
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic stability, phase separation and Ag grading in (Ag,Cu)(In,Ga)Se2 solar absorbers†
Kostiantyn V.
Sopiha
 *a,
Jes K.
Larsen
a,
Olivier
Donzel-Gargand
a,
Faraz
Khavari
a,
Jan
Keller
a,
Marika
Edoff
a,
Charlotte
Platzer-Björkman
a,
Clas
Persson
bc and
Jonathan J. S.
Scragg
a
*a,
Jes K.
Larsen
a,
Olivier
Donzel-Gargand
a,
Faraz
Khavari
a,
Jan
Keller
a,
Marika
Edoff
a,
Charlotte
Platzer-Björkman
a,
Clas
Persson
bc and
Jonathan J. S.
Scragg
a
aSolar Cell Technology, Department of Materials Science and Engineering, Uppsala University, Box 534, SE-75121 Uppsala, Sweden. E-mail: kostiantyn.sopiha@gmail.com
bDepartment of Materials Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
cCenter for Materials Science and Nanotechnology, Department of Physics, University of Oslo, P.O. Box 1048 Blindern, NO-0316 Oslo, Norway
First published on 13th April 2020
Abstract
Gallium alloying and grading in Cu(In,Ga)Se2 (CIGS) are well-established strategies for improving performance of thin-film solar cells by tailoring band profiles within the absorber. Similarly, Ag incorporation is considered to be an effective complementary route towards further advancement of the field. Herein, we explore thermodynamics of the formation of (Ag,Cu)(In,Ga)Se2 (ACIGS) alloy. Using first-principles methods, we reveal the existence of a miscibility gap in the Ga-rich alloys at temperatures close to those employed for the co-evaporation growth. We demonstrate that this property can result in phase separation and the formation of Ag gradients throughout the film thickness. We prove experimentally that the phase separation can indeed occur during low-temperature growth and/or post-deposition treatments. Furthermore, we uncover the anticorrelation between Ag and Ga contents, and demonstrate thermodynamically-driven formation of [Ag]/([Ag] + [Cu]) gradients in films with a steep [Ga]/([Ga] + [In]) profile. Finally, we discuss how these phenomena can influence solar cell devices. The presented results are expected to provide fundamental insight into the physics of growth and processing of ACIGS absorbers, which could be utilized to further boost the efficiency of thin-film solar cells.
Introduction
Among the absorber materials for thin-film photovoltaics, Cu(In,Ga)Se2 (CIGS) emerges as one of the best-performing and most promising candidates due to the unique combination of chemical stability, high absorption coefficient, non-toxicity, long carrier lifetimes, and excellent transport properties in the polycrystalline form.1 Although this material has been studied for nearly 50 years, recent boosts in solar cell efficiency2–4 prove that further advancements of the technology are still possible. To make the next progressive step, many research teams are looking closely at various types of functional alloying in CIGS.The main idea behind alloying as a method is that fundamental electronic properties of semiconductors can be deliberately manipulated, which is incredibly useful for developing thin-film photovoltaics technology in particular. From one side, a homogeneous change in absorber composition can be used to adjust the band gap for higher fundamental efficiency limits, whether it be for the single-junction5 or tandem architecture.6 In addition, shifting band edges by alloying allows tailoring band offsets at the absorber/buffer interface to minimize interface recombination.7,8 From the other side, formation of a compositional gradient can further promote photogenerated carrier separation. An effective utilization of these effects is best exemplified by CIGS itself, which combines optimum [Ga]/([Ga] + [In]) (GGI) level of 0.2–0.4 in the main absorbing region and GGI grading promoting carrier selectivity at the back surface to achieve efficiencies over 20%.9,10 The problem with Ga–In alloying, however, is that it mainly widens the band gap of CIGS by lifting its conduction band minimum,11 which can result in so-called “cliff”-type conduction band offset in wide-gap absorbers, and thus, potentially increase the Voc deficit.12
Besides the well-established case of Ga–In intermixing, two other strategies are gaining increasing attention, namely Ag–Cu (ACIGS) and S–Se (CIGSSe) alloying. Both of them have already been shown to improve certain aspects of the absorber materials and device performance.5,6,9,10,13,14 Alloying of S on the Se sublattice has been performed by sulfurizing CIGS in H2S13–16 or S-rich17–19 atmosphere. The latter approach results in the formation of a stoichiometric CuInS2 surface with underlying CuIn(S,Se)2 mixed crystals and S in-diffusion in Cu-depleted grain boundaries further underneath.18,19 Such an elemental distribution, on the one hand, creates a double gradient promoting the photogenerated carrier separation and suppressing the interface recombination,9 while on the other hand, the S-rich layer can act as an electron transport barrier limiting the performance.15,17 The competition between these effects requires an accurate control of S incorporation for optimum performance, which is difficult to achieve in practice. For instance, controllable S alloying during co-evaporation is a challenge due to the high partial pressure of elemental S at the baseline growth conditions.20 In contrast, Ag incorporation on the (I) sublattice (i.e. group-I element position in the chalcopyrite lattice) can be easily achieved during the co-evaporation stage,5,6 providing more control over the concentration and distribution of the elements. With the addition of Ag, it was made possible to fabricate wide band gap ACIGS-based solar cells that had reduced interface recombination and smaller Voc deficit as compared to the CIGS devices with high GGI.5,6,8 For single-junction ACIGS devices, the optimum performance has been achieved at [Ag]/([Ag] + [Cu]) ≈ 0.2 and GGI ≈ 0.4, with recently reported efficiencies of 19.9%21 and 21%.5 Among wide-gap ACIGS absorbers for tandem applications, prominent results in terms of efficiency are 13.0% for a band gap of 1.6 eV,22 14.8% for a band gap of 1.5 eV,6 and 15.1% for a band gap of 1.45 eV.8 One explanation for these improvements is that co-alloying Ga and Ag allows engineering conduction band offset at the absorber/buffer interface in addition to the band gap tuning achieved by the Ga–In intermixing.8 Furthermore, since Ag-based ternary chalcopyrites have lower melting temperatures than their Cu-based counterparts,23 Ag alloying is also considered as a way to promote crystallisation of the absorbers and open a realm for developing lower-temperature routes for fabricating solar cells on flexible polymeric substrates.24
For alloying to be effective in tuning the semiconductor properties, the relevant alloy compositions should be (a) miscible during the deposition and (b) stable (either kinetically or thermodynamically) during the post-deposition processing and/or storage. Otherwise, formation of a homogeneous alloy may not be feasible, or decomposition of an unstable alloy could lead to segregation of impurity phases that might be harmful to the device performance.25,26 For instance, CIGS is an alloy with a complete miscibility above 400 K,27 which is much lower than typical temperatures used during the co-evaporation. In principle, the existence of a miscibility gap below 400 K could lead to phase segregation in CIGS at near-room temperature, but this effect does not occur in practice due to slow transformation kinetics at ambient temperatures. Similarly, previous first-principles calculations indicate that (Ag,Cu)GaSe2 has a miscibility gap that extends to much higher temperatures than in CIGS.28 Indeed, a limited miscibility was reported for ACIGS ingots grown at 600–800 °C,29 although more recent investigations on thin-films show complete miscibility.30,31 The discrepancy can be attributed to the film off-stoichiometry, although other factors, such as the sample geometry and grain morphology, may also play a role. Indications of phase segregation have been found in ACIGS films deposited at lower temperatures (350 °C) though.32 Taking into account that diffusion on the (I) sublattice is expected to be faster than that on the (III) sublattice (i.e. group-III element position in the chalcopyrite lattice),33 the miscibility gap in the Ag-rich alloys may even be expected to manifest itself during baseline processing. In contrast, there are widespread reports of successful synthesis and characterisation of ACIGS, which stem from a common belief that the stability issues related to the miscibility gap do not occur in thin films.
