
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and development of 3D hierarchical ultra-microporous CO2-sieving carbon architectures for potential flow-through CO2 capture at typical practical flue gas temperatures†
Xin
Liu
 a,
Jingjing
Liu
a,
Chenggong
Sun
*a,
Hao
Liu
a,
Wenlong
Wang
b,
Emily
Smith
c,
Long
Jiang
d,
Xinyong
Chen
d and
Colin
Snape
a
a,
Jingjing
Liu
a,
Chenggong
Sun
*a,
Hao
Liu
a,
Wenlong
Wang
b,
Emily
Smith
c,
Long
Jiang
d,
Xinyong
Chen
d and
Colin
Snape
a
aFaculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: cheng-gong.sun@nottingham.ac.uk
bSchool of Energy and Power Engineering, Shandong University, Jinan, P. R. China
cSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK
dSchool of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK
First published on 5th August 2020
Abstract
Developing effective carbon materials for post-combustion CO2 capture (PCC) has received great attentions over many recent years, owing to their desirable adsorption–desorption performance and exceptional thermo-oxidative stability compared to virtually any other capture materials typically the wide array of amine-based sorbent materials. However, due to the nature of physical adsorption, virtually none of the carbon materials reported so far can be practically used for PCC applications without deep flue gas cooling to ambient or even lower temperatures in order to achieve appreciable levels of CO2 uptake capacities at low CO2 partial pressures. Here, we present a category of 3D hierarchical molecular sieving carbon architectures that are able to operate at realistic flue gas temperatures with exceedingly high reversible CO2 capacities. The breakthrough CO2-sieving carbon materials are prepared from using a cost-effective and commercially widely available precursor of polymeric polyisocyanurates with a facile one-step compaction–activation methodology. Tested at sensible flue gas temperatures of 40–70 °C and a low CO2 partial pressure of 0.15 bar, the best performing materials are found to have exceedingly high reversible CO2 capacities of up to 2.30 mmol g−1 at 40 °C and 1.90 mmol g−1 at 70 °C. Advanced characterisations suggest that the unique geometry and chemistry of the easily available precursor material coupled with the characteristics of the compaction–activation protocol used are responsible for the CO2-sieving structures and capacities of the 3D carbon architectures. The findings essentially change the general perception that carbon-based materials can hardly find applications in post-combustion capture due to their low CO2 uptake capacity at low CO2 partial pressures and realistic flue gas temperatures.
1 Introduction
CO2 emissions from the consumption of fossil fuels have been considered as the main cause of climate change. Over 40% of the global anthropogenic CO2 emissions comes from large stationary emission sources such as coal-fired power plants.1,2 Despite the efforts over recent decades to speed up the development and deployment of renewable energy technologies, fossil energy will continue to dominate the energy consumption landscape, giving rise to a continual increase in carbon emissions from fossil fuels utilisation in the foreseeable futures. Carbon capture and storage (CCS) has been widely regarded as being one of the most efficient and viable strategies in the short to medium term to reduce the greenhouse carbon emissions without threatening global energy security and socioeconomic development.1,3,4 Typically, the flue gas from a coal-fired power plant contains CO2 (12–15%), N2 (73–77%) and H2O (5–7%) at about 40–50 °C.5,6 Capturing CO2 from such kind of flue gas streams can be very challenging in terms of cost and energy penalty. Today's state-of-the-art aqueous amine scrubbing technology, which is initially developed for use in oil and gas industries,7 can facilitate high capture efficiencies and produce high purity CO2 streams ready for storage and/or utilisation, but its high energy penalty and CAPEX and OPEX requirements as well as a range of associated environmental and operational issues have been the well-known hard-to-overcome performance barriers for implementation to post-combustion carbon capture in power plants.8,9 Consequently, developing alternative capture technologies have received enormous attention over recent years.10–12 Among the alternative capture technologies under development, adsorption-based or dry sorbent scrubbing technological systems have been the most investigated category of capture technologies, offering potentially significantly improved process efficiency at much reduced energy penalty, lower capital and operational costs and smaller plant footprints. This has stimulated the development of a variety of CO2-capturing adsorbent materials, such as zeolites,13–15 metal–organic frameworks (MOFs),16–22 covalent organic frameworks (COFs),23,24 microporous polymers (MOPs),25 zeolitic imidazolate frameworks (ZIFs),26 porous carbons27 and amine-functionalized solid adsorbents.28–30In recent years, carbon materials have gained the research community's great persistent interest as a promising category of materials for CO2 capture, due to their intriguing chemical, physical and adsorptive properties that are usually not easily possessed by the aforementioned materials, such as their easy-to-achieve exemplar surface areas and porosities, fast adsorption kinetics, excellent thermal and chemical stability and desirable surface re-addressability for specific target applications,31–33 However, due to the nature of physical adsorption, the CO2 adsorptive properties of carbon materials are very sensitive to both the CO2 partial pressure and temperature of a flue gas stream, with their CO2 capacities usually sharply decreasing with increasing flue gas temperatures or decreasing partial pressures. Consequently, in order to achieve high capture capacities with active carbon materials, this will essentially require the flue gas streams to be deeply cooled down to ambient temperatures or even lower, which is not only a slow but also a very costly and energy-intensive process.34,35 Therefore, numerous efforts have been made in recent years to tailor the surface textural properties and modify the surface chemistries of carbon materials in order to substantially improve their performance for carbon capture at low partial pressures and sensible flue gas temperatures. It has been found that ultra-microporous carbon materials with pore diameters being <0.5 