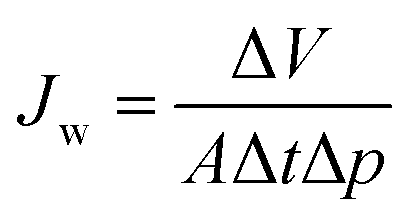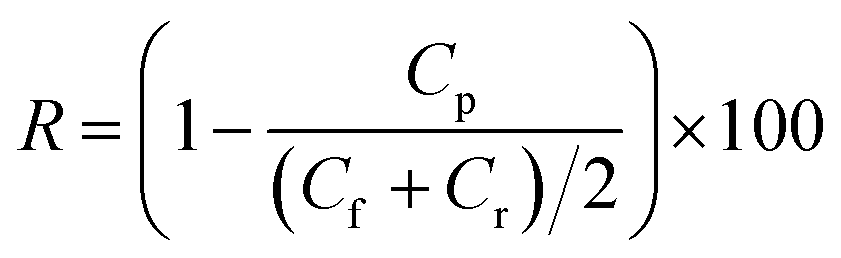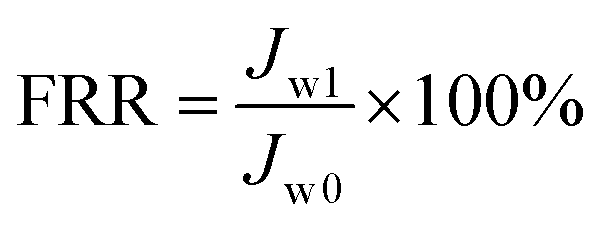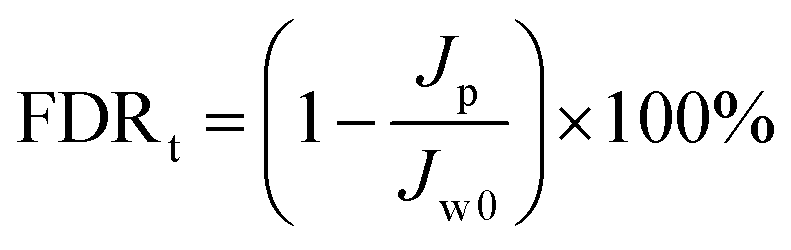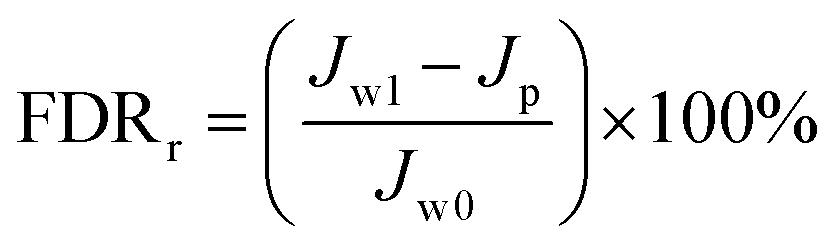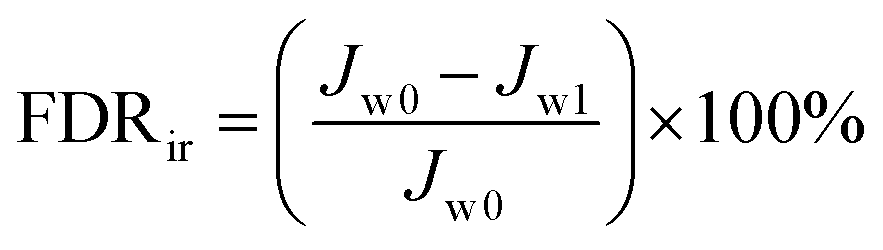High-performance asymmetric isoporous nanocomposite membranes with chemically-tailored amphiphilic nanochannels†
Received
25th January 2020
, Accepted 16th March 2020
First published on 16th March 2020
Abstract
Tailoring the well-defined nanochannels of ultrafiltration membranes from self-assembled block copolymers (BCPs) toward the nanofiltration regime can expand their potential applications in the fractionation or separation of small molecules. One big issue is to fabricate a nanofiltration membrane with high permeance, good selectivity and excellent fouling resistance. Here such a membrane is presented using a tailor-made diblock copolymer composed of a hydrophobic major block and an amphiphilic minor block. A scalable integral asymmetric isoporous membrane is fabricated by the solvent evaporation induced co-assembly of BCP and in situ formed inorganic titanium dioxide nanoparticles combined with nonsolvent induced phase separation. The membrane nanopores are readily post-functionalized using negatively charged moieties by straightforward in situ gas–solid reactions. The potential to use the post-functionalized membrane for separation of small organic molecules having 1–2 nm lateral dimensions (having molecular weights in the range of 300–1500 g mol−1) is demonstrated. The negatively charged membrane displays high flux, excellent antifouling properties with a low permeation flux decline and nearly complete flux recovery. This type of membrane is a promising candidate for a new generation of nanofiltration membranes.
1. Introduction
Membrane-based separation offers a scalable, economically and environmentally friendly tool for numerous applications, e.g., biochemical and pharmaceutical processing, water purification, and wastewater treatment.1–5 Although the interest in developing high-performance membranes has been growing from diverse inorganic, organic, and composite materials,6–13 a long-standing goal in membrane technology is still to pursue an ideal membrane with a high permeance, good selectivity and strong fouling resistance.
Block copolymers (BCPs), an intriguing class of hybrid macromolecules, provide a very strong platform for the preparation of next generation membranes with high porosity and narrow pore size distribution, attributed to their well-known self-assembly into a variety of well-defined nanostructures.14–19 Such membranes based on BCP self-assembly have been generated through selective removal of the minority blocks20–22 or blend partners3,23 from bulky BCP thin films with multiple treatment steps. A fascinating one-step scalable approach to translate the periodic ordered nanostructure of BCPs into high-performance membranes employs the evaporation induced self-assembly together with the non-solvent induced phase separation (SNIPS).24,25 SNIPS membranes typically possess a rather thin (<200 nm) selective layer with a high porosity (>1014 pores per m2) of ordered vertically-aligned cylindrical pores above a macroporous sublayer.26–31 Such integral asymmetric isoporous structure can achieve a high permeance attributed to the rather thin selective layer and the macroporous substructure, while ensuring a good selectivity due to the uniform pore size from BCP self-assembly. However, most BCP membranes to date have been fabricated with pore sizes in the ultrafiltration regime, ascribed to the limitation of the intrinsic length scale of the microphase-separated nanostructure (ca. 10–100 nm). This is due to limitations in lowering the chain length of block copolymers, when they should still microphase separate upon removal of solvent. Although microphase separation can still occur in short block copolymers with very strong repulsive interactions between the dissimilar blocks, the mechanical properties generally will become very poor due to lack of entanglements. Therefore the development of ultrafiltration membranes toward the nanofiltration regime, which can differentiate between small molecules would expand their potential applications in the separation of chemical,32,33 pharmaceutical34 and biological35 molecules having dimensions of ca. 0.5–5 nm. A recent promising trend is to obtain soft nanochannels by taking advantage of the swelling of the pore-forming block at the hydrated state that are confined within the mesopores of SNIPS membrane.36–39 The water permeance of the resulting SNIPS membranes with swelled soft nanochannels reported so far is in a range of 0.6–15 L m−2 h−1 bar−1. There is a critical need to prepare SNIPS membranes with a higher permeance and good selectivity in the nanofiltration regime. In a previous work we have demonstrated that the hydrated channels of a quaternized polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) membrane can be tuned from the ultra- to the nanofiltration regime gradually by changing the quaternization agent and the degree of functionalization.40
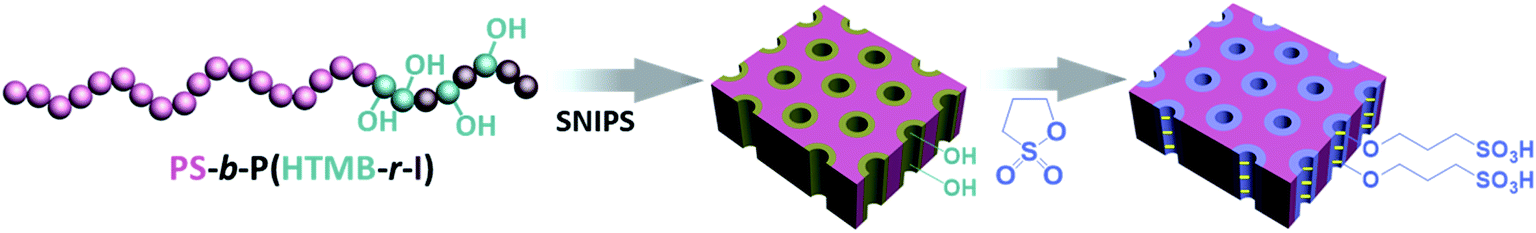
SNIPS membranes are mostly fabricated from amphiphilic block copolymers with at least one hydrophobic and one or more hydrophilic segment,41e.g. PS-b-P4VP,24,42,43 polyisoprene-block-polystyrene-block-poly(4-vinylpyridine) (PI-b-PS-b-P4VP),44,45 polyisoprene-block-polystyrene-block-poly(N,N-dimethylacrylamide),36,37 polystyrene-block-poly(4-vinylpyridine)-block-poly(solketal methacrylate),46 polystyrene-block-poly(4-vinylpyridine)-block-poly(ethylene oxide).47 In these cases, the pore-forming moieties consist of completely hydrophilic units. Recently we reported on a PI-b-PS-b-P4VP triblock terpolymer with partially functionalized PI blocks, which leads to a membrane with multifunctional pores built by the two mixed end blocks.48 In this study, we prepared a novel tailor-made diblock copolymer, polystyrene-block-poly(4-(2-hydroxyethyl-thio)-2-methyl butene-random-4-(2-hydroxyethyl-thio)-3-methyl butene-random-isoprene) (PS-b-P(HTMB-r-I)). It consists of a hydrophobic major block PS and a minor block P(HTMB-r-I) with randomly distributed isoprene (I), 4-(2-hydroxyethyl-thio)-2-methyl butene and 4-(2-hydroxyethyl-thio)-3-methyl butene (HTMB) units. HTMB units are relatively hydrophilic due to hydroxyl groups while I units are hydrophobic. Due to this feature, the minor block P(HTMB-r-I) itself can be considered an amphiphilic block. Herein, we report the preparation of an integral asymmetric isoporous membrane via SNIPS using the PS-b-P(HTMB-r-I) which has a hydrophobic major block and an amphiphilic minor block instead of the conventional block copolymers used for SNIPS, e.g. PS-b-P4VP and PS-b-PEO49 which have a hydrophobic major block and a hydrophilic minor block. In addition, we address the often encountered problem of a rather dense interface between the bottom of the block copolymer membrane and the substrate by introducing a titania precursor solution into the block copolymer solution prior to membrane casting. The in situ formed inorganic nanofillers lead to an open porous substructure of high mechanical stability, while other methods, e.g. the addition of poly(ethylene glycol), a typical porogen, did not lead to the desired result. The –OH groups of P(HTMB-r-I) along the pore walls were converted to sulfonic acid moieties using an in situ scalable gas–solid interface reaction (Fig. 1). The sulfonated membrane exhibit a high water permeance and the potential to separate anions having 1–2 nm lateral dimensions from each other. Additionally, antifouling performance was assessed in static adsorption and dynamic filtration, respectively, demonstrating the excellent fouling resistance of the prepared membranes. Our aim here is not only to provide a new nanofiltration membrane for efficient separation of small molecules, but also to broaden the range of block copolymers suitable for the SNIPS process which can further promote the application of SNIPS membranes.