In light of the recent progress in developing ACIGS-based solar cells, a more detailed analysis is needed to establish the true stability of absorbers formed by co-alloying on both the (I) and (III) sublattices simultaneously, which is a more practically relevant scenario. Motivated by this, we combine first-principles and experimental analyses to explore the thermodynamics of ACIGS alloy system. The obtained results are expected to guide further design of ACIGS-based solar cells and promote understanding of semiconductor alloys in general.
Methods
We consider ACIGS as a substitutional alloy within the regular solution model,34–36 which defines the free energy of mixing as Gmix = Hmix − TSmix, where Hmix is the mixing enthalpy for the studied system, Smix is the configurational entropy of mixing for ideal solution, and T is the temperature. For a binary alloy, the mixing enthalpy is equivalent to the bowing parameter (parabolic fit) of the formation energy versus composition that can be computed using first-principles methods. Since in ACIGS alloys the (I) sublattice is shared by Ag and Cu atoms, whereas the (III) sublattice is co-occupied by In and Ga, the mixing enthalpy can be generalized as:35| Hmix = ω1yx(1 − x) + ω2(1 − y)x(1 − x)+ω3xy(1 − y) + ω4(1 − x)y(1 − y) | (1) |
| Hmix = Etot(x,y) − (Axy + Bx + Cy + D) | (2) |
By fitting the values computed with eqn (2) by the function given in eqn (1), the interaction parameters can be determined. Further, in the case of two independent sublattices, the mixing entropy within the regular solution approximation is the sum of the mixing entropies for its sublattices, as given by the following analytical expression:
Smix = kB(x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(x) + (1 − x)ln(1 − y)+yln(y) + (1 − y)ln(1 − y)) ln(x) + (1 − x)ln(1 − y)+yln(y) + (1 − y)ln(1 − y)) | (3) |
To calculate ω1, ω2, ω3, and ω4, we sampled the entire composition range (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) into a 10 × 10 grid yielding 100 composition points. For each of them, a 216-atom-supercell with random occupation of the cation sublattice sites was generated using the special quasi-random structure (SQS) algorithm,37 as implemented in the Alloy-Theoretic Automated Toolkit (ATAT) package.38 Then, first-principles calculations within density functional theory were carried out using the Vienna Ab initio Simulation Package (VASP).39–41 The projector augmented wave (PAW) pseudopotentials42,43 with valence electron configurations of 3d104s1 for Cu, 4d105s1 for Ag, 4d105s25p1 for In, 3d104s24p1 for Ga, and 4s24p4 for Se were employed to model the effect of core electrons. The total energy calculations were performed with the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional.44 The Brillouin-zone integrations were performed using 2 × 2 × 2 Γ-centred Monkhorst–Pack grid45 and a cut-off energy of 350 eV. The ionic relaxation threshold was set to 0.01 eV Å−1. The cell geometry optimization was enabled to exclude possible impacts of strain.
To assess thermodynamic stability, binodal lines were calculated from the common tangent construction;35,36 in this conception, an alloy with a given composition (x, y) is stable if a tangent plane to the free energy surface ΔG(x,y) at the given composition point does not cross the surface at any other composition. The spinodal curve for the alloy was constructed by solving the following equation:36
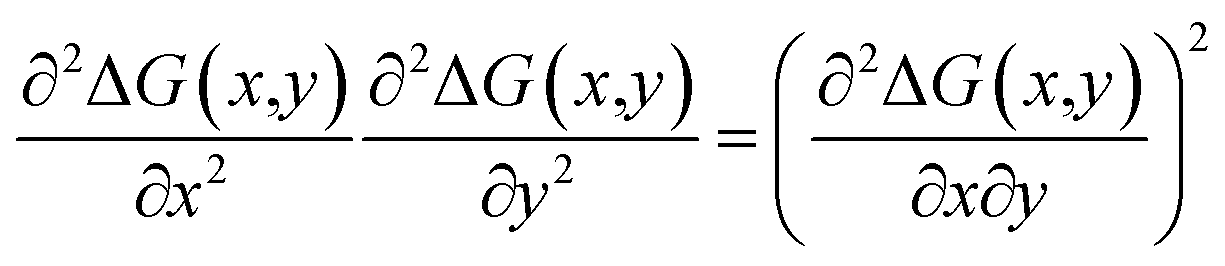 | (4) |
Notably, the total free energy of the alloy ΔG(x,y) is connected to its free energy of mixing via the relation:
| ΔG(x,y) = Gmix(x,y) + G0(x,y) | (5) |
Following the convention for Hmix given by eqn (1) and (2), G0(x,y) should also be a function of the form:
| G0(x,y) = Exy + Fx + Gy + H | (6) |
Importantly, the first term on the right-hand side is non-linear, thus its mixed second-order partial derivative is non-zero. Therefore, the expression for G0(x,y) contributes to the computed binodal and spinodal lines. In eqn (6), we calculate E, F, G, and H values based on the formation enthalpies of the ternary compounds obtained by first-principles calculations for the primitive cells (see Table S1 in the ESI†), yielding 43.8, −146.8, 60.6, and −1781.9 meV/formula (per four atoms) for the corresponding constants.