nm, which are only slightly larger than the kinetic diameter of CO2 molecule (0.33 nm), have been reported as the most preferable for low partial pressure CO2 adsorption due to the stronger adsorption potential produced by the adjacent pore walls.31,36–40 It was found that the CO2 capacity of some ultra-microporous carbons derived from carefully selected precursors, such as potassium hydrogen phthalate,41 phenolic resin spheres42 or ZIF-8,43 could reach 1.4 to 1.6 mmol g−1 at 298 K and 0.15 bar CO2, being higher than those of general carbon materials reported, which have CO2 capacities typically ≤1 mmol g−1.44 However, it has proven to be very difficult to further increase the CO2 capacity because of the limit of ultra-microporosity that can possibly be achieved in activation.45 Surface modification by some heteroatom functional groups, such as nitrogen or oxygen-containing functionalities that may potentially increase the surface basicity and hence the CO2 affinity of carbon materials, has also been widely investigated as a means to increase the CO2 capacity at low partial pressures and relatively high flue gas temperatures.40,41,46–51 Different types of heteroatom-functionalised ultra-microporous carbons have been synthesized and the adsorption capacities of the ‘designer’ microporous carbons were further increased to 2.0–2.1 mmol g−1 at 25 °C and 0.15 bar CO2, which was considerably higher than those of their non-functionalised counterparts. For instance, benzimidazole derived ultra-microporous carbon, which have nitrogen contents varying from 7.9 to 17.6 wt% N, was found to achieve the highest adsorption capacities of up to 2.10 mmol g−1 reported so far,31 whilst the zeolitic potassium-intercalated ultra-microporous bio-carbons produced from using nitrogen-free rice husk biomass waste were found to be able to achieve 2.00 mmol g−1 at 25 °C and 0.15 bar CO2,40 however, none of these best-performing carbon materials reported so far are able to achieve appreciable levels of capture capacities at the realistic flue gas temperatures of 40–50 °C and typical CO2 partial pressures of ≤0.15 bar, with capacities being typically below 1.0 mmol CO2 per gram of sorbent.19,49,50,52–55 This essentially rule out the suitability of virtually any carbon materials reported so far for post-combustion carbon capture without the deep cooling of the huge volumes of flue gas streams. CO2 has large electric quadrupole moment and high polarizability but a smaller kinetic diameter than nitrogen molecule, so abundance of ultra-micropores and strong polarising surfaces are of great importance for carbon materials to achieve high capture capacities at practical flue gas temperatures. Bearing this in mind and using what could be a simplest approach-integrated compaction–combined carbonisation–activation, we have successfully developed a new category of CO2-capturing carbon materials with unique 3D hierarchical CO2-sieving carbon architectures and favourable surface chemistries, which are able to achieve fully reversible and high flow-through capture capacities at realistic flue gas temperatures and low CO2 partial pressures. We think our research essentially changes the general perception that carbon-based capture materials cannot possibly find their applications for CO2 capture at practical flue gas temperatures and low CO2 partial pressures. Further importantly, compared to virtually any other ‘designer’ capture materials reported so far, the cost of producing this new category of carbon-based capture materials can be dramatically reduced as the precursors used can be obtained from recycled waste as the hard-to-degrade precursors are widely used in large quantities as packing materials and/or insulation materials in different commercial and industrial sectors.
2 Materials and methods
2.1 Synthesis of porous carbon
The polyisocyanurate (PIR) polymer obtained from local merchant was pre-oxidized in horizontal tube furnace at 260 °C for 6 hours in 50 mL min−1 of airflow. The oxidized sample was then mixed with 20 mL KOH solution (KOH/polyisocyanurate weight ratio of 1) for overnight and then all samples were dried in an oven at 105 °C. The dried samples were compacted into pellets with a diameter of 13 mm under different loads of 2, 5 and 10 metric tons respectively; each pellet was then heated in a horizontal tube furnace from ambient to a pre-selected temperature at 5 °C min−1 under the protection of N2 flow (1 L min−1) and maintained at the temperature for one hour. The activated samples were washed with DI water. The obtained samples were labelled in the form of Pxy_z, where x, y, z represents the activation temperature divided by 100, KOH/AC mass ratio and compaction load, respectively.2.2 Characterization
Scanning electron microscopy (SEM) was obtained using a FEI Quanta 600 microscope with a 5 kV accelerating voltage and 25 pA current. Transmission electron microscopy (TEM) was performed on a JEOL2100F. In addition, nanofocus Computed Tomography (nano CT), GE Nanotom S, was employed to analyze the 3D structure of selected sample. The pore structure of each prepared sample was determined by nitrogen adsorption and desorption at 77 K on a Micrometrics ASAP 2420 using ultrahigh purity grade adsorbates. Before each experiment, the sample was degassed at 120 °C for 16 h. Brunauer–Emmett–Teller (BET) method was employed to calculate the specific surface area from the nitrogen adsorption isotherm in the relative pressure range of 0.01–0.1. The total pore volume (Vtotal) was determined from the amount of nitrogen adsorbed at a relative pressure of 0.99. Because nitrogen molecule could not get access to the narrow micropores, a combined analysis of N2 adsorption isotherms at −196 °C and CO2 adsorption isotherms at 0 °C by non-local density functional theory (NLDFT) model was used to calculate the volume of narrow micropores and pore size distribution (PSD). The X-ray photoelectron spectrum (XPS) of all samples was measured by a Kratos AXIS Ultra DLD X-ray photoelectron spectrometer. The XRADIA Versa XRM-500 was used to non-destructively measure the 3D structure of the selected samples at 0.5 μm resolution. Surface potential measurements were performed on a Multimode 1 AFM (Bruker Instrument) using AM-KPFM mode. An AFM probe (Bruker RTESPA-150) with a spring constant of 6 N m−1 and a typical tip radius of 8 nm was used for the imaging of morphology and the surface potential. P81_2T and P81_2T_48H were characterized by using the same tip and 3 different areas with a size (1 μm × 1 μm) in each sample were measured. Surface potential of the sample was finally obtained as an average of the surface potential of each area.2.3 CO2 adsorption performance