 |
| | Fig. 1 Schematic representation of the top layer of an integral asymmetric isoporous membrane with amphiphilic pores and the in situ integration of negatively charged moieties along the pore walls. | |
2. Experimental
2.1 Synthesis of diblock copolymers PS-b-P(HTMB-r-I)
Diblock copolymers PS-b-P(HTMB-r-I) were prepared by living anionic polymerization with subsequent thiol–ene click chemistry. First, a poly(styrene-block-isoprene) (PS-b-PI) diblock copolymer was synthesized by sequential living anionic polymerization following a previously published procedure.50,51 The polyisoprene (PI) block of PS-b-PI was subsequently hydroxylated by a thiol–ene click reaction to obtain the P(HTMB-r-I) with various degrees of hydroxylation (DH) following a reported procedure.48 Detailed description is shown in the Section 1.2 of expanded experimental part (ESI†).
2.2 Preparation of polymer films and membranes
Thin films were prepared via spin-coating of a 2 wt% polymer solution in CHCl3 on a silicon wafer at 3000 rpm for 1 min. Integral asymmetric isoporous membranes were prepared by using the SNIPS technique following a published procedure,48 described in the ESI.† Titanium dioxide (TiO2) sol nanoparticles (NPs) as an inorganic additives were in situ prepared by hydrolysis of TiO2 precursors (titanium(IV) isopropoxide, TTIP) in an aqueous environment, following a reported procedure.52,53
2.3 Sulfonation post-functionalization of membranes
The membranes were further in situ post-functionalized using 1,3-propane sultone vapor, following our reported procedure.48 Briefly, the sulfonation reaction was accomplished by placing the PS-b-P(HTMB-r-I) derived membranes into a evacuated desiccator with a predetermined amount of 1,3-propane sultone at 50 °C for 14 h.
2.4 Characterization
1H NMR measurements were performed on a Bruker Advance 300 NMR spectrometer at 300 MHz using deuterated chloroform (CDCl3) as a solvent. Molecular weights and dispersity indices of the polymers were determined by gel permeation chromatography (GPC). Fourier transform infrared spectroscopy (FTIR) was conducted using a Bruker Alpha (diamond-ATR unit). Transmission electron microscopy (TEM) was performed on a Tecnai G2 F20 (FEI, Eindhoven, The Netherlands) with an acceleration voltage of 120 kV in bright-field mode. The samples were cut at room temperature to approx. 50 nm thin sections using a Leica EM UCT ultra-microtome (Leica Microsystems, Wetzlar, Germany) equipped with a diamond knife. The porous membranes were embedded in epoxy resin before cutting into ultrathin slides. Atomic force microscopy (AFM) was carried out on a Bruker MultiMode 8 AFM (NanoScope IV controller) operated in tapping mode at ambient conditions, using commercial silicon RTESP-150 tips (Bruker, USA). Scanning electron microscopy (SEM) was carried out on a LEO Gemini 1550 VP (ZEISS, Oberkochen, Germany) at a voltage of 3 kV or 5 kV. The samples were coated with ca. 2.0 nm platinum. Cross sections of the membranes were prepared by dipping the membrane in isopropanol, freezing in liquid nitrogen and finally cracking. Backscattered electron (BSE) images were taken on a Merlin (ZEISS) at a voltage of 3 kV. In this case the samples were coated with 6.0 nm carbon. Average pore size values were determined using the software analySIS (Olympus) on the basis of the SEM results. Elemental analysis of the membranes was performed by energy disperse X-rays (EDX) with the Merlin (ZEISS) scanning electron microscope. Thermogravimetric analysis (TGA) was accomplished using a TGA-DSC2 Thermogravimetric Analyzer (Mettler-Toledo) over the range of 25 to 1000 °C with a heating rate of 10 K min−1 in an argon atmosphere. Differential scanning calorimetry (DSC) was carried out with a differential scanning calorimeter DSC1 (Mettler-Toledo) in a temperature range between −50 °C and 200 °C under nitrogen atmosphere and at a heating rate of 20 K min−1. Membrane surface zeta potential was determined using a SurPASS 3 electrokinetic analyzer (Anton Paar, Austria) with a background of 1 mM NaCl solution. Dynamic water contact angle was measured with 1 μL water droplets on a KRUESS Drop Shape Analysis System DSA 100.
2.5 Membrane performance test
Water permeance measurements were performed in dead-end mode using a home-made automatic testing device at a transmembrane pressure (Δp) of 1 bar at room temperature. The normalized water permeance (Jw) was calculated by normalizing the flux by the transmembrane pressure as follows:| | 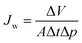 | (1) |
Additionally, in order to assess the mechanical stability of the porous structure of inorganic–organic hybrid membranes, different transmembrane pressures were employed to test the water flux from 0.25 to 2.9 bar at room temperature.
Separation performance was evaluated using a stirred test cell (EMD Millipore™ XFUF04701, effective membrane area 1.77 cm2) at a transmembrane pressure of 1 bar at room temperature. Aqueous solutions of model small molecules, i.e. orange II (OR−) and reactive green 19 (RG6−), were used at the concentrations of 0.1 mM. The concentration of the solutes in the feed solutions Cf (mg L−1), permeate solutions Cp (mg L−1) and retentate solutions Cr (mg L−1) was determined by a UV-vis spectrophotometer (GENESYS 10S, Thermo Scientific) and the retention (R, %) of the solutes was calculated using eqn (2):
| | 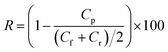 | (2) |
To quantify the figure of merit for molecular separation, we calculated the selectivity ψ, defined as the ratio of transmission of two species using eqn (3):
| | 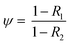 | (3) |
where
R1 and
R2 are the observed retention values of the two different solutes.
Antifouling performance was determined by the static adsorption and dynamic filtration using OR− and RG6− aqueous solutions as foulants. Adsorption measurements were accomplished following the published procedure48 (the detailed procedure is reported in the ESI†). The dynamic filtration of 0.1 mM aqueous solutions of the individual foulants was performed using the aforementioned dead-end stirred test cell at a transmembrane pressure of 1 bar at room temperature. Pure water initially passed through the membrane for 30 min and the average permeance was calculated as Jw0. The feed solution was then changed to the model foulant solution (0.1 mM OR− and RG6− aqueous solutions). The filtration of the foulant solution was conducted for 3 h while the corresponding permeate permeance (Jp) was recorded every 10 min. After filtration of the foulant solution, the membranes were washed with demineralized water two times for 20 min, then the pure water permeance of the cleaned membrane (Jw1) was measured again for 30 min. To evaluate the antifouling property in detail, several ratios were defined, i.e. the flux recovery ratio (FRR, %) in eqn (4), the total flux decline ratio (FDRt, %) in eqn (5), the reversible flux decline ratio (FDRr, %) in eqn (6) and the irreversible flux decline ratio (FDRir, %) in eqn (7), as shown below:
| | 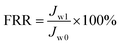 | (4) |
| | 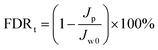 | (5) |
| | 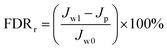 | (6) |
| | 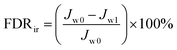 | (7) |
3. Results and discussion
3.1 Synthesis of diblock copolymers PS-b-P(HTMB-r-I)
For the synthesis of diblock copolymers PS-b-P(HTMB-r-I), first a hydrophobic diblock copolymer PS-b-PI was prepared by living anionic polymerization with a total molecular weight of 104 kg mol−1 and a narrow dispersity index of 1.05 (Table 1, Fig. S1 and S2, ESI†). Subsequently, the hydrophobic polyisoprene (PI) block was partially hydroxylated by mercaptoethanol via thiol–ene click reaction and transformed into the amphiphilic P(HTMB-r-I) block successfully, which was confirmed by 1H NMR spectrum of PS-b-P(HTMB-r-I) (Fig. S1, ESI†).48,54 To fabricate the desired SNIPS membrane, four different polymers of P1, P2, P3 and P4 were prepared with the degree of hydroxylation of 35 mol%, 44 mol%, 55 mol% and 65 mol%, respectively (Table 1). Notably, the narrow dispersity index of the parent polymer PS-b-PI was always persisted after functionalization, indicating the good control over thiol–ene click reaction (Table 1 and Fig. S2, ESI†).