In principle, eqn (2), (5), and (6) could be grouped into a single expression:
 | (7) |
A notable limitation of the employed approach is that vibrational free energy is neglected, suggesting that the computed critical (consolute) temperature is likely to be overestimated.46 Another error in the critical temperature can be caused by the basic assumption of randomness of the alloy, which implies (a) averaging out all interatomic interactions with the parabolic function in eqn (1) and (b) neglecting entropic correlation between atomic clusters in eqn (3).47 Resolving these limitations is necessary to reproduce the stability of ordered defect compounds and their alloys, such as (Ag,Cu)(In,Ga)3(S,Se)5 and (Ag,Cu)(In,Ga)5(S,Se)8, but it is not critical for random alloys, such as stoichiometric ACIGS. For random alloys, it was reported that the regular solution model provides about 20% higher consolute temperatures compared to the results of Monte Carlo simulations.48 As such, these errors are not expected to affect conclusions of this study qualitatively, whereas quantitatively they would be partially cancelled out if a more accurate treatment of exchange-correlation interactions was employed. Specifically, our tests revealed that using the hybrid Heyd–Scuseria–Ernzerhof functional (HSE06) with 25% fraction of exact exchange49 yields systematically higher mixing enthalpies for ACIGS, resulting in up to 40% larger interaction parameters. A focused analysis of the exchange-correlation interactions is needed in order to pinpoint the exact origin of such a strong dependence on the functional.
To determine the equilibrium elemental distribution within ACIGS films, we employed a Sequential Least SQuares Programming (SLSQP) algorithm finding the optimum AAC profile that corresponds to a minimum of free energy for a film with a fixed GGI profile and a given integral (average) AAC value at a constant temperature. This approach allows deduction of the elemental distributions in ACIGS films at equilibrium without considering the kinetics of the underlying processes. The analysis was performed in one-dimensional approximation within which the film was divided into 25 cells unless specified, with the objective function for the minimisation defined as:
 | (8) |
| ΔGi = nαiΔG(xαi,yαi) + nβiΔG(xβi,yβi) + nγiΔG(xγi,yγi) | (9) |
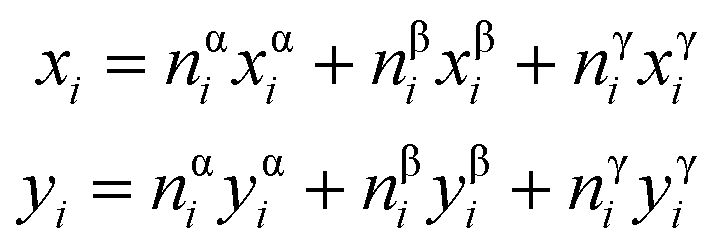 | (10) |
Experimental verification of the decomposition-related phenomena was performed on ACIGS films with varying integral AAC and similar GGI profiles. The absorber films were deposited following a three-stage co-evaporation process described in our previous works.5,8,50 In short, the elemental precursors were co-evaporated and deposited on two types of substrates, i.e. standard soda-lime glass (SLG) and high-strain-point K-rich/Na-poor glass (KRG), in each case coated with DC-sputtered Mo back contact layers. The first and third co-evaporation stages were poor in group-I elements, which was compensated by an excess of group-I elements during the second stage. The initial stage also involved increased Ga and reduced In deposition rates to realize a steep GGI gradient. The initial substrate temperature was set to 420 °C when depositing the Ga-rich layer and increased linearly while decreasing GGI deposition rate until reaching 550 °C at the final stages of the deposition. A thin NaF layer of 10–15 nm was evaporated on top of the Mo before the ACIGS deposition. The integral compositions of the films were measured using X-ray fluorescence (XRF) in a Spectro X-Lab 2000 instrument. The elemental distributions within the films were measured by Glow-Discharge Optical Emission Spectroscopy (GDOES) using a Spectruma Analytik GDA 750 HR system, which provides concentration profiles averaged over an area of about 5 mm2. The integral compositions from the XRF analysis were used to quantify the GDOES profiles. The cation compositions of as-grown films, their band gaps, and typical device characteristics are summarised in Table S2,† where one can see that the efficiencies of our cells are at the 17–18% level for narrow-gap ACIGS. The corresponding cation deposition protocols for each sample are presented alongside the GDOES data in the ESI.† Cross-section imaging was performed using a probe-corrected FEI Titan Themis XFEG scanning transmission electron microscope (STEM) equipped with a SuperX energy dispersive X-ray spectroscopy detector (EDS) and a Zeiss Merlin scanning electron microscope (SEM).
The phase decomposition within the ACIGS films was induced by performing a series of prolonged post-deposition anneals in a chemically-inert atmosphere (350 mbar Ar pressure) using a custom-built furnace. The annealing temperatures and times varied depending on the purpose of the anneal and are specified individually in the discussion below. After each anneal, the samples were cooled rapidly without changing the atmosphere, thus reaching room temperature within a minute. The same XRF data and thicknesses of as-grown samples were used to quantify the GDOES profiles for the annealed films as no significant material loss was expected during the annealing.
