscale, 2015, 7, 19653–19662 RSC.
- D. L. Jacobs and L. Zang, Chem. Commun., 2016, 52, 10743–10746 RSC.
- A. Ummadisingu and M. Grätzel, Sci. Adv., 2018, 4(2), e1701402 CrossRef PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Z. Song, S. C. Watthage, A. B. Phillips, B. L. Tompkins, R. J. Ellingson and M. Heben, Chem. Mater., 2015, 27, 4612–4619 CrossRef CAS.
- A. Dunlap-Shohl and Y. Zhou, Chem. Rev., 2019, 119(5), 3193–3295 CrossRef PubMed.
- Z. Liu, J. Hu, H. Jiao, L. Li, G. Zheng, Y. Chen, Y. Huang, Q. Zhang, C. Shen, Q. Chen and H. Zhou, Adv. Mater., 2017, 29, 1606774 CrossRef PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. Snaith, J. Sci., 2013, 342, 341–344 CAS.
- A. D. Wright, R. L. Milot, G. E. Eperon, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2017, 27, 1700860 CrossRef.
- Y. Yamada, T. Nakamura, M. Endo, A. Wakamiya and Y. Kanemitsu, J. Am. Chem. Soc., 2014, 136, 11610–11613 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1997919, 1936930 and 1998017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ta02494e |
| This journal is © The Royal Society of Chemistry 2020 |