Thermogravimetric analysis (TGA) has been employed as a proven technique to study the effect of adsorption temperatures, gas compositions and thermal regeneration of solid adsorbents at simulated flue gas condition.9,22,49,52 In this paper, CO2 adsorption tests were carried out on a thermogravimetric analyser (TGA, Q500, TA instruments). In a typical test, the sample was first degassed at 150 °C for 30 min in N2 before cooling down to the designed temperature, and then the simulated flue gas (15%/85% CO2/N2) was introduced into the sample chamber with a flow of 100 mL min−1 and the sample was held isothermally for 60 min. The influence of temperature on the CO2 adsorption capacity of prepared samples was tested as follows: after 1 h exposure to simulated flue gas (15%/85% CO2/N2) at 25 °C, the sample was heated up to 100 °C at a heating rate of 0.5 °C min−1 with the same gaseous atmosphere, allowing for equilibrium adsorption capacity to be attained at each temperature. Up to 40 adsorption–desorption cycles were done to verify the stability and cyclability of the samples. CO2/N2 selectivity of selected sample was also tested on a thermogravimetric analyser (TGA, Q500, TA instruments). In a typical test, the sample was first degassed at 150 °C for 30 min in helium before cooling down to the designed temperature, and then the simulated flue gas (15%/85% CO2/He) or pure nitrogen was introduced into the sample chamber with a flow of 100 mL min−1 and the sample was held isothermally for 60 min. In addition, a TG-DSC instrument (Setaram SENSYS evo) was used to measure the heat of adsorption.3 Results and discussion
3.1 Preparation of the 3D hierarchical carbon materials
Heteroatom-doped carbon materials with 3D hierarchical structure is desirable for CO2 capture,27 as the macroscopic networks benefit the CO2 diffusion with reduced mass-transfer resistance while the ultra-microporous structure and strong surface chemistry facilitate the CO2 adsorption at low CO2 partial pressure and high flue gas temperature. However, integrating all those features together into one carbon material is challenging to synthesize. Here, our strategy is to use a cost-effective and commercially available nitrogen-rich polyisocyanurate foam with unique 3D connected pentagonal and hexagonal ring structure as precursor to prepare carbon adsorbents, using a facile one-step compaction–activation–potassium intercalation methodology (Fig. 1a). The morphology of the prepared samples was first analysed by scanning electron microscopy (SEM) and the results were shown in Fig. 1b–e. It can be seen from Fig. 1b and c that the carbon sample prepared from non-compacted carbonisation/activation (sample P71) has highly interconnected porous macro-structures. To further understand the microporous structure, TEM analysis of selected sample was also performed. As shown in Fig. 1f and g, slit-like ultra-micropores can be clearly seen in the TEM images, and the ultra-microporosity is dominantly distributed across the porous network skeleton of the PIR carbon. The 3D micro-CT scanning as shown in Fig. 1h and i reveals that the carbon network is characteristically architectured with cross-linked pentagonal and hexagonal ring structures, which are very similar to the original molecular geometry of the precursor feedstock from which the activated carbons are derived. As a result, the activated carbon prepared looks more like the ‘fossil’ or imprint of the precursor chemical used. However, after compaction, as shown in Fig. 1c and d, the cross-linked ring structures were forged to yield the much the smaller structural units that form the new densely textured carbon networks with highly developed sub-nano porosity. | ||
| Fig. 1 Morphology of prepared PIR carbons: (a) schematic of the synthesis route of PIR carbons; SEM of sample P71 (b and c) and P81_2T (d and e); TEM images of sample P71_2T (f and g); micro CT analysis of P71 (h and i). | ||
Fig. 2 shows the nitrogen isotherms obtained at −196 °C for all samples. It can be seen that with a compaction load being lower than 1.5 tons per cm2, the samples prepared at temperatures below 700 °C all show type I isotherms highly characterised by the sharp knees observed at relative pressures (P/P0) lower than 0.1 and virtually no additional adsorption of N2 took place at higher relative pressures as indicated by the plateaus observed, indicating the superior narrow ultra-microporosity of the materials. When the integrated carbonisation–activation temperature increased to 800 °C or the compaction load exceeded 5 tons, mesoporosity started to evolve in the resultant carbon materials as shown by the emerging of the hysteresis loops at higher relative pressures >0.4.
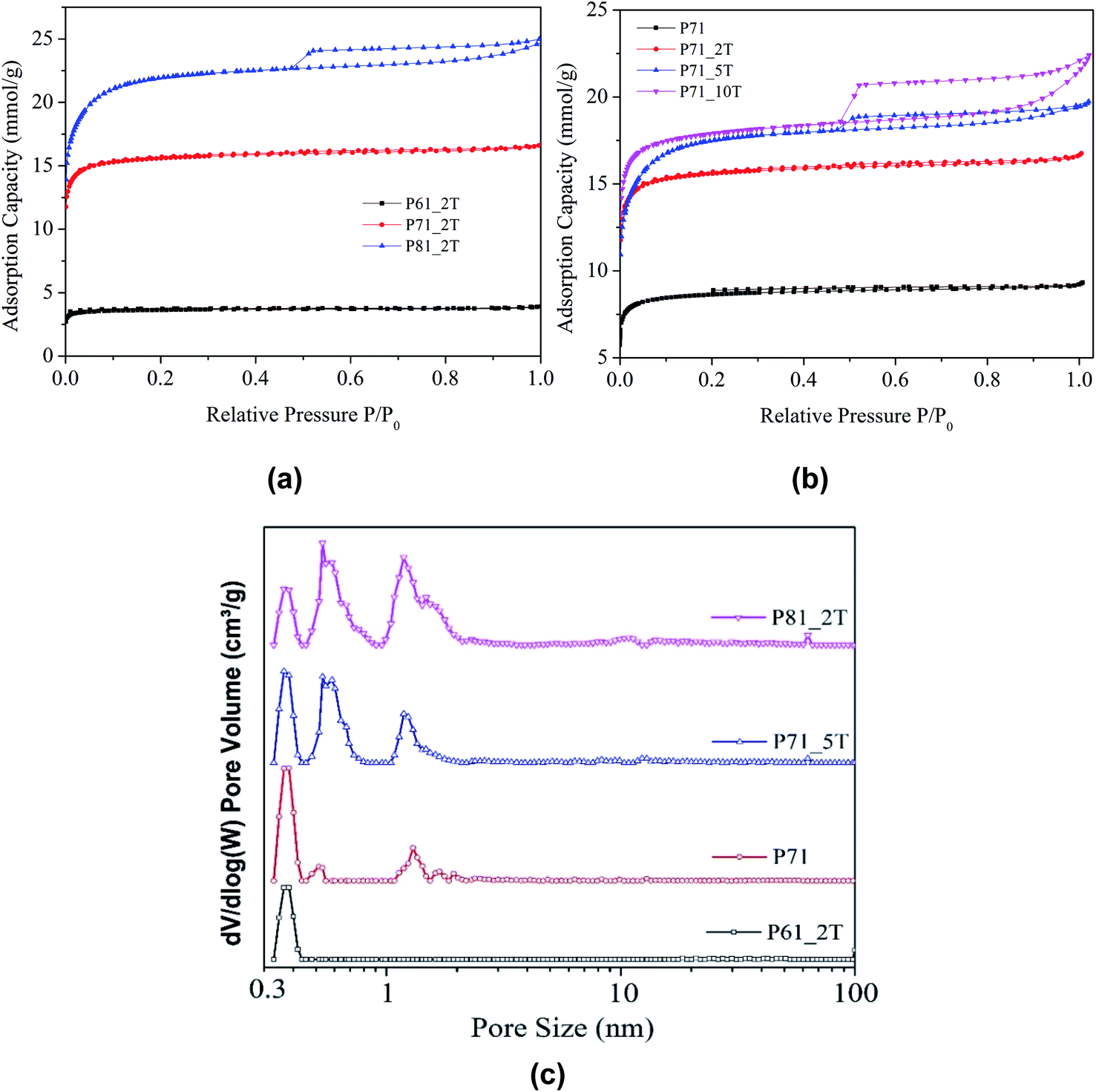 | ||
| Fig. 2 Textural properties of the prepared PIR carbons: (a and b) N2 adsorption isotherms obtained at −196 °C; (c) NLDFT pore size distributions for samples prepared at 700 °C with different compaction load. | ||
Table 1 shows the surface textural properties of the samples. It can be observed that the carbon materials have modest surface areas ranging from 328 to 1906 m2 g−1 and total pore volumes from 0.124 to 0.797 cm3 g−1, which both showed an increase with the temperature and compaction load used in the integrated compaction, carbonisation and activation protocol. The pore size distributions shown in Fig. 2c exhibit that the carbon materials derived from polyisocyanurates have exceptionally narrow ultra-microporosities with pore sizes centred at 0.37 nm, 0.53 nm and 1.2 nm, with the sample P61_2T having a pure single pore size centred at 0.37 nm, which has been rarely observed before for activated carbon materials. Previous investigations have revealed that the narrow ultra-micropores smaller than <0.7 nm is of primary importance in CO2 adsorption of carbonaceous sorbent materials.37,38 It can be found from Table 1 that increasing activation temperature and compaction pressure led to an increase in the ultra-micropore volume calculated for pores smaller than 0.7 nm. However, the ultra-micropore volume as a fraction of total micropore volume decreased slightly with increasing activation temperature, due to the pore widening effect as a result of the enhanced activation.