Results and discussion
The computed mixing enthalpy of ACIGS is given in Fig. 1. The first thing to note is that the parabolic fit provides a good approximation, and therefore, the introduction of higher order terms to the expression for mixing enthalpy would be redundant in this system. The computed values of ω1, ω2, ω3, and ω4 in eqn (1) are 49.2, 72.7, 130.2, and 77.9 meV/formula, respectively. These values are in a good agreement with the previously reported interaction parameters computed for quaternary alloys (i.e. ω2 and ω3 for CIGS and (Ag,Cu)GaSe2, respectively).28,51 As it was also noticed for chalcopyrite compounds earlier,11,28 the interaction parameters increase with a lattice mismatch (Δa/ā) between the terminal phases, which we estimate to be 2.4%, 3.5%, 6.5%, and 5.4% for the corresponding quaternary alloys (based on the optimized lattice parameters in Table S1†). The greatest computed bowing parameter among the quaternary compounds corresponds to (Ag,Cu)GaSe2, which indicates that alloying Ag into Ga-rich CIGS has a tendency to lower its stability, and thus, make it more prone to decomposition.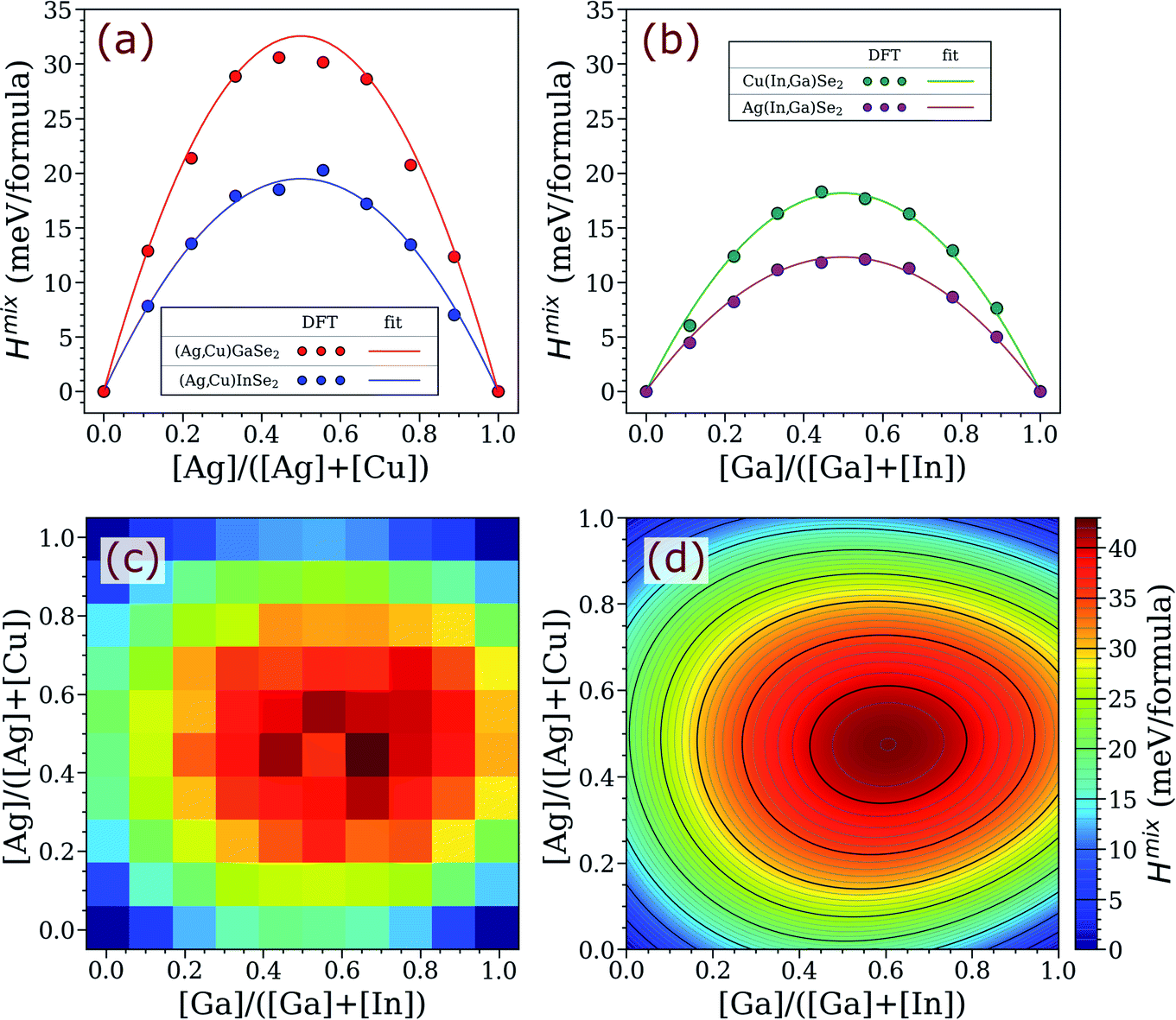 | ||
| Fig. 1 Mixing enthalpy of ACIGS alloy. (a and b) Computed values and the regular solution fits from eqn (1) projected for alloying on one cation sublattice (i.e. for the quaternary alloys). Heatmaps for (c) computed and (d) regular solution fit for the mixing enthalpy in case of alloying on both cation sublattices. | ||
In addition to the ground state energy, the stability of alloys at different processing temperatures can also be assessed within the regular solution approximation using the computed interaction parameters, as described in the methods section. Here, we are particularly interested in finding binodal and spinodal lines, where the former encompasses the composition range for all unstable alloys, including metastable ones that span the space between the former and the latter. From a practical perspective, all alloys under the binodal line are prone to decomposition, but only the metastable ones require thermally-activated nucleation, which is then manifested in inter-grain character of phase separation. In contrast, all alloys under the spinodal line can decompose spontaneously without nucleation, resulting in intra-grain phase separation (so-called spinodal decomposition).52
The computed binodal (black) and spinodal (red) lines for ACIGS overlaid on top of heatmaps for the free energy of mixing at 350 °C (typical temperature of post-deposition alkali treatments) and 50 °C (operation temperature) are plotted in Fig. 2a and b. As one can see, the area encompassed by the binodal line at 350 °C is relatively wide and centres around Ga-rich compositions (GGI → 1.00) with moderate Ag content (AAC ≈ 0.50). This area expands significantly at 50 °C. Thus, all ACIGS systems within this wide region tend to separate into either two or three stable phases. For instance, one can see that a homogeneous ACIGS alloy with GGI = 0.85 and AAC = 0.50 (magenta marker) is neither stable at 50 °C nor at 350 °C, undergoing a phase separation into two phases, one being Ag-rich/Ga-depleted and the other Ag-poor/Ga-enriched (yellow markers).