Table 1 Molecular characterization of the parent polymer PS-b-PI and thiol–ene click reaction modified polymers PS-b-P(HTMB-r-I)
| Block copolymer |
PSa [wt%] |
PIa [wt%] |
PHTMBa [wt%] |
M
n
[kg mol−1] |
M
w
[kg mol−1] |
Dispersity indicesb |
DHc (mol%) |
|
The composition of polymers calculated from 1H NMR spectra.
Molecular weight and dispersity index determined by GPC.
Degree of hydroxylation (DH) calculated by 1H NMR measurement.
|
| PS-b-PI |
81 |
19 |
0 |
100 |
104 |
1.05 |
0 |
| P1 |
76 |
11 |
13 |
113 |
124 |
1.06 |
35 |
| P2 |
74 |
10 |
16 |
115 |
125 |
1.09 |
44 |
| P3 |
73 |
7 |
20 |
124 |
135 |
1.09 |
55 |
| P4 |
71 |
6 |
23 |
132 |
165 |
1.25 |
65 |
3.2 Solvent evaporation induced self-assembly of the block copolymers during spin-coating and SNIPS
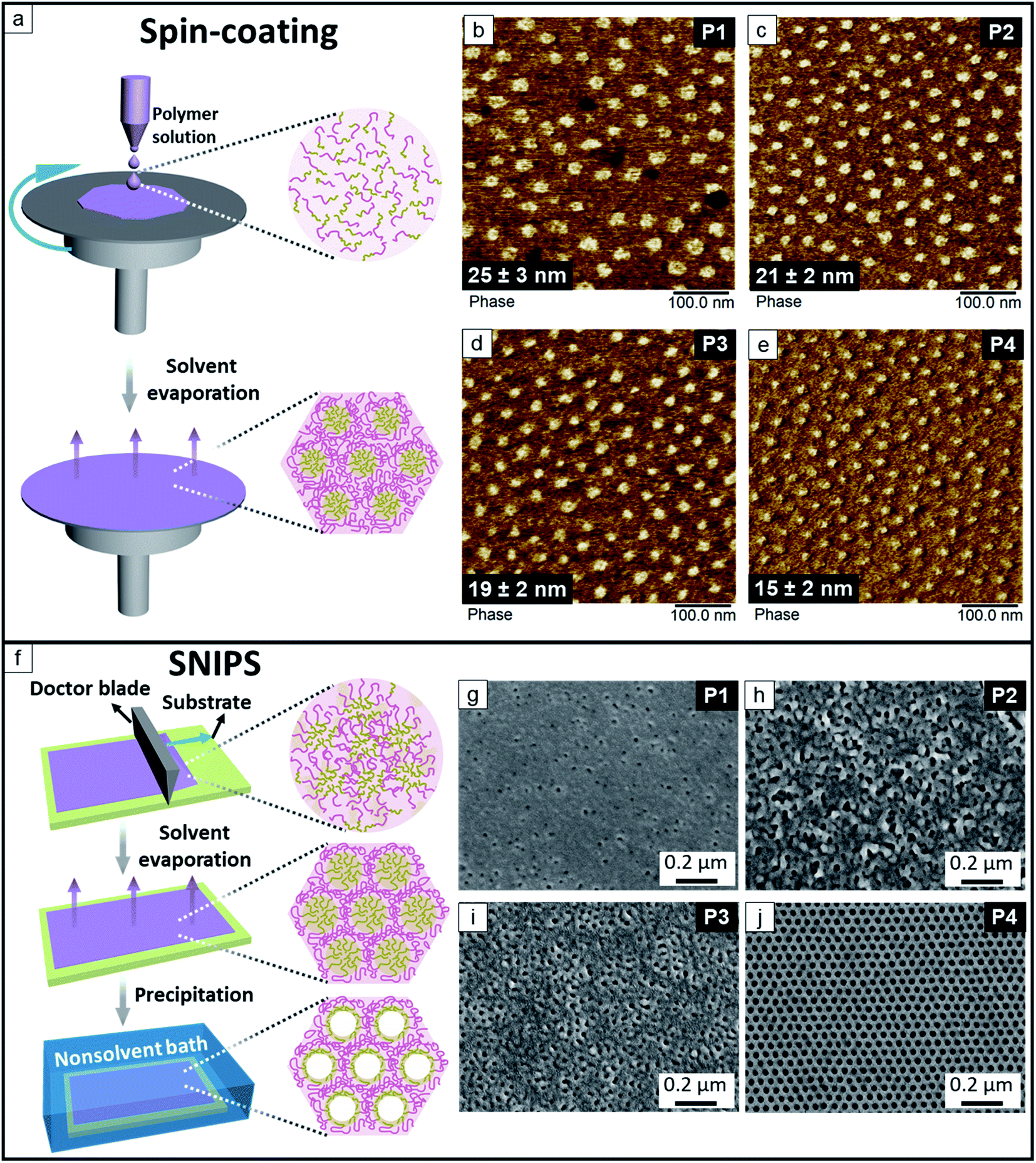
Fig. 2 shows the kinetically trapped surface morphologies of the spin-coated thin films and SNIPS membranes prepared from P1, P2, P3 and P4, respectively. In the AFM phase maps (Fig. 2b–e) of the spin-coated films the discrete P(HTMB-r-I) domains appear as bright regions in a darker continuous PS matrix. The P(HTMB-r-I) domain size is expected to increase with the degree of hydroxylation as the total P(HTMB-r-I) content increases (Table 1). However, the average sizes of the P(HTMB-r-I) domains gradually decrease with increasing degree of hydroxylation. It implies CHCl3 is not a neutral solvent for these polymers i.e., it does not distribute itself equally between the PS and P(HTMB-r-I) microdomains during the drying of the films. In the 2 wt% spin-coating solutions the polymer chains are rather isolated. During the evaporation, as the polymer concentration increases, the PS and P(HTMB-r-I) blocks start to segregate and CHCl3 distributes favourably towards the P(HTMB-r-I) domains. Upon evaporation of CHCl3 the PS segments collapse earlier than the swelled P(HTMB-r-I) segments and the morphology is trapped. With increasing degree of hydroxylation i.e. increasing hydrophilicity, the P(HTMB-r-I) blocks adopt a less stretched conformation at the point when PS blocks collapse around them (Fig. 2a). Consequently, in the completely dried spin-coated films the sizes of the P(HTMB-r-I) domains follow the sequence of P1 > P2 > P3 > P4.
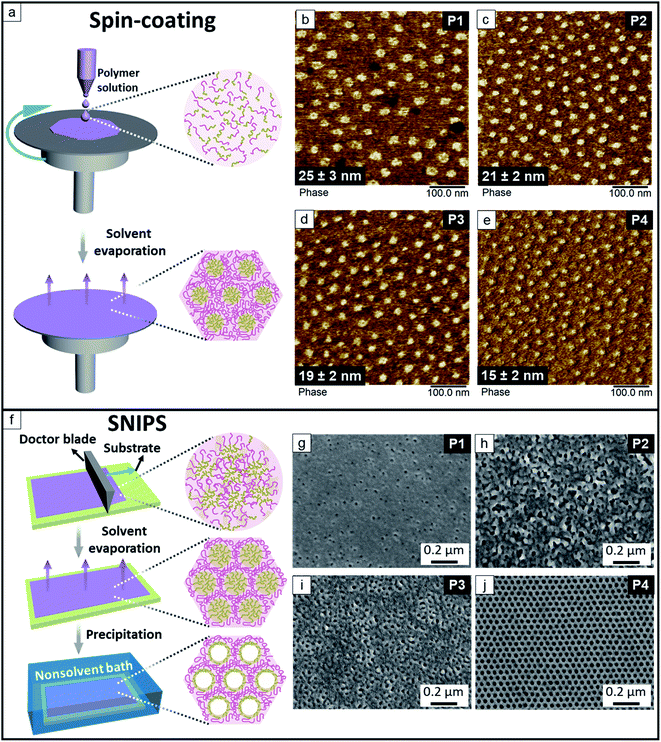 |
| | Fig. 2 (a) Schematic representation of the spin-coating process. AFM phase maps of the spin-coated dense films prepared from CHCl3: (b) P1, (c) P2, (d) P3, (e) P4. (f) Schematic representation of the SNIPS technique. Secondary electron (SE) images of SEM of the representative membranes cast from: (g) 28 wt% P1 solution in THF/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt%, the evaporation time t = 2 s, (h) 20 wt% P2 solution in THF/DMF 3:2 wt%, the evaporation time t = 25 s, (i) 20 wt% P3 solution in THF/DMF 3:2 wt%, the evaporation time t = 20 s, (j) 24 wt% P4 solution in THF/DMF/DOX 2:1:1 wt%, the evaporation time t = 10 s. :1 wt%, the evaporation time t = 2 s, (h) 20 wt% P2 solution in THF/DMF 3:2 wt%, the evaporation time t = 25 s, (i) 20 wt% P3 solution in THF/DMF 3:2 wt%, the evaporation time t = 20 s, (j) 24 wt% P4 solution in THF/DMF/DOX 2:1:1 wt%, the evaporation time t = 10 s. | |
Formation of the isoporous top layer of a SNIPS membrane also requires the selective swelling of the minor block due to unequal distribution of solvents and earlier collapse of the major block of an asymmetric diblock copolymer. Additionally the solvents used for SNIPS must be miscible with the nonsolvent to ensure a fast quenching of the membrane. In this work a series of the casting solutions of P1, P2, P3 and P4 in the binary solvent system THF/DMF or ternary solvent system THF/DMF/DOX were employed to cast the membranes on a neat glass plate or a non-woven (Fig. 2, S6 and S7, ESI†). The concentration of the polymer solution and the evaporation time were varied to find out the optimum condition. Fig. 2 shows the representative membrane surface morphology prepared from the four polymers. The membrane prepared from P1 (Fig. 2g) has very few pores, while the membrane from P2 (Fig. 2h) has irregular pores with a large size distribution. The pores of the membrane from P3 (Fig. 2i) has higher symmetry and relatively narrow size distribution compared to those from P1 and P2. The desired hexagonally packed isoporous morphology is visible for the membrane prepared from a 24 wt% P4 solution in THF/DMF/DOX 2:1:1 wt% (Fig. 2j). Among the solvents THF and DOX are more selective for the matrix-forming PS block and DMF is more selective for the pore-forming P(HTMB-r-I) block.43,55 After casting the viscous solution the more volatile THF evaporates and directs the self-assembly of BCP domains perpendicular to the surface. Upon evaporation of THF the segregation of the blocks becomes stronger and a preferential swelling of the P(HTMB-r-I) domains takes place due to partitioning of the high boiling DMF. Eventually the matrix-forming PS chains acquire a rather collapsed conformation around the highly swollen P(HTMB-r-I) chains. While the solvent evaporation induces BCP self-assembly at the surface vicinity a gradient of polymer concentration builds up along the whole thickness of the cast layer. By quenching into a nonsolvent bath, the self-assembled highly swollen P(HTMB-r-I) chains collapse to form the open pores on a macroporous sublayer (Fig. 2f).