| Sample | S BET (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V micro <0.7 nm (cm3 g−1) | XPS | |
|---|---|---|---|---|---|---|
| N (at%) | K (at%) | |||||
| P61_2T | 328 | 0.124 | 0.124 | 0.124 | 3.6 | 5.1 |
| P71 | 773 | 0.298 | 0.280 | 0.254 | 1.8 | 6.5 |
| P71_2T | 1407 | 0.535 | 0.511 | 0.378 | 1.6 | 4.8 |
| P71_5T | 1392 | 0.540 | 0.498 | 0.372 | 1.2 | 5.7 |
| P71_10T | 1606 | 0.715 | 0.572 | 0.407 | 1.1 | 4.0 |
| P81_2T | 1906 | 0.797 | 0.702 | 0.342 | 0.5 | 8.7 |
| P81_5T | 1514 | 0.622 | 0.557 | 0.297 | 1.3 | 8.9 |
3.2 CO2 adsorption and desorption performances
To evaluate the CO2 adsorption performance under simulated flue gas conditions (15% CO2 in N2) at different temperatures varying from 40 °C to 100 °C, temperature-programmed adsorption tests were first carried out at an extremely slow heating rate of 0.5 °C min−1. The use of such a low heating rate is to achieve the near-equilibrium CO2 uptake capacity at each individual temperature in the temperature region examined. It can be seen that the PIR carbon materials prepared under compacted carbonization/activation conditions exhibited extraordinary CO2-capturing performance at low CO2 partial pressures across a range of favourable adsorption temperatures, with adsorption capacities in 15% CO2/N2 reaching an unprecedented level of 2.30 mmol g−1 at 40 °C, 2.13 mmol g−1 at 50 °C and 1.90 mmol g−1 at 70 °C, respectively. As physical adsorbents, the CO2 adsorption capacity of carbon materials is usually expected to decrease sharply with increasing adsorption temperatures, which limit their applicability at practical flue gas temperatures. As shown in Fig. 3a and S1,† no such unfavourable sharp deceases in adsorption capacity were observed for the PIR carbon adsorbent materials prepared from the compacted carbonisation/activation conditions, this indicating that the use of compaction before activation helped to achieve the high reversible CO2 capacities at high adsorption or flue gas temperatures. Of all the samples, the PIR carbon (P81_2T) prepared at 800 °C with a compaction load of 1.5 tons per cm2 showed the highest capacities at 40–70 °C. A comprehensive comparison with virtually all the previously reported carbon adsorbent materials, over 140 activated carbons (Table S1†), at different adsorption temperatures shows that the adsorption capacity of PIR carbons outperforms all carbons reported so far particularly at high adsorption temperatures (≥40 °C). At adsorption temperature of 70 °C, the adsorption capacity of P81_2T was found to be still able to accomplish a marvellous CO2 uptake of ca. 1.90 mmol g−1, outperforming the solid-supported polyamine chemical adsorbents which are generally recognised as being the most suitable sorbents for use at practical flue gas temperatures56,57 to guarantee the required CO2 capture capacities. This indicates the extraordinary favourable surface affinity of the PIR carbons to CO2 molecules.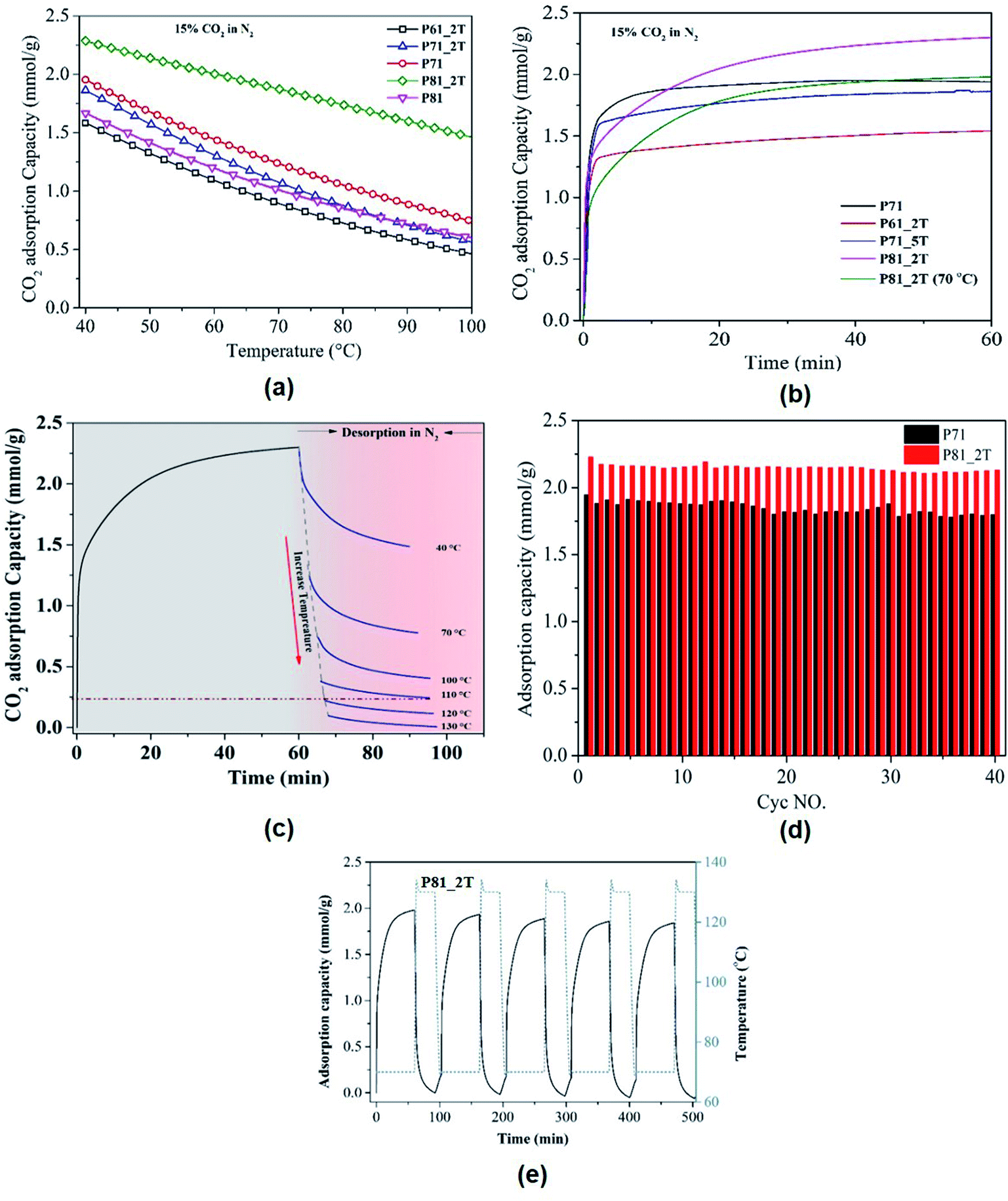 | ||
| Fig. 3 CO2 adsorption profiles: (a) the change of CO2 adsorption capacity of PIR carbons with increasing adsorption temperature in simulated flue gas condition (15% CO2 + 85% N2); (b) kinetic adsorption curves of different PIR carbons (adsorption at 40 °C and 0.15 bar CO2); (c) CO2 desorption profiles of P81_2T in N2 at different temperatures; cyclic temperature-swing adsorption–desorption performance of selected PIR carbons in 15% CO2/N2 at 40 °C (d) and at 70 °C (e). | ||
Fig. 3b shows the adsorption and desorption profiles of selected carbon materials obtained from TGA at a temperature of 40 °C and 70 °C (P81_2T) and CO2 partial pressure of 0.15 bar. It can be found that all samples exhibit fast adsorption kinetics with the times to achieve 80% and 90% of their equilibrium capacities being just around 1 min and 5 min in the TGA conditions, respectively. It is interesting to note that the sample P81_2T, which shows the highest CO2 capacity, exhibits an adsorption profile very similar to those of polyamine-based chemical adsorbents, which usually require longer time to reach their adsorption equilibria compared to physical adsorbents due to the effect of steric hindrance.21,58 To evaluate the CO2 desorption properties of the PIR carbons, the CO2 desorption profiles of a selected PIR carbon sample (P81_2T) was examined at different desorption temperatures in N2, with the results shown in Fig. 3c. It can be seen that while the amount of desorbed CO2 