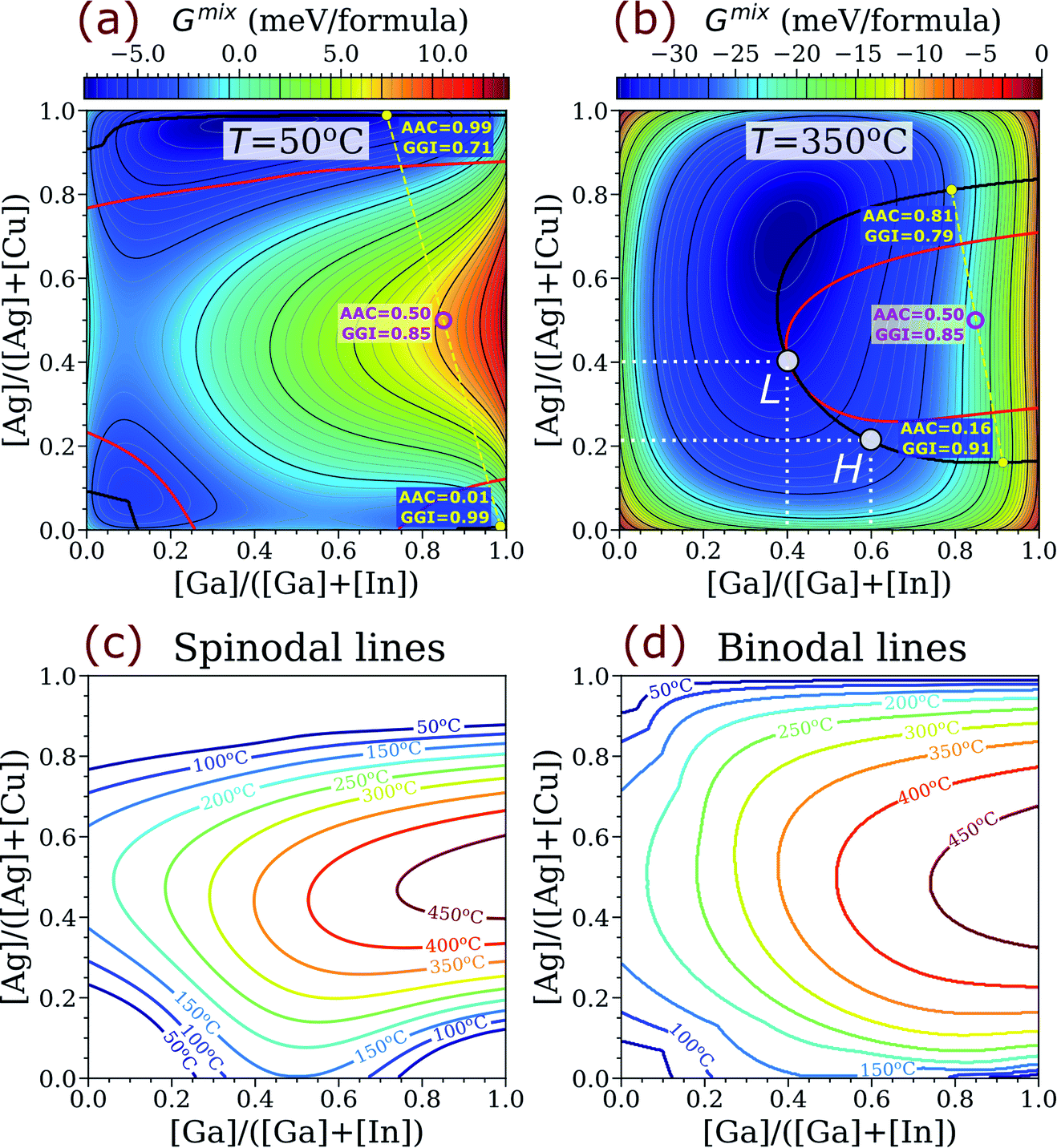 | ||
| Fig. 2 Stability of ACIGS alloy at different temperatures. Heatmaps constructed for free energy of mixing as functions of the alloy composition at (a) 50 °C and (b) 350 °C; the red and black solid curves represent spinodal and binodal lines. The yellow markers indicate computed compositions of stable phases formed upon decomposition of ACIGS alloy with AAC = 0.50 and GGI = 0.85; the open magenta marker shows overall composition of the phase mixtures. The H and L points indicate limiting AAC in ACIGS with model high (0.60) and model low (0.40) GGI levels, respectively. Temperature dependence for the (c) spinodal and (d) binodal lines. | ||
The tendency for decomposition in ACIGS as such is of high practical importance, since 350 °C is within the temperature range for post-deposition treatments.50,53 The kinetics of phase separation, however, are determined by the rates of diffusion, which might be too slow at 350 °C to manifest during baseline processing. This is particularly important for metastable alloys due to the need for thermally-activated nucleation. For example, despite of the existence of the miscibility gap in the CIGS system at room temperature,27 experimentally, decomposition on the (III) sublattice is not observed due to slow Ga–In interdiffusion.33 In this regard, relatively fast Ag–Cu inter diffusion is more likely to mediate the decomposition on the (I) sublattice. Furthermore, if the film composition falls in the wide instability region encompassed by the spinodal line, the decomposition could proceed without nucleation even at room temperature. The resulting intra-grain compositional inhomogeneities could induce spatial band profile perturbations, and hence, influence charge carrier mobility. Even if cation diffusion would limit the size of such inhomogeneities to only a few nanometers, they could still be causing detrimental ageing effects in solar cells. Finally, the miscibility gap can also hinder formation of homogeneous ACIGS absorbers on polymeric substrates, which naturally require lower processing temperatures,24 thus limiting further advances on that front.
To explore the evolution of alloy stability upon heating/cooling, spinodal and binodal curves akin to those in Fig. 2a and b were constructed for temperatures ranging from 50 °C (operation condition) to 550 °C (typical deposition condition). The obtained phase diagrams are presented in Fig. 2c and d. As one can see, the areas under the curves decrease gradually upon heating, as the entropy contribution to the free energy increases. The instability region eventually vanishes at 482 °C, which marks the critical temperature at which ACIGS alloy becomes miscible throughout the entire composition spectrum. Noteworthy, since this consolute point is close to typical substrate temperatures during the ACIGS co-evaporation,5,30,31 the homogeneity of alloys with AAC ∼0.50 and GGI ∼1.00 could be affected thermodynamically even at the deposition stage. Conversely, at near-room temperature, the instability region spans the majority of ACIGS compositions, except for the regions near the ternary phases (i.e. the corners of the composition grid). Hence, spinodal decomposition during the operation/storage could be a reasonable concern for most ACIGS absorbers. Naturally, however, experimental verification is needed to assess kinetics of the predicted decomposition.