From Fig. 2 it is clear that the composition of the amphiphilic P(HTMB-r-I) block plays a crucial role in partitioning of the solvents. Increasing the degree of hydroxylation leads to a lower partitioning of the hydrophobic CHCl3 and higher partitioning of the hydrophilic DMF towards the P(HTMB-r-I) block. Additionally, we used TEM to investigate the bulk morphology of the P1, P2, P3 and P4 films prepared from two different solvent systems – CHCl3 and CHCl3/DMF 95:5 vol% under slow evaporation (Section 2.3, ESI†). TEM investigation confirms that the P(HTMB-r-I) blocks swell significantly more in presence of DMF compared to pure CHCl3 regardless of the –OH group content. The hydroxyl content of P4 is above the threshold to allow a sufficient partitioning of DMF to bring about the formation of an isoporous top layer via SNIPS. Therefore, among the synthesized polymers P4 is selected for the fabrication of the desired membrane. P4 endows a highly ordered isoporous structure in a wide processing window, i.e., the optimum polymer concentration can be tuned from 22 wt% to 30 wt% and the evaporation time is from 5 s up to 20 s (Fig. S8 and S9, ESI†).
3.3 Organic–inorganic hybrid nanocomposite PS-b-P(HTMB-r-I) isoporous membrane with TiO2 nanoparticles
The cross section of the membranes prepared from P4 consists of vertically-aligned porous cylinders connected with the disordered macropores and big caverns of the sublayer (Fig. S12a and b, ESI†). During the phase inversion process, a typical integral asymmetric structure with a coarse porous network substructure under the denser surface is a result of the spinodal decomposition of the viscous layer having a concentration gradient due to exchange of solvent and nonsolvent.25,56 However, the membranes contain a completely dense interface between the bottom of the PS-b-P(HTMB-r-I) membrane and the substrate (denoted as bottom interface) without any open pores (Fig S12c, ESI†). To our knowledge, there is to date no report to discuss the dense bottom interface structure formed during the SNIPS process. In order to solve the problem, at first we investigated the influence of the exchange rate of solvent and non-solvent on bottom interface formation. Therefore, temperature and composition of non-solvent bath and solvent system were varied to enhance and decrease the exchange rate. No influence on the bottom interface structure was observed (Fig. 3a, b, d, e and S13, ESI†). Water-soluble polymers are usually used as pore-forming agents (porogens) for the fabrication of porous polymeric membranes such as polysulfone (PSF), or polyethersulfone (PES) via non-solvent induced phase separation (NIPS), like poly(ethylene glycol) (PEG),57,58 poly(sodium 4-styrene sulfonate)59 and poly(vinyl pyrrolidone).60,61 Therefore, PEG of molecular weight 400 g mol−1 (PEG400) was introduced as a porogen by blending with P4 in the casting solution. However, in spite of varying the PEG400 content between 6–20 wt% with respect to the weight of P4 an open bottom interface was not obtained (Fig. 3c, f, S13k and n, ESI†).
 |
| | Fig. 3 Secondary electron (SE) images of SEM: top surface and bottom interface of P4 membranes cast from 25 wt% solution in THF/DMF/DOX 2:1:1 wt% (a and d) in the 32 °C H2O bath, (b and e) in the non-solvent bath of methanol/H2O (2:8 vol%), (c and f) with 6 wt% (as P4) PEG400 as the porogen. All of the evaporation time is t = 5 s. | |
Gu et al. reported that due to introduction of inorganic titanium dioxide (TiO2) nanoparticles (NPs), the bottom interface of the PI-b-PS-b-P4VP membranes exhibited big open pores with diameters in the range of 10–30 μm.52 Therefore, different amounts of a TiO2 sol solution prepared by a hydrolytic sol–gel route were added into the P4 casting solutions, i.e., 6 wt%, 8 wt%, 10 wt% and 20 wt% with respect to the weight of P4. Fig. 4 shows a comparison of the purely organic membrane without TiO2 NPs and the membranes with different amounts of TiO2 NPs. All the membranes were cast from the same concentration of the polymer solutions, i.e. 16 wt%. The purely organic membrane without TiO2 NPs displays the completely dense bottom interface and an irregular porous top surface (Fig. 4a and f), since the polymer solution of 16 wt% is too diluted to microphase separate in a well ordered way in the top layer. Notably, the corresponding hybrid nanocomposite membranes possess an open porous bottom interface with macroscale structural features, meanwhile maintaining a highly ordered isoporous top surface except the hybrid nanocomposite membrane with 20 wt% TiO2 as P4 (Fig. 4b–e and g–j). Through the hydrolytic sol–gel process, the resulting TiO2 sol NPs have the Ti–OH groups on their surface.52 Owing to the hydrogen-bonding interactions among –OH groups, TiO2 sol NPs are expected to selectively incorporate into the pore-forming block P(HTMB-r-I), increase the volume fraction of P(HTMB-r-I), and thus drive BCP-TiO2 co-assembly52 and consequently the formation of an isoporous structure at a relatively low polymer concentration. It is remarkable that highly ordered isoporous membranes are achieved by the incorporation of such significant amounts of inorganic TiO2 NPs into the casting solution, i.e. 6–10 wt% with respect to P4. To our knowledge an isoporous top layer having such hexagonally packed vertically aligned pores has never been reported in a nanocomposite SNIPS membrane.
 |
| | Fig. 4 Secondary electron (SE) images of SEM: top surface and bottom interface of P4 membranes from 16 wt% solution in THF/DMF/DOX 2:1:1 wt% with different amount of TiO2 NPs as the additives: (a and f) without TiO2 NPs, the evaporation time t = 15 s, (b and g) 6 wt% TiO2 NPs, the evaporation time t = 15 s, (c and h) 8 wt% TiO2 NPs, the evaporation time t = 15 s, (d and i) 10 wt% TiO2 NPs, the evaporation time t = 10 s, (e and j) 20 wt% TiO2 NPs, the evaporation time t = 5 s. | |
To figure out the optimum processing window, different concentrations of polymer solutions were evaluated in the wide range of 14–25 wt%. Overall, the desired membrane with a highly ordered isoporous top structure and an open macroporous bottom interface can be successfully obtained in the wide range of 15–22 wt% polymer solutions with 6–8 wt% TiO2 NPs as P4 (Fig. 5). Compared to the 22–30 wt% optimum window of the purely organic system, the optimum window of the hybrid nanocomposite system with TiO2 NPs shifts to lower range, verifying the hydrogen-bonding interaction between TiO2 NPs and P(HTMB-r-I) and thus BCP-TiO2 co-assembly. As we know, there are very few reports to show a BCP SNIPS membrane with such wide processing window.
 |
| | Fig. 5 Secondary electron (SE) images of SEM: top surface and bottom interface of P4 membranes from different concentration of casting solutions in THF/DMF/DOX (2:1:1 wt%) with 6–8 wt% of TiO2 NPs as the additives: (a and e) 14 wt%, the evaporation time t = 15 s, (b and f) 15 wt%, the evaporation time t = 15 s, (c and g) 16 wt%, the evaporation time t = 15 s, (d and h) 18 wt%, the evaporation time t = 15 s, (i and m) 19 wt%, the evaporation time t = 10 s, (j and n) 20 wt%, the evaporation time t = 10 s, (k and o) 22 wt%, the evaporation time t = 10 s, (l and p) 25 wt%, the evaporation time t = 15 s. The inset images are the overview of the cross section of the membranes. | |
Moreover, the purely organic and hybrid nanocomposite membranes were analyzed by back-scattered electron imaging of SEM (Fig S14, ESI†) and the cross section of a hybrid nanocomposite membrane was investigated by TEM (Fig. S16, ESI†). The results show that TiO2 sol NPs are distributed all over the surface and cross-section of the membrane. A part of the TiO2 sol NPs might have been washed away during membrane fabrication (detailed discussion is provided in Fig. S14–S16, ESI†). Therefore, although the P4 membranes (Fig. 5) are prepared from casting solutions containing 6–8 wt% TiO2 NPs, the final concentration of TiO2 NPs in the obtained membranes might be lower.