increased with increasing temperature and all the CO2 can be desorbed completely at a final desorption temperature of 130 °C, it is noteworthy that nearly 90% of the adsorbed CO2 can be desorbed at a relatively low temperature of 110 °C, leading to potentially significantly energy savings in sorbent regeneration compared to the supported polyamine chemical adsorbents that usually need considerably higher temperatures to regenerate the capture materials or desorb the adsorbed CO2.59,60Fig. 3d and e presents the temperature swing cyclic adsorption–desorption testing results for the PIR carbons, which highlights the superior adsorption reversibility and durability of the PIR carbon materials for CO2 capture.
It is generally believed that the narrow micropores and surface heteroatoms of carbon materials favors the CO2 adsorption of carbon materials at high temperatures and low partial pressures, due to the enhanced interaction force between CO2 and carbon surface.36–38 It has been found previously that there exist critical pore sizes that determine the CO2 adsorption capacity of carbon materials at specific temperatures and/or CO2 partial pressures, with the critical pore size for CO2 adsorption decreasing with increasing adsorption temperatures or decreasing CO2 partial pressures. For instance, pores smaller than 0.44 nm are responsible for CO2 adsorption at 0.15 bar and 25 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31,38,40 while they are reduced to 0.40 nm as the adsorption temperature increases to 50 °C.31 Indeed, a linear relationship was obtained between the ultra-micropore (≤0.40 nm) volumes and the CO2 adsorption capacity at 40 °C and 0.15 bar (see Fig. S2†) for the PIR carbons prepared at 600–700 °C, which agrees well with previous findings on carbon materials. However, similar relationship was not found the PIR carbons prepared at 800 °C, which have the lowest ultra-micropore volumes but showed substantially higher CO2 capacities, implying that the porous structures alone cannot possibly account for the unusually high CO2 uptakes obtained for the PIR carbons.
31,38,40 while they are reduced to 0.40 nm as the adsorption temperature increases to 50 °C.31 Indeed, a linear relationship was obtained between the ultra-micropore (≤0.40 nm) volumes and the CO2 adsorption capacity at 40 °C and 0.15 bar (see Fig. S2†) for the PIR carbons prepared at 600–700 °C, which agrees well with previous findings on carbon materials. However, similar relationship was not found the PIR carbons prepared at 800 °C, which have the lowest ultra-micropore volumes but showed substantially higher CO2 capacities, implying that the porous structures alone cannot possibly account for the unusually high CO2 uptakes obtained for the PIR carbons.
To understand the nature of the functional groups on the carbons, XPS was performed. It can be seen from Table 1 and Fig. S3 and S4† that nitrogen containing groups61–63 and surface potassium oxide species64,65 exist in PIR carbons. Fig. S2† shows the correlation between CO2 adsorption capacities and the contents of different heteroatom functionalities. Apparently, no direct relationship is evident between the CO2 adsorption capacity and the content of nitrogen or nitrogen-containing functionalities but instead, the high adsorption capacity of these PIR carbon materials appears to be directly linked to the presence of potassium in the carbons, which is believed to present in intercalated forms within the carbon matrices. For instance, sample P81_2T and P81_5T, which have the highest potassium contents, was found to have the highest CO2 uptakes among all the PIR carbons. To verify the role of the intercalated potassium on CO2 adsorption, sample P81_2T, P71 and P71_5T were subjected to Soxhlet extraction with hot water for 48 h as a means to enhance the removal of the intercalated potassium. As shown in Fig. 4, the CO2 capacity of the Soxhlet-extracted PIR carbons at 40 °C in 15% CO2/N2 was found to decrease drastically by >50%, from 2.30 to 1.05 mmol g−1 for P81_2T_48H and from 1.96 to 0.75 mmol g−1 for P71_48H (Fig. 4b), as the content of intercalated potassium was reduced sharply from 8.70 to 2.13 at% for P81_2T and from 6.45 to 1.7 at% for sample P71. Similar trend was also observed for sample P71_5T. This highlights the vital importance of the intercalated potassium in determining the unusually high performance of the ultra-microporous PIR carbons for CO2 adsorption.