While the near-room-temperature decomposition awaits verification, another phenomenon induced by the miscibility gap in ACIGS films during the post-deposition treatment can be pointed out. Specifically, unlike in the discussed model, the elemental distribution in CIGS absorber films is inhomogeneous, particularly due an intentional GGI grading with higher Ga content towards the back contact. Such a design is adopted to promote photogenerated carrier separation by forming a gradient of conduction band minimum throughout the film.9,10 The results presented in Fig. 2, however, indicate that stability of the alloys in Ga-rich and Ga-poor parts of the film diverges with addition of Ag, which results in different local equilibrium compositions throughout the absorber. This tendency can be better explained in a model ACIGS film with an integral AAC = 0.30 undergoing post-deposition treatment at 350 °C. Taking GGI values near the Ga-rich back and Ga-poor front contacts of 0.60 and 0.40, respectively, the AAC miscibility limits (AAC values at which the alloy composition hits the binodal line) are estimated to be 0.21 and 0.40, respectively. These limiting compositions correspond to the H and L points in the phase diagram in Fig. 2b. Notably, the model integral AAC = 0.30 exceeds the limiting value for ACIGS near the back contact (AAC = 0.21), and thus, the alloy in that region becomes unstable and would stabilise itself by either decomposing locally or distributing the excess matter throughout the film thickness. While the exact route of such transitions depends on both kinetics and thermodynamics of the competing processes, the stabilisation is likely to be mediated by Ag–Cu interdiffusion because the atoms on the (III) sublattice are relatively slow.33 As such, the stabilisation could cause a thermodynamically-driven formation of an AAC gradient, which could be useful for designing future thin-film solar cell devices.
To explore the possibility of thermodynamically-driven AAC grading in ACIGS films, a one-dimensional model was developed based on a minimization algorithm applied to the total free energy of the profile, as described in the methods section. The model GGI profile and the computed energy-minimized AAC distributions for different systems are presented in Fig. 3. As one can see, the computed AAC profiles have dips near the back contact that coincide with a hump in the GGI profile, revealing a thermodynamically-driven anticorrelation between them. The AAC profiles are flattened with temperature (see Fig. 3b), reflecting the retreat of the binodal line seen in Fig. 2. However, the AAC profiles do not level out completely even at 600 °C, which is significantly higher than the computed consolute temperature of 482 °C. Furthermore, this temperature is also higher than typical ACIGS co-evaporation, suggesting that AAC gradients can form unintentionally during baseline processing in absorbers with inhomogeneous GGI distribution.
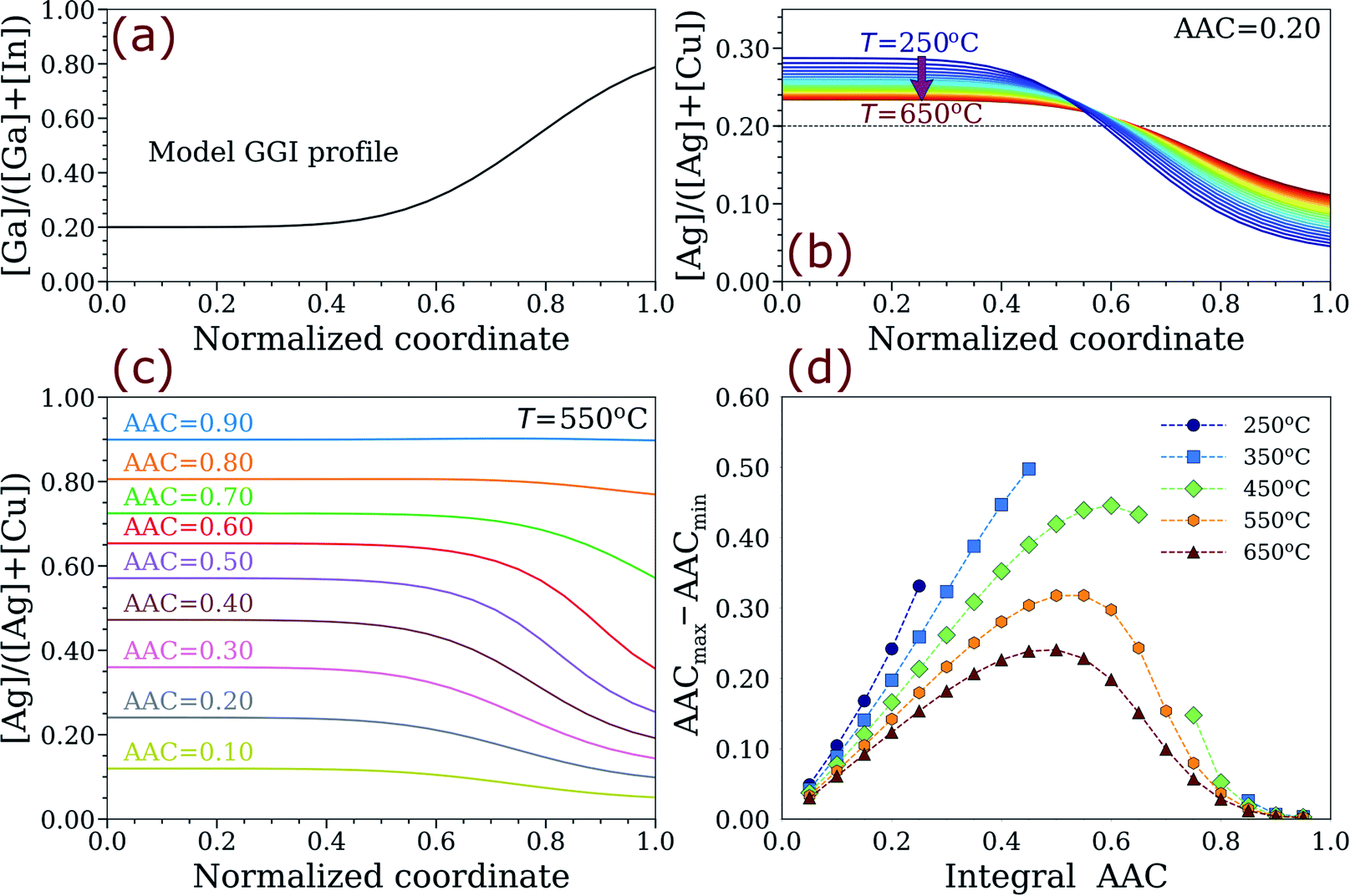 | ||
| Fig. 3 Thermodynamically-driven formation of AAC gradient in ACIGS films. (a) Model GGI profile (fixed). Computed equilibrium AAC profiles in ACIGS with (b) integral AAC = 0.20 at different temperatures and (c) different model integral AAC at 550 °C. (d) Calculated difference between the maximum and minimum of the optimized AAC profiles versus integral AAC at different temperatures. The omitted datapoints in (d) are to avoid quantifying profiles with occurring local phase decomposition (see Fig. S2 and S3†). | ||