All the cross sections of the hybrid nanocomposite membranes consist of an isoporous top layer and underneath finger-like macrovoids instead of typical spongy porous support structure (Fig. 5). It is crucial to assess the mechanical stability of the corresponding integral asymmetric structure under pressure driven condition. Therefore, the ultrapure water fluxes of the hybrid membrane were measured at different transmembrane pressures from 0.25–2.9 bar. The ultrapure water fluxes increase linearly with the transmembrane pressure (Fig. S17, ESI†). It reveals that our prepared P4/TiO2 NPs hybrid nanocomposite isoporous membranes are mechanically stable up to 2.9 bar pressure, whereas in the study by Gu et al. it was reported that the PI-b-PS-b-P4VP/TiO2 hybrid membranes was stable in a pressure range of 0.01–0.06 bar.52
3.4 Sulfonation of the organic–inorganic hybrid nanocomposite isoporous membranes
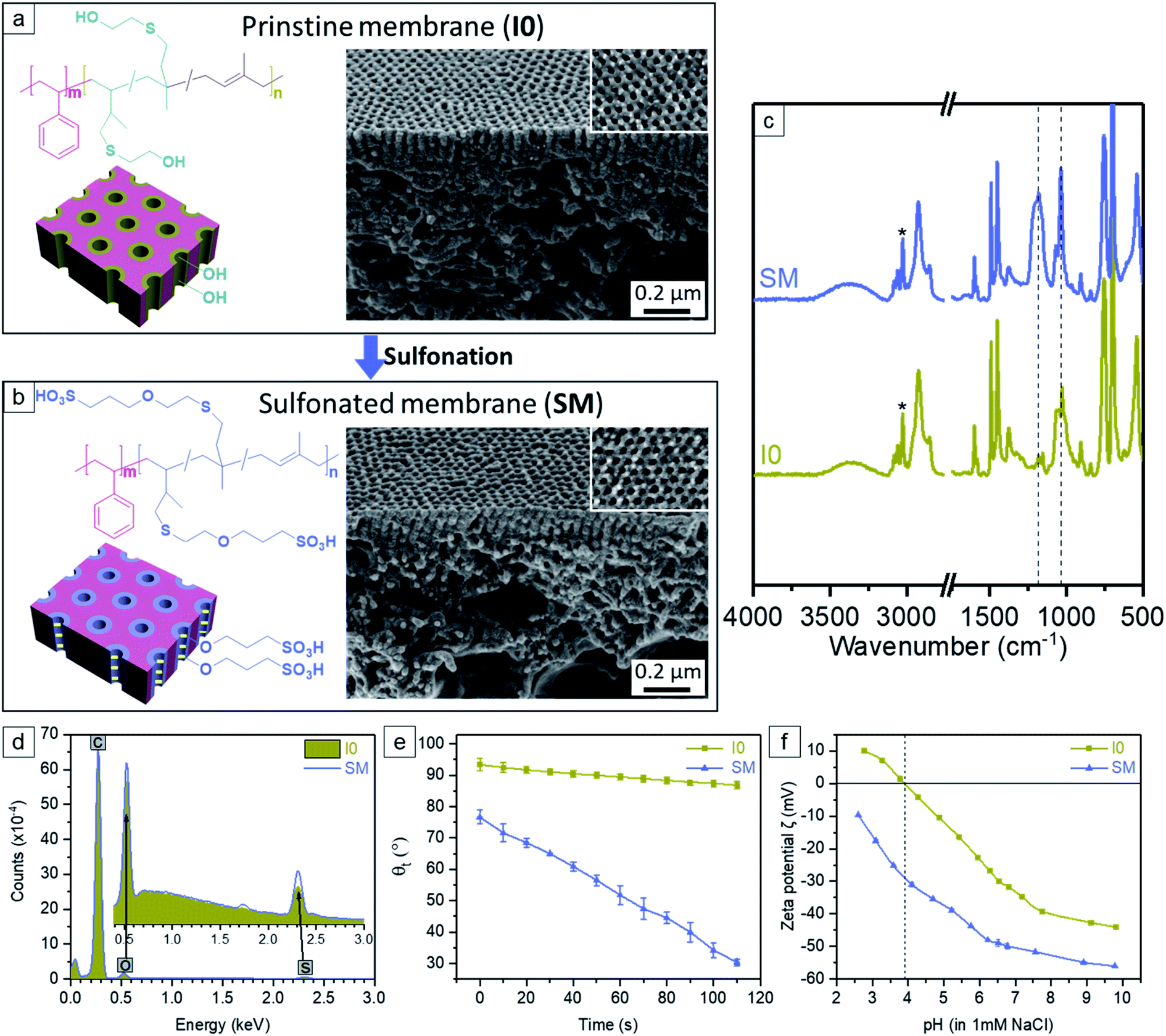
The P4/TiO2 NPs hybrid membranes were post-functionalized with 1,3-propane sultone using a straightforward in situ scalable gas–solid interface reaction. The appearance of two new characteristic vibrations at ∼1036 cm−1 and ∼1181 cm−1 in the sulfonated membrane (SM) spectrum of ATR-FTIR is ascribed to the stretch vibration of the –HSO3 groups, indicating the ring opening of 1,3-propane sultone and the successful covalent attachment of the sulfonic acid moieties (Fig. 6c).48 Additionally it is clear that compared to the pristine membrane I0, the content of sulfur (S) and oxygen (O) in the SM spectrum of EDX increases (Fig. 6d), in agreement with the success of sulfonation reaction. The comparison of SEM images confirms that SM retains the kinetically trapped integral asymmetric isoporous structure of I0 (Fig. 6a, b and S18, ESI†). Notably, no big change of the pore size of SM (i.e. 22.5 nm) is observed compared to the pore size 22.9 nm of I0. The morphology of their cross sections is also rather similar. Fig. 6e shows that I0 has a higher initial water contact angle (θ0 = 94°) than SM (θ0 = 76°) while the sinking rate of a water droplet through I0 is prominently slower than that of SM. Our previous study reported that after introduction of the hydrophobic moieties (i.e., 2-ethylcarbamoyloxy) within the hydrophilic pore walls of a polystyrene-block-poly(2-hydroxyethyl methacrylate) (PS-b-PHEMA) membrane, the initial contact angle θ0 was changed from 63.8° to 94.5°, and also the changes of contact angles with time showed a very slow decrease, similar to the current trend of membrane I0.31 Additionally, the reported PS-b-P4VP membrane with positively charged nanochannels exhibited a similar sinking rate of a water droplet as that of SM.40 Therefore, these results reveal that I0 is a strong hydrophobic membrane, whereas SM turns into a hydrophilic membrane which is ascribed to the introduction of the sulfonate groups along the pore wall. The surface zeta potential (ζ) of I0 is negative in the pH range of 3.9–10, likely due to the hydrophobic character and preferred absorption of hydroxide ions. The isoelectric point (around pH 3.9) and the shape of curve fit with typical hydrophobic polymeric membranes without dissociating groups (Fig. 6f).62–64 Compared to I0, SM displays a much higher negative ζ in the whole pH range of 2.5–10 without isoelectric point, which demonstrates the presence of acidic groups and their corresponding dissociation (Fig. 6f).65
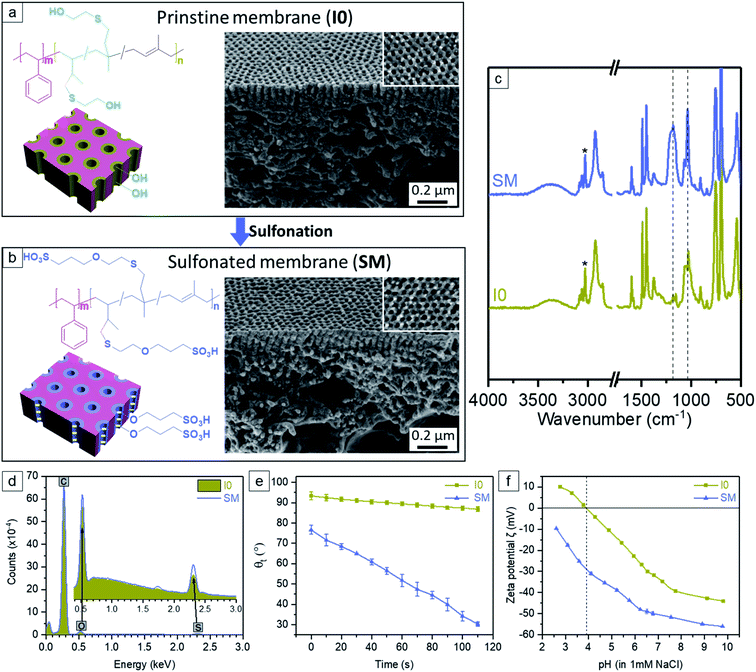 |
| | Fig. 6 Schematic representation, chemical structure and SEM images of (a) the pristine membrane I0, (b) the sulfonated membrane SM. (c) FTIR spectra of I0 and SM. The relative intensities were normalized using the characteristic aromatic C–H stretches (*) of the unreactive polystyrene segment between 3100 and 3000 cm−1. (d) Comparison of EDX spectra of I0 and SM. (e) Dynamic water contact angle of I0 and SM as a function of time. (f) The surface zeta potential of I0 and SM as a function of pH (2.5–10). Experiments were performed with a background electrolyte of 1 mM NaCl. | |