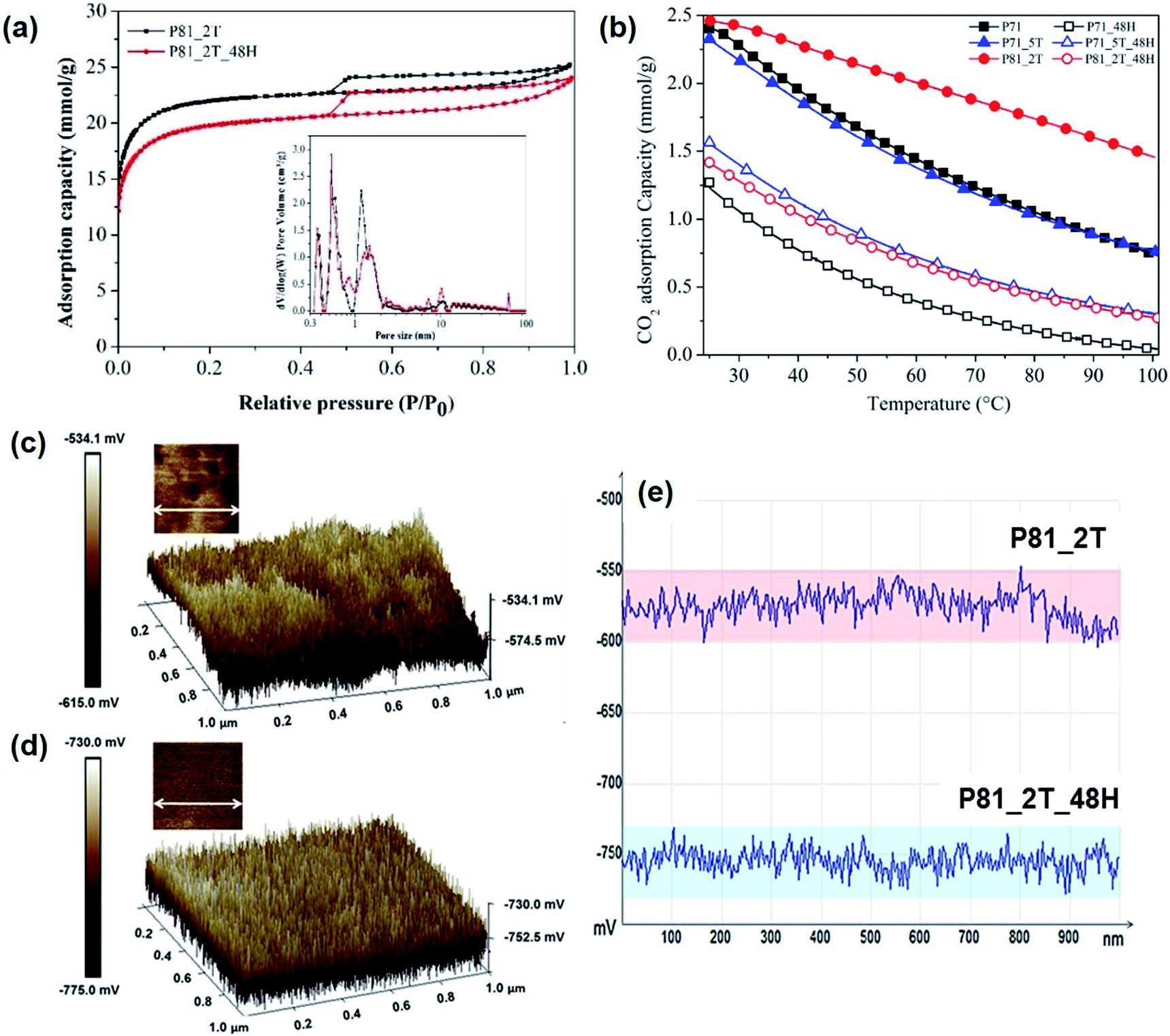 | ||
| Fig. 4 The underlying role of surface chemistry and porous structures of the PIR carbons in CO2 adsorption: (a) the surface textural properties of selected PIR carbons (P81_2T and P81_2T_48H); (b) CO2 adsorption profiles of selected PIR carbons before and after the 48 h Soxhlet extraction; KPFM electric surface potential map of selected PIR carbons before and after enhanced removal of intercalated potassium (c) P81_2T carbon, (d) P81_2T_48H; (e) the variations of surface potential distribution of selected PIR carbons along the selected horizontal line. | ||
It is believed that the vital role of the intercalated potassium in CO2 adsorption observed for the PIR carbons is accomplished via the presence of extra framework potassium ions, which appears to be significantly enhanced due to the ultra-microporous structure and hence the density of the edge sites in the PIR carbons, rather than a mechanism via the formation of carbonate and/or bi-carbonate. This is because the formation of carbonate is obviously irreversible at such low desorption temperatures whereas the carbonate–bicarbonate cycle cannot possibly proceed under the dry flue gas conditions. Previous studies tend to suggest that a significant proportion of the intercalated potassium present in alkali-activated carbon materials may present in the form of extra framework ions,40,51 similar to those commonly observed in MOFs and zeolite materials that essentially determine their adsorptive properties for CO2.66–68 The extra-framework ions can create strong local electric fields that can polarize adsorbate molecules and hence provide either additional active adsorption sites and/or enhance the interaction of the adsorbate with the adsorption surface. As a result, gases like CO2 with large electrical quadrupole moment will be preferentially polarised and adsorbed onto the surface, thus giving rise to enhanced performance for selective CO2 adsorption. However, no direct evidence has been revealed so far regarding the presence of the extra framework ions in carbon materials as a result of potassium intercalation during alkali-activation. Therefore, kelvin probe force microscopy (KPFM) was used in this investigation to examine the presence of extra framework ions and hence their role in CO2 adsorption by measuring the electric surface potential of the PIR carbons, which is obtained as the potential difference (CPD) between the probe tip and the carbon surface (Φsample − Φtip).69,70Fig. 4c–e, S5 and Table S2† show the measurement results for the selected P81_2T carbon sample before and after the 48 hours' continuous Soxhlet extraction. It is evident that the P81_2T and P81_2T_48H carbons both exhibit high levels of heterogeneities in electric field distribution both on the basal surfaces and along the edges of the textured structures, with the degree of electrical heterogeneity along both the lateral and vertical directions being significantly greater for the non-extracted P81_2T carbon. More importantly, it was found that the overall electric surface potential of the P81_2T sample, which has a significantly higher content of intercalated potassium, was significantly greater than that of the Soxhlet-extracted P81_2T_48H carbon by as much as 153 ± 20 mV. This highlights the vital role of the intercalated potassium in the PIR carbon materials in creating strong local electrostatic fields, which are well known for their importance in CO2 adsorption due to their strong preferential polarizing effect on CO2 over N2 molecules.66–68
In order to better understand the interaction strength between CO2 and the carbon surface with the existence of potassium heteroatoms, the heat of CO2 adsorption of the PIR carbons was measured by TG-DSC under a simulated flue gas condition at 40 °C and 15% CO2/85% N2, and the results were shown in Fig. S6.† It can be found that the heat of adsorption for the prepared samples ranged from 33.0 kJ mol−1 to 40.7 kJ mol−1 and showed a general decrease with increasing activation temperatures, which corresponds well to the increased porosity development and hence the increased role of surface textural properties in adsorption. The adsorption heat obtained by TG-DSC, which represents the average heat of adsorption, differs from the isosteric heat of adsorption often used in previous studies, which is a function of surface coverage usually calculated from using adsorption isotherms. It was found that the heat of adsorption of the best-performing PIR carbons, which varied from 36 to 40.7 kJ mol−1 CO2, is clearly significantly higher than those of any other carbon materials,19,40,50,51 and many zeolites and MOFs reported so far,14 being indicative of the strong interaction strength of the adsorbed CO2 phase with the carbon surface. As shown in Fig. S6†, the 48 hours' Soxhlet extraction with hot water led to a decrease in the heat of adsorption by nearly 15%, from ca. 36 kJ mol−1 to 31 kJ mol−1 for samples P71 and P71_5T, indicating the contribution of intercalated potassium. However, the heat of the adsorption does not appear to correlate directly with the bulk intercalated potassium contents but instead linearly with the density of the surface distribution of the potassium, which is defined as the ratio of the bulk potassium content to the surface area of the PIR carbons (Fig. S6†). This suggests that for a given potassium content, the interaction strength of CO2 molecules with the intercalated potassium atoms in the PIR carbons is stronger in smaller pores than in larger pores. The y-intercept of 31 kJ mol−1 CO2 obtained from the linear relationship, which indicates the contributions of the surface textural properties, is also significantly higher than those of other microporous carbon materials reported (15 kJ mol−1 to 25 kJ mol−1),44 being indicative of the stronger adsorption force field created by the exceedingly high ultra-microporosity of the PIR carbons. In general, it is evident that the high unimodal ultra-microporosity structure coupled with the surface chemistry arising from potassium intercalation is responsible for the high CO2 capacities achieved by the PIR carbons at high flue gas temperatures and low CO2 partial pressures.
To be a desirable candidate sorbent for CO2 capture, the sorbent must also have both high selectivity in addition to adsorption capacity. In this work, the dynamic CO2/N2 selectivity of selected carbon sample P81_2T at 40 and 70 °C was evaluated by using 15% CO2 in helium and pure nitrogen as feed gas on TGA and the results were shown in Fig. 5. The CO2/N2 selectivity was calculated as the ratio between CO2 adsorption capacity in 15% CO2/N2 and nitrogen adsorption capacity in pure nitrogen. The calculations demonstrate that in addition to the record high CO2 capacities at low CO2 partial pressure and high flue gas temperatures, the best-performing PIR carbon (P81_2T) also achieved high CO2/N2 selectivites of up to 115:1 and 117:1 at 40 and 70 °C in the first minute, respectively. With increasing adsorption time to 50 min, the CO2/N2 selectivity continuously decreased to about 40:1, due to the fact that the nitrogen adsorption capacity linearly increased with adsorption time whereas the adsorption rate of CO2 was much faster than that of N2. A comparison with previous investigations on carbon materials for CO2 capture shows that showed that the CO2/N2 selectivity of P81_2T was among the highest ever reported for carbon materials.27,31,50 A careful examination of the results suggest that the high selectivity of the PIR carbon materials originates from a combination of their narrow ultra-microporosity, which have the pore sizes close to the dynamic molecular diameter of CO2 molecules, and highly CO2-preferential polarising surfaces as a result of the enhanced formation of surface extra-framework cations, which occurred as a co-benefit of the integrated carbonisation and alkali-activation under the compacted conditions.
 | ||
| Fig. 5 Evaluation of CO2/N2 selectivity: (a) the adsorption curves of P81_2T in simulated flue gas (15% CO2 + 85% He) and pure nitrogen at 40 and 70 °C, respectively; (b) CO2/N2 selectivity of P81_2T at 40 and 70 °C. | ||
4 Conclusions
Activated carbon materials able to achieve high CO2 capture capacity and selectivity at high flue gas temperatures and low CO2 partial pressure have been developed via a facile integrated compaction–combined carbonisation–chemical activation protocol, using waste PIR materials available in large quantities. The PIR carbon materials for carbon capture are highly characterised not only by their essentially high ultra-microporosities but also by their highly CO2-preferential polarising surfaces as a result of the enhanced formation of extra-framework ions within the ultra-microporous carbon networks, which occurred naturally as co-benefit of the integrated carbonisation and alkali-activation protocol under compacted conditions. At a low CO2 partial pressure of 0.15 bar and high flue gas temperatures of 40–70 °C, the materials exhibit extraordinary reversible CO2 uptake capacities of up to 2.3 mmol g−1, which represent the highest CO2 uptakes ever reported for any activated carbon, MOF and many polyamine-based chemical adsorbent materials. In addition to the exceedingly high capacities, the materials also show high CO2/N2 selectivities, which can reach up to 115:1 and 117:1 at 40 and 70 °C, respectively. The results highlight the importance of a combination of the ultra-microporosity and surface chemistries in determining the performance of carbon-based capture materials at high flue gas temperatures and low CO2 partial pressures. The performance of the PIR carbon materials essentially changes the perception that active carbon materials as physical adsorbents cannot be used for highly efficient CO2 capture at low CO2 partial pressures and high flue gas temperatures.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant number EP/R001308/1], UK and the UK Carbon Capture and Storage Research Centre (EP/K000446/1 and flexible funded research programme). The UKCCSRC is supported by the EPSRC as part of the UKRI Energy Programme. We thank Dr Elisabeth Steer and Dr Nicola Weston for her kind assistance with SEM measurements and Dr Martin Roe for his help with TEM measurement. We also thank Dr Martin Corfield for his kind assistance with nano-CT analysis.References
- IEA, CO2 emissions from fuel combustion Highlights 2016, 2016 Search PubMed.
- IPCC, Climate Change 2014: Mitigation of Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014 Search PubMed.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Carbon dioxide capture in metal–organic frameworks, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. C. Ferrari, R. Gross, J. P. Hallett and R. S. Haszeldine, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell and B. Vlaisavljevich, Nature, 2015, 519, 303 CrossRef CAS PubMed.
- L. C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633 CrossRef CAS PubMed.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- J. G. Vitillo, B. Smit and L. Gagliardi, Chem. Rev., 2017, 117, 9521–9523 CrossRef CAS PubMed.
- T. H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 RSC.
- N. Konduru, P. Lindner and N. M. Assaf-Anid, AIChE J., 2007, 53, 3137–3143 CrossRef CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339–8344 CrossRef CAS.
- Z. Zhang, Z. Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS.
- Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Martineau, F. Taulelle, P. L. Llewellyn and H. Jobic, Angew. Chem., Int. Ed., 2013, 125, 10506–10510 CrossRef.
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov, O. Terasaki and K. B. Yoon, Science, 2015, 350, 302–306 CrossRef CAS PubMed.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H. C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- J. W. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W. G. Bae, L. Pan, J. B. H. Tok and J. Wilcox, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed.
- K. Min, W. Choi, C. Kim and M. Choi, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. Sci., 2014, 116, 306–316 CrossRef CAS.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A. H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 RSC.
- B. Ashourirad, P. Arab, T. Islamoglu, K. A. Cychosz, M. Thommes and H. M. El-Kaderi, J. Mater. Chem. A, 2016, 4, 14693–14702 RSC.
- M. G. Plaza, A. S. González, J. J. Pis, F. Rubiera and C. Pevida, Appl. Energy, 2014, 114, 551–562 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- M. Terhan and K. Comakli, Appl. Therm. Eng., 2016, 100, 1007–1015 CrossRef.
- D. Che, Y. Liu and C. Gao, Energy Convers. Manage., 2004, 45, 3251–3266 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 RSC.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523–2529 RSC.
- V. Presser, J. McDonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS PubMed.
- X. Liu, Y. Sun, J. Liu, C. Sun, H. Liu, Q. Xue, E. Smith and C. Snape, ACS Appl. Mater. Interfaces, 2017, 9, 26826–26839 CrossRef CAS PubMed.
- B. Adenira and R. Mokaya, Chem. Mater., 2016, 28, 994–1001 CrossRef.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 82, 297–303 CrossRef CAS.
- S. Gadipelli and Z. X. Guo, ChemSusChem, 2015, 8, 2123–2132 CrossRef CAS.
- S. Himeno, T. Komatsu and S. Fujita, J. Chem. Eng. Data, 2005, 50, 369–376 CrossRef CAS.
- Q. Cai, Z. H. Huang, F. Kang and J. B. Yang, Carbon, 2004, 42, 775–783 CrossRef CAS.
- W. Shen and W. Fan, J. Mater. Chem. A, 2013, 1, 999–1013 RSC.
- D. L. Sivadas, S. Vijayan, R. Rajeev, K. N. Ninan and K. Prabhakaran, Carbon, 2016, 109, 7–18 CrossRef CAS.
- W. Xing, C. Liu, Z. Zhou, L. Zhang, J. Zhou, S. Zhuo, Z. Yan, H. Gao, G. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323–7327 RSC.
- M. Sevilla, P. Valle-Vigón and A. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- J. H. Lee, H. J. Lee, S. Y. Lim, B. G. Kim and J. W. Choi, J. Am. Chem. Soc., 2015, 137, 7210–7216 CrossRef CAS PubMed.
- J. Liu, N. Sun, C. Sun, H. Liu, C. Snape, K. Li, W. Wei and Y. Sun, Carbon, 2015, 94, 243–255 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 RSC.
- X. Fan, L. Zhang, G. Zhang, Z. Shu and J. Shi, Carbon, 2013, 61, 423–430 CrossRef CAS.
- X. Ma, Y. Li, M. Cao and C. Hu, J. Mater. Chem. A, 2014, 2, 4819–4826 RSC.
- G. K. Parshetti, S. Chowdhury and R. Balasubramanian, Fuel, 2015, 148, 246–254 CrossRef CAS.
- K. Li, J. Jiang, S. Tian, F. Yan and X. Chen, J. Mater. Chem. A, 2015, 3, 2166–2175 RSC.
- M. W. Hahn, J. Phys. Chem. C, 2015, 119, 4126–4135 CrossRef CAS.
- K. S. N. Kamarudin and N. O. R. M. A. Alias, Fuel Process. Technol., 2013, 106, 332–337 CrossRef CAS.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. J., 2014, 251, 293–303 CrossRef CAS.
- Y. Sun, X. Liu, C. Sun, W. Al-Sarraf, K. Z. Foo, Y. Meng, S. Lee, W. Wang and H. Liu, J. Mater. Chem. A, 2018, 6, 23587–23601 RSC.
- J. M. Gu, W. S. Kim, Y. K. Hwang and S. Huh, Carbon, 2013, 56, 208–217 CrossRef CAS.
- W. T. Zheng, K. Z. Xing, N. Hellgren, M. Lögdlund, Å. Johansson, U. Gelivs, W. R. Salaneck and J. E. Sundgren, J. Electron Spectrosc. Relat. Phenom., 1997, 87, 45–49 CrossRef CAS.
- A. Adenier, E. Cabet-Deliry, A. Chaussé, S. Griveau, F. Mercier, J. Pinson and C. Vautrin-Ul, Chem. Mater., 2005, 17, 491–501 CrossRef CAS.
- J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer, Eden Prairie, MN, 1992 Search PubMed.
- A. V. A. K. Shchukarev and D. Korolkov, ChemistryOpen, 2004, 2, 347–362 CAS.
- G. D. Pirngruber, P. Raybaud, Y. Belmabkhout, J. Čejka and A. Zukal, Phys. Chem. Chem. Phys., 2010, 12, 13534–13546 RSC.
- A. Pulido, P. Nachtigall, A. Zukal, I. Domínguez and J. Čejka, J. Phys. Chem. C, 2009, 113, 2928–2935 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. A. C. H. I. N. Chavan, G. T. Palomino, C. O. Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185–11191 CrossRef CAS.
- V. Palermo, M. Palma and P. Samorì, Adv. Mater., 2006, 18, 145–164 CrossRef CAS.
- J. Lv, L. Eng, R. Bennewitz, E. Meyer, H. J. Güntherodt, E. Delamarche and L. Scandella, Surf. Interface Anal., 1999, 27, 368–373 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta01417f |
| This journal is © The Royal Society of Chemistry 2020 |