The AAC grading appears to become more pronounced when the integral AAC is set closer to 0.50 (see Fig. 3c). To inspect this tendency, we also plotted the difference between the maximum and minimum of the optimized AAC profiles versus integral AAC at different temperatures, as shown in Fig. 3d. Indeed, the constructed graph generalizes the earlier conclusions, such that (a) AAC profiling is amplified upon cooling irrespective of Ag content, and (b) the strongest gradients form at integral AAC of 0.40–0.60, with the exact values depending on temperature. It is also worth noting that the AAC difference approaches zero faster when integral AAC → 1.00 than AAC → 0.00, indicating weaker anticorrelation in Ag-rich ACIGS films. This conclusion is particularly evident from Fig. S1,† which shows the relative AAC difference obtained by dividing the values in Fig. 3d by integral AAC (if AAC < 0.50) or 1 − AAC (if AAC > 0.50). This result could not be expected from analysing limiting AAC values based on the binodal lines in Fig. 2, which would predict a correlation between AAC and GGI instead of the discussed anticorrelation. This observation signifies the importance of the developed one-dimensional profile model.
A simple explanation for the observed AAC grading phenomenon can be given based on the partial derivatives of free energy, which are related to chemical potentials (μ) in ACIGS as:
 | (11) |
Since the grading characterises equilibrium AAC distributions at a fixed GGI profile, μAg − μCu must always be constant throughout the sample. As can be seen in Fig. S2,† the model cell compositions indeed follow iso-∂ΔG(x,y)/∂y curves irrespective of the integral AAC and temperature. This result validates the presented one-dimensional profile model and provides a shortcut for the future analyses of thermodynamically-driven grading in alloys.
One can also notice that some datapoints in Fig. 3d are omitted. This is done to avoid quantifying profiles with local phase decomposition, which is the scenario characterised by formation of two-phase mixture in some cells of the model profiles (see Fig. S2c and d† for more details). Such a phenomenon was observed for all integral AAC values shown in Fig. 3c, although inducing it required much lower temperatures for the profiles with low Ag content. Despite being thermodynamically favourable, such a decomposition might by hindered in practice owing to slow kinetics of nucleation, which is a necessary initial step on a way towards phase separation in metastable systems. A comprehensive description of the alloy evolution would require employment of kinematic models,36,54 which is outside the scope of this study.
To verify the unintentional formation of AAC gradients in standard ACIGS absorbers, we investigated a series of samples with different integral AAC levels but similar GGI profiles. The films were deposited following a baseline three-stage co-evaporation route on different glass substrates and later processed into solar cells (see Methods section and/or our previous works5,8 for details). Noteworthy, for all samples, the substrate temperature at the final co-evaporation stage was set to 550 °C, which is slightly above the predicted consolute temperature of the alloy (482 °C). Typical GGI and AAC profiles measured by GDOES for a film with target integral AAC = 0.10 are presented in Fig. 4a.
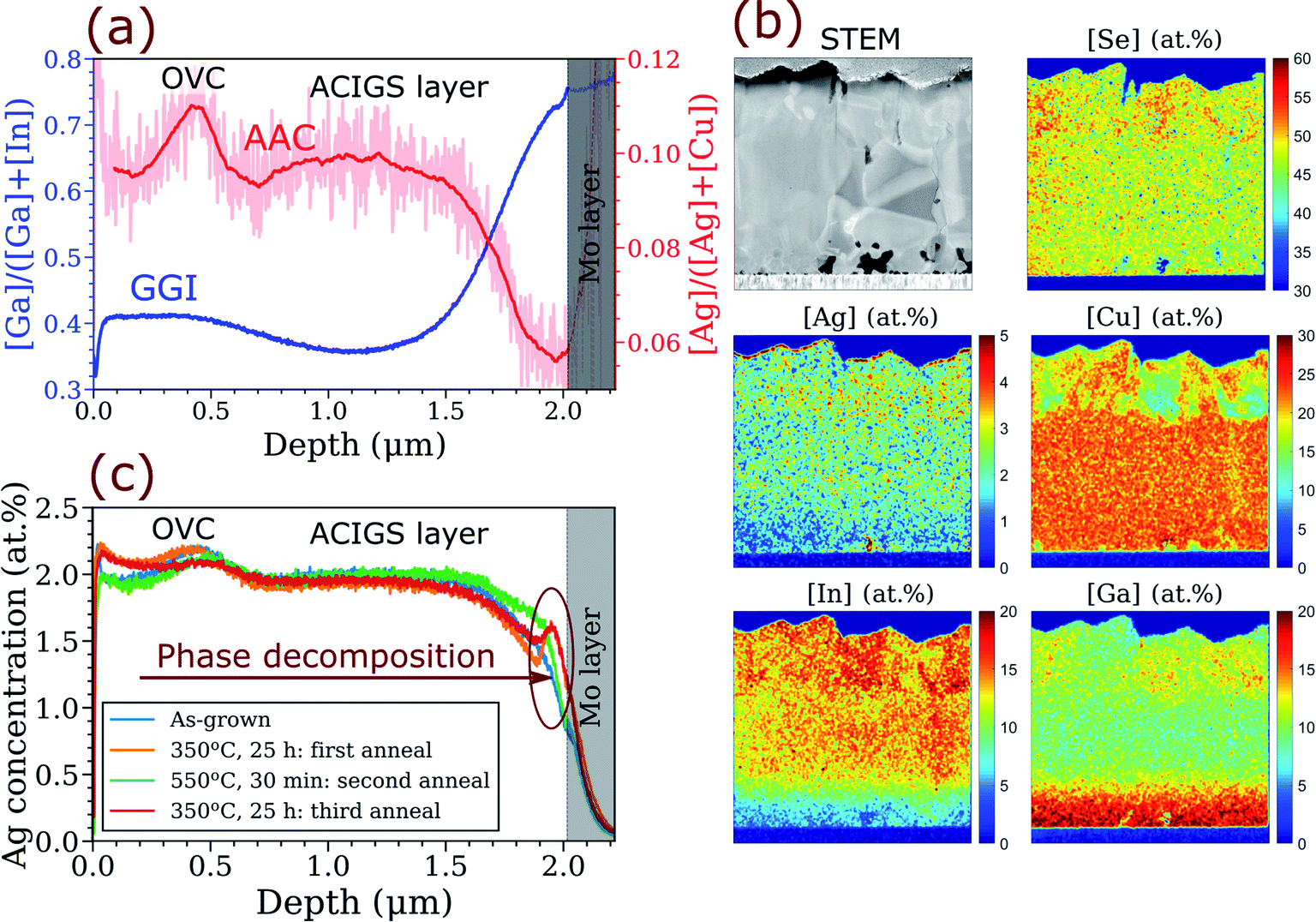 | ||
| Fig. 4 Elemental distribution in ACIGS film with AAC = 0.10 and strong GGI gradient deposited on K-rich/Na-poor glass substrate (sample AAC10@KRG in Table S2†). (a) Apparent anticorrelation between GGI and AAC profiles revealed by GDOES in as-grown ACIGS films (see Fig. S4 and S5† for more samples). (b) Cross-section STEM-EDS imaging showing slight Ag depletion in the Ga-rich region near the Mo back contact; the measurement is performed on the sample after the first anneal. (c) Repeatable formation of Ag-enriched region near Mo layer induced by annealing at 350 °C. | ||