3.5 Membrane performance
3.5.1 Membrane permeability and selectivity.
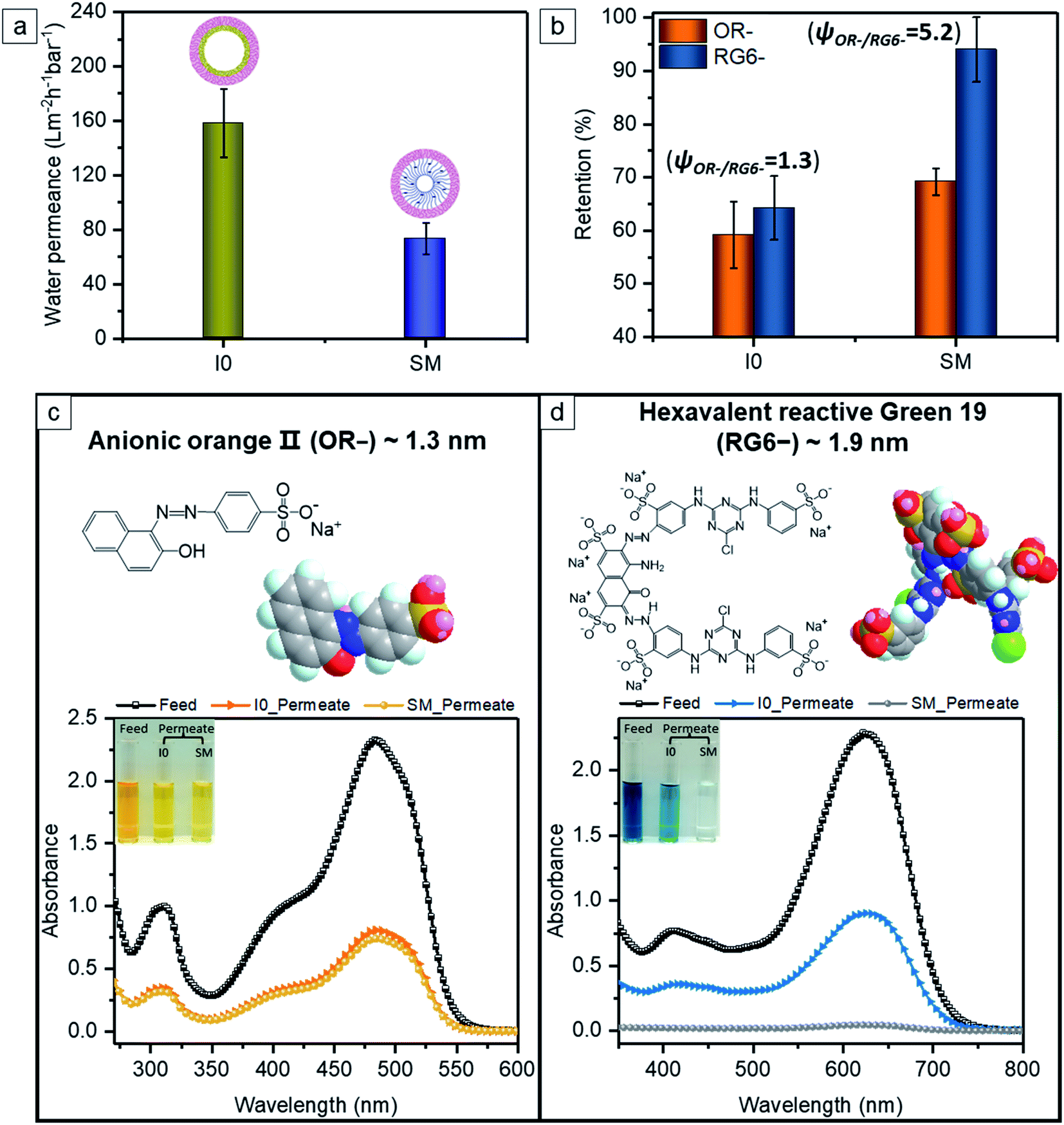
I0 exhibits a constant water permeance of 159 L m−2 h−1 bar−1 (Fig. 7a) in the pressure range 0.25–2.9 bar. Thus the effective pore size of the I0 membrane is big enough to overcome the unfavorable wetting of the hydrophobic surface of I0 to allow fast water transport even at 0.25 bar transmembrane pressure. The negatively charged membrane SM has a water permeance of 74 L m−2 h−1 bar−1 (Fig. 7a), which is significantly higher than reported SNIPS BCP membrane with polyelectrolyte swelling nanopores (0.6–15 L m−2 h−1 bar−1).36–39 The amphiphilic pore-forming block P(HTMB-r-I) having the randomly distributed hydrophilic HTMB and hydrophobic I repeating units offers molecular-scale compositional heterogeneity.66 Due to such molecular-level compositional heterogeneity, the negatively charged pore-forming block of SM does not assume a fully stretched conformation but swells to some extent in a hydrated state. Therefore in a hydrated state SM contains charged nanopores which are significantly smaller compared to those of I0. In spite of having higher hydrophilicity (Fig. 6e) the water permeance of SM is significantly lower than I0 (Fig. 7a).
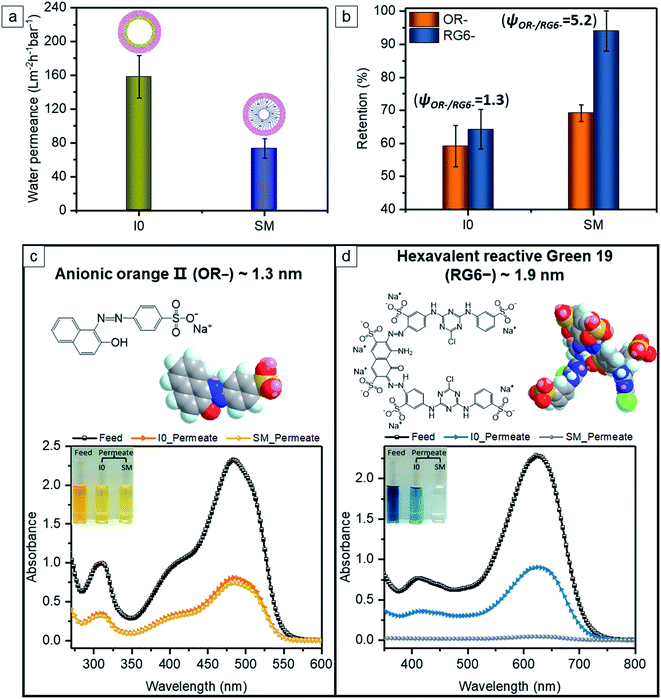 |
| | Fig. 7 (a) Comparison of water permeance of the pristine membrane I0 and the sulfonated membrane SM under trans-membrane pressure of 1 bar. (b) The separation behavior of small organic molecules (i.e. orange II (OR−) and reactive green 19 (RG6−)) using the membranes I0 and SM. (c and d) Molecular structure and space-filling model of small molecules, and the corresponding UV-vis spectra and a color change of the solution: (c) OR− and (d) RG6−. | |
To demonstrate the separation efficiency of the membranes, we employed two hydrophilic water-soluble model molecules with negatively charged functional group and different molecular weights (350.32 and 1418.93 g mol−1, respectively) – monovalent orange II (OR−) and hexavalent reactive green 19 (RG6−). The aqueous solutions of OR− and RG6− permeate through I0 with a similar moderate extent of retention, i.e. 59% and 64% (Fig. 7b–d). It presumably arises from the hydrophilic–hydrophobic repulsive interaction, since hydrophilic molecules (i.e. OR− and RG6−) are unfavorable to be close to the rather hydrophobic surface of I0 (Fig. 6e) and to reach the entrance of the nanochannels. The pH of OR− and RG6− aqueous solutions are 6.4 and 4.6, respectively. Although at pH 6.4 I0 has a much stronger negative charge than that at pH 4.6, the pores are too big to exert the electrostatic repulsion on OR− and RG6−. The resulting selectivity ψOR−/RG6− is thus only 1.3. In the case of SM, 69% of OR− is retained from the aqueous solution (Fig. 7b and c). The negative charge at the surface of SM is very strong at pH 6.4 (Fig. 6f). Although the hydrophobicity is obviously decreased by sulfonation post-functionalization (Fig. 6e), the negatively charged nanochannels of SM endow an additional electrostatic repulsion between the like charges to reject OR− at a slightly higher extent than I0. At pH 4.6 SM also possesses a strong negative charge and the nanopores of SM are narrow enough to exert a strong electrostatic repulsion on the bigger hexavalent RG6− molecules leading to a 94% retention (Fig. 7b and d). It is clear that RG6− is more hindered to enter the charged soft nanochannels of SM due to its higher energy barrier of electrostatic repulsion and bigger molecular dimensions compared to OR−. It is remarkable that the selectivity ψOR−/RG6− = 5.2 of SM is four fold higher than that of I0.
3.5.2 Fouling resistance of organic solutes.
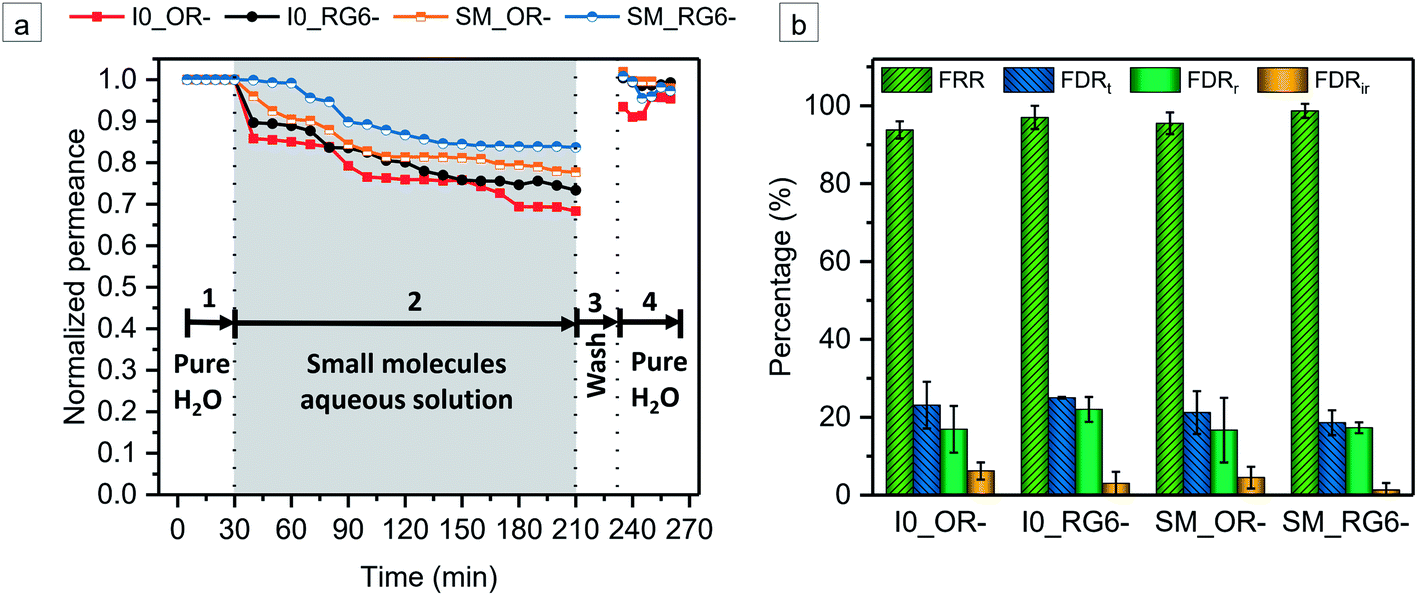
The model small molecules OR− and RG6− were employed as foulants to evaluate the antifouling ability of the membranes in static adsorption and dynamic filtration, respectively. There is no static adsorption of OR− and RG6− onto both I0 and SM (Table S1†). After 24 h exposure, the surfaces of the membranes do not show any color staining (Fig. S19†). Fig. 8 displays the time-dependent normalized permeance variations during foulant solution filtration and several parameters related to antifouling property, i.e., flux recovery ratio (FRR), total flux-decline ratio (FDRt), reversible flux-decline ratio (FDRr), irreversible flux-decline ratio (FDRir). A higher value of FRR and a lower value of FDRt, implies better antifouling property of membrane. For I0, the permeate fluxes of OR− and RG6− aqueous solutions are decreased compared with the initial pure water flux while FDRt is around 23.1% and 25.0%, respectively. However, the pure water permeance is recovered to a high extent, i.e., FRROR− = 93.8%, FRRRG6− = 97%, indicating a good antifouling property of I0 (Fig. 8). It has been reported that the incorporation of TiO2 NPs on the membrane surface can mitigate fouling.67–69 Thus, the addition of TiO2 NPs not only facilitates the formation of the porous bottom interface, it may also impart the fouling resistance. Additionally, the molecular-scale compositional heterogeneity of the amphiphilic random copolymer may discourage thermodynamically favorable interactions between foulant and the surface, leading to a low adsorption of OR− and RG6− on I0.66,70,71 In the case of SM, we observe an enhanced fouling resistance. The corresponding FRR values of SM are increased to FRROR− = 95.5%, FRRRG6− = 98.7% while FDRt values are decreased to FDROR−t = 21.2%, FDRRG6−t = 18.6% (Fig. 8). Besides the SM possesses a much higher negative charge than I0 at the pH of foulant aqueous solutions (Fig. 6f), which induces a strong electrostatic repulsive forces to prevent the adsorption of anionic foulants. Synergistically, a strong hydration layer forms along the sulfonated polyelectrolyte decorated pore walls of SM which acts as a physical and energy barrier to inhibit the adsorption of the foulants on the pore wall.72–74 The surfaces of all the membranes almost display no color staining after fouling filtration measurements (Fig. S20, ESI†). It is particularly noteworthy that owing to the excellent antifouling performance, the SM membrane maintains a rather high permeate flux during filtration, around 60 L m−2 h−1 bar−1, meanwhile ensuring a good selectivity ψOR−/RG6− = 5.2. Compared to recently reported charged membranes, SM demonstrates excellent antifouling property with a high permeate flux, which can potentially minimize the energy consumption of separation. Therefore, it is clear that SM is a very promising membrane for both the high-performance separation of small molecules and the dye wastewater treatment.
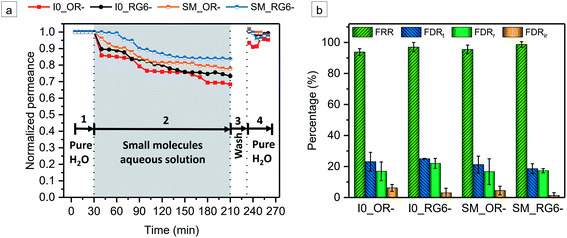 |
| | Fig. 8 (a) Time-dependent normalized permeance variations of the pristine membrane I0 and the sulfonated membrane SM during the filtration process using OR− and RG6− as the permeate solutes, respectively. The operation process included four steps: (1) 30 min pure water filtration, (2) 3 h filtration of small molecules aqueous solution, (3) 20 min water washing, (4) 30 min pure water filtration after washing. (b) A summary of the corresponding FRR, FDRt, FDRr, FDRir values of I0 and SM. | |
4. Conclusion
In summary, we report a novel asymmetric isoporous membrane having embedded hydrophilic hydroxyl groups along the amphiphilic pores, which is derived from a well-designed diblock copolymer PS-b-P(HTMB-r-I). In this work for the first time we have demonstrated that the incorporation of in situ formed TiO2 NPs facilitates the formation of a macroporous bottom interface, resulting in an organic–inorganic hybrid nanocomposite integral asymmetric isoporous membrane. The highly accessible hydroxyl groups on the interior of the pore walls allows straightforward scalable gas–solid interface post-functionalization to integrate the negatively charged moiety within the pores. The molecular-scale compositional heterogeneity of amphiphilic pore-forming block P(HTMB-r-I) results in a moderate swelling of negatively charged polyelectrolyte along the pore walls. As a result, the membrane with well-defined soft nanochannels in a hydrated state exhibits a significantly high water permeance in nanofiltration regime and also the potential for a good separation of 1–2 nm small anionic molecules (having molecular weights in the range of 300–1500 g mol−1) from each other. The static absorption and dynamic filtration studies demonstrate the excellent antifouling performance of the prepared membranes. Especially the negatively charged membrane shows a rather low permeation flux decline and a high flux recovery, mainly due to the electrostatic repulsion and the physical and energy barrier of hydration layer. This concept of molecular-level manipulation of the composition and function of the pore-forming block by bottom-up design of a block copolymer provides a suitable platform to develop next generation high-performance nanofiltration membranes for a broad range of selective transport.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Anke-Lisa Höhme and Erik Schneider for support in TEM and SEM measurements, Evgeni Sperling for AFM investigations, Maren Brinkmann and Silvio Neumann for polymer characterization, and Barbara Bajer for support in the membrane performance characterization.
Notes and references
- I. Romanenko, M. Lechner, F. Wendler, C. Hörenz, C. Streb and F. H. Schacher, J. Mater. Chem. A, 2017, 5, 15789–15796 RSC.
- G. Jeon, S. Y. Yang and J. K. Kim, J. Mater. Chem., 2012, 22, 14814 RSC.
- S. Y. Yang, I. Ryu, H. Y. Kim, J. K. Kim, S. K. Jang and T. P. Russell, Adv. Mater., 2006, 18, 709–712 CrossRef CAS.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- M. Geise Geoffrey, H. S. Lee, J. Miller Daniel, D. Freeman Benny, E. McGrath James and R. Paul Donald, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1685–1718 CrossRef.
- H. Fan, J. Gu, H. Meng, A. Knebel and J. Caro, Angew. Chem., Int. Ed. Engl., 2018, 57, 4083–4087 CrossRef CAS PubMed.
- X. You, H. Wu, R. Zhang, Y. Su, L. Cao, Q. Yu, J. Yuan, K. Xiao, M. He and Z. Jiang, Nat. Commun., 2019, 10, 4160 CrossRef CAS PubMed.
- S. Abdikheibari, W. Lei, L. F. Dumée, N. Milne and K. Baskaran, J. Mater. Chem. A, 2018, 6, 12066–12081 RSC.
- J. Shen, R. Zhang, Y. Su, B. Shi, X. You, W. Guo, Y. Ma, J. Yuan, F. Wang and Z. Jiang, J. Mater. Chem. A, 2019, 7, 18063–18071 RSC.
- P. H. H. Duong, K. Daumann, P. Y. Hong, M. Ulbricht and S. P. Nunes, Langmuir, 2019, 35, 1284–1293 CrossRef CAS PubMed.
- A. Dundua, S. Franzka and M. Ulbricht, Macromol. Rapid Commun., 2016, 37, 2030–2036 CrossRef CAS PubMed.
- P. van Rijn, M. Tutus, C. Kathrein, N. C. Mougin, H. Park, C. Hein, M. P. Schürings and A. Böker, Adv. Funct. Mater., 2014, 24, 6762–6770 CrossRef CAS.
- Y. Yang Seung, J. Park, J. Yoon, M. Ree, K. Jang Sung and K. Kim Jin, Adv. Funct. Mater., 2008, 18, 1371–1377 CrossRef.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434 CrossRef CAS PubMed.
- E. A. Jackson and M. A. Hillmyer, ACS Nano, 2010, 4, 3548–3553 CrossRef CAS PubMed.
- J. C. Brendel and F. H. Schacher, Chem.–Asian J., 2018, 13, 230–239 CrossRef CAS PubMed.
- P. van Rijn, M. Tutus, C. Kathrein, L. Zhu, M. Wessling, U. Schwaneberg and A. Boker, Chem. Soc. Rev., 2013, 42, 6578–6592 RSC.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS.
- M. Radjabian and V. Abetz, Prog. Polym. Sci., 2020 DOI:10.1016/j.progpolymsci.2020.101219.
- W. A. Phillip, J. Rzayev, M. A. Hillmyer and E. L. Cussler, J. Membr. Sci., 2006, 286, 144–152 CrossRef CAS.
- W. A. Phillip, B. O'Neill, M. Rodwogin, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2010, 2, 847–853 CrossRef CAS PubMed.
- C. G. Gamys, J. M. Schumers, C. Mugemana, C. A. Fustin and J. F. Gohy, Macromol. Rapid Commun., 2013, 34, 962–982 CrossRef CAS PubMed.
- C. G. Gamys, A. Vlad, O. Bertrand and J.-F. Gohy, Macromol. Chem. Phys., 2012, 213, 2075–2080 CrossRef CAS.
- K. V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- V. Abetz, Macromol. Rapid Commun., 2015, 36, 10–22 CrossRef CAS PubMed.
- S. Schöttner, H.-J. Schaffrath and M. Gallei, Macromolecules, 2016, 49, 7286–7295 CrossRef.
- S. P. Nunes, M. Karunakaran, N. Pradeep, A. R. Behzad, B. Hooghan, R. Sougrat, H. He and K.-V. Peinemann, Langmuir, 2011, 27, 10184–10190 CrossRef CAS PubMed.
- H. Yu, X. Qiu, N. Moreno, Z. Ma, V. M. Calo, S. P. Nunes and K.-V. Peinemann, Angew. Chem., 2015, 127, 14143–14147 CrossRef.
- R. Shevate, M. Karunakaran, M. Kumar and K.-V. Peinemann, J. Membr. Sci., 2016, 501, 161–168 CrossRef CAS.
- H. Yu, X. Qiu, S. P. Nunes and K. V. Peinemann, Angew. Chem., Int. Ed. Engl., 2014, 53, 10072–10076 CrossRef CAS PubMed.
- J. Wang, M. M. Rahman, C. Abetz, S. Rangou, Z. Zhang and V. Abetz, Macromol. Rapid Commun., 2018, 39, 1800435 CrossRef PubMed.
- E. N. Savariar, K. Krishnamoorthy and S. Thayumanavan, Nat. Nanotechnol., 2008, 3, 112–117 CrossRef CAS PubMed.
- E. N. Savariar, M. M. Sochat, A. Klaikherd and S. Thayumanavan, Angew. Chem., Int. Ed. Engl., 2009, 48, 110–114 CrossRef CAS PubMed.
- S. B. Lee, D. T. Mitchell, L. Trofin, T. K. Nevanen, H. Soederlund and C. R. Martin, Science, 2002, 296, 2198–2200 CrossRef CAS PubMed.
- B. B. Lakshmi and C. R. Martin, Nature, 1997, 388, 758–760 CrossRef CAS PubMed.
- Y. Zhang, R. A. Mulvenna, S. Qu, B. W. Boudouris and W. A. Phillip, ACS Macro Lett., 2017, 6, 726–732 CrossRef CAS.
- R. A. Mulvenna, J. L. Weidman, B. Jing, J. A. Pople, Y. Zhu, B. W. Boudouris and W. A. Phillip, J. Membr. Sci., 2014, 470, 246–256 CrossRef CAS.
- Y. Gu and U. Wiesner, Macromolecules, 2015, 48, 6153–6159 CrossRef CAS.
- J. I. Clodt, V. Filiz, S. Rangou, K. Buhr, C. Abetz, D. Höche, J. Hahn, A. Jung and V. Abetz, Adv. Funct. Mater., 2013, 23, 731–738 CrossRef CAS.
- Z. Zhang, M. M. Rahman, C. Abetz, B. Bajer, J. Wang and V. Abetz, Macromol. Rapid Commun., 2019, 40, 1800729 CrossRef PubMed.
- Q. Zhang, Y. Gu, Y. M. Li, P. A. Beaucage, T. Kao and U. Wiesner, Chem. Mater., 2016, 28, 3870–3876 CrossRef CAS.
- M. Radjabian and V. Abetz, Adv. Mater., 2015, 27, 352–355 CrossRef CAS.
- S. Rangou, K. Buhr, V. Filiz, J. I. Clodt, B. Lademann, J. Hahn, A. Jung and V. Abetz, J. Membr. Sci., 2014, 451, 266–275 CrossRef CAS.
- W. A. Phillip, R. M. Dorin, J. Werner, E. M. Hoek, U. Wiesner and M. Elimelech, Nano Lett., 2011, 11, 2892–2900 CrossRef CAS PubMed.
- R. M. Dorin, W. A. Phillip, H. Sai, J. Werner, M. Elimelech and U. Wiesner, Polymer, 2014, 55, 347–353 CrossRef CAS.
- S. Saleem, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Polymers, 2019, 12, 41 CrossRef PubMed.
- A. Jung, V. Filiz, S. Rangou, K. Buhr, P. Merten, J. Hahn, J. Clodt, C. Abetz and V. Abetz, Macromol. Rapid Commun., 2013, 34, 610–615 CrossRef CAS PubMed.
- Z. Zhang, M. M. Rahman, C. Abetz, A. L. Höhme, E. Sperling and V. Abetz, Adv. Mater., 2020, 32, 1907014 CrossRef CAS PubMed.
- J. Hahn, V. Filiz, S. Rangou, J. Clodt, A. Jung, K. Buhr, C. Abetz and V. Abetz, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 281–290 CrossRef CAS.
- P. Georgopanos, S. Rangou, T. Gil Haenelt, C. Abetz, A. Meyer, V. Filiz, U. A. Handge and V. Abetz, Colloid Polym. Sci., 2014, 292, 1877–1891 CrossRef CAS.
- P. Georgopanos, V. Filiz, U. A. Handge and V. Abetz, Macromol. Chem. Phys., 2016, 217, 1293–1304 CrossRef CAS.
- Y. Gu, R. M. Dorin and U. Wiesner, Nano Lett., 2013, 13, 5323–5328 CrossRef CAS PubMed.
- P. Docampo, M. Stefik, S. Guldin, R. Gunning, N. A. Yufa, N. Cai, P. Wang, U. Steiner, U. Wiesner and H. J. Snaith, Adv. Energy Mater., 2012, 2, 676–682 CrossRef CAS.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Polymer, 2005, 46, 12057–12064 CrossRef CAS.
- M. Radjabian, C. Abetz, B. Fischer, A. Meyer and V. Abetz, ACS Appl. Mater. Interfaces, 2017, 9, 31224–31234 CrossRef CAS PubMed.
- C. Stegelmeier, V. Filiz, V. Abetz, J. Perlich, A. Fery, P. Ruckdeschel, S. Rosenfeldt and S. Förster, Macromolecules, 2014, 47, 5566–5577 CrossRef CAS.
- Y. Ma, F. Shi, J. Ma, M. Wu, J. Zhang and C. Gao, Desalination, 2011, 272, 51–58 CrossRef CAS.
- B. Chakrabarty, A. K. Ghoshal and M. K. Purkait, J. Membr. Sci., 2008, 309, 209–221 CrossRef CAS.
- N. Noor, J. Koll, N. Scharnagl, C. Abetz and V. Abetz, Membranes, 2018, 8, 54 CrossRef PubMed.
- H. Matsuyama, T. Maki, M. Teramoto and K. Kobayashi, Sep. Sci. Technol., 2003, 38, 3449–3458 CrossRef CAS.
- M. Son, H. Kim, J. Jung, S. Jo and H. Choi, Chemosphere, 2017, 179, 194–201 CrossRef CAS PubMed.
- Y. Liao, T. P. Farrell, G. R. Guillen, M. Li, J. A. T. Temple, X.-G. Li, E. M. V. Hoek and R. B. Kaner, Mater. Horiz., 2014, 1, 58–64 RSC.
- M. E. A. E. Childress, J. Membr. Sci., 1996, 119, 253–268 CrossRef.
- G. M. Geise, B. D. Freeman and D. R. Paul, Polymer, 2010, 51, 5815–5822 CrossRef CAS.
- S. Temmel, W. Kern and T. Luxbacher, Prog. Colloid Polym. Sci., 2006, 132, 54–61 CAS.
- S. H. Baxamusa and K. K. Gleason, Adv. Funct. Mater., 2009, 19, 3489–3496 CrossRef CAS.
- X. Zhao, Y. Su, J. Cao, Y. Li, R. Zhang, Y. Liu and Z. Jiang, J. Mater. Chem. A, 2015, 3, 7287–7295 RSC.
- M.-L. Luo, J.-Q. Zhao, W. Tang and C.-S. Pu, Appl. Surf. Sci., 2005, 249, 76–84 CrossRef CAS.
- R.-X. Zhang, L. Braeken, T.-Y. Liu, P. Luis, X.-L. Wang and B. Van der Bruggen, Appl. Sci., 2017, 7, 81 CrossRef.
- Z. Zhao, H. Ni, Z. Han, T. Jiang, Y. Xu, X. Lu and P. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 7808–7818 CrossRef CAS PubMed.
- C. A. Amadei, R. Yang, M. Chiesa, K. K. Gleason and S. Santos, ACS Appl. Mater. Interfaces, 2014, 6, 4705–4712 CrossRef CAS PubMed.
- C. Liu, C. Wang, Y. Guo, J. Zhang, Y. Cao, H. Liu, Z. Hu and C. Zhang, J. Mater. Chem. A, 2019, 7, 6695–6707 RSC.
- L. Shen, C. Cheng, X. Yu, Y. Yang, X. Wang, M. Zhu and B. S. Hsiao, J. Mater. Chem. A, 2016, 4, 15575–15588 RSC.
- G. Jiang, S. Zhang, Y. Zhu, S. Gao, H. Jin, L. Luo, F. Zhang and J. Jin, J. Mater. Chem. A, 2018, 6, 2927–2934 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta01023e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Md. Mushfequr
Rahman
a,
Md. Mushfequr
Rahman