The first thing to note in the GDOES profiles is the perturbations in AAC profile near the surface, which reflect the presence of ordered vacancy compounds (OVC) that were identified by EDS as patches of Cu-depletion with group-III and Se enrichments (see Fig. 4b). The formation of surface OVC was found in all the films deposited on K-rich/Na-poor substrates, whereas the absorbers deposited on soda-lime glass were free of OVC, highlighting a strong influence of alkalis on crystallization of ACIGS. Both GDOES and STEM-EDS show that the OVC grains have higher Ag concentration than the underlying ACIGS grains, for a reason that is currently unknown. Another distinct feature is a steep GGI profile from about 0.40 near the surface to 0.75 near the back contact. More importantly, there is a slight but clear AAC gradient formed throughout the film thickness, with the highest value of about 0.10 in the middle and the lowest value of 0.06 near the back contact (here we exclude the region containing OVC). Notably, for this sample, Ag was only introduced during the first stage of deposition, meaning that its concentration would be higher near Mo if no thermodynamic interaction between Ag and Ga was taking place.
The anticorrelation between AAC and GGI was observed consistently in all investigated films irrespective of the integral AAC (see Fig. S4†) and of the stage at which Ag was introduced (see Fig. S5†), except for Ag-rich ACIGS with integral AAC = 0.80 (not shown), which exhibited a flat AAC profile, in line with the predictions in Fig. 3d. Moreover, the anticorrelation can also be recognized in some earlier works.8,55,56 The independence of the AAC profile on the stage at which Ag is introduced during deposition implies that Ag diffusion is considerably faster than that of Ga, affirming the key assumption of our one-dimensional profile model. Ag depletion near the Mo layer was also observed by EDS, as shown in Fig. 4b. The STEM-EDS imaging also suggests the intra-grain nature of the AAC grading, although further high-resolution investigation would be beneficial to ascertain this claim. Nonetheless, the presented characterisation not only proves the existence of the predicted anticorrelation between GGI and AAC in ACIGS but also provides strong experimental evidence for the thermodynamic origin of the effect.
Since the regular solution model predicted that the extent of grading depends on temperature, we further investigated Ag redistribution in ACIGS during a post-deposition heat treatment. In order to observe a noticeable change in AAC profile, the temperature of such a heat treatment must, from one side, be sufficiently low for the alloy to become unstable while, from the other side, still maintain relatively fast diffusion of Ag to mediate the process. Hence, we performed the heat treatment at 350 °C for 25 hours in a chemically-inert atmosphere (350 mbar Ar pressure). The measured elemental profiles for ACIGS with integral AAC = 0.10 before and after the heat treatment are compared in Fig. 4c. As one can see, the annealing indeed induced a significant redistribution of Ag within the ACIGS film. However, instead of altering the AAC gradient as expected from the results in Fig. 3b, the major change occurred near the Mo layer where the formation of a peak in Ag concentration is seen. Most likely, formation of such a peak reflects local decomposition of ACIGS alloy in the region with the highest GGI. Similar enrichment near the back contact was found in most analysed samples (see Fig. S6†), although the shape of the Ag peak and its intensity varied among them. The peak intensity was higher in the samples deposited on K-rich/Na-poor substrates, once again demonstrating the impact of alkalis on crystallization of ACIGS and drawing more attention to this important topic. Noteworthy, some Ag accumulation also occurred near the ACIGS surface, as shown by both GDOES and EDS in Fig. 4. However, this feature was observed inconsistently (see Fig. S6†), and is therefore disregarded in the discussion henceforth.
Special attention should be paid to the fact that location of the Ag-enriched peak after the annealing in Fig. 4c is different from the prediction of our one-dimensional profile model (see Fig. S2c and d†). Specifically, the model suggests that local decomposition should first occur in the middle of the films with AAC ≤ 0.50, and not near the back contact as observed in the experiments. One could attribute this discrepancy to the fact that GDOES averages the composition over certain volume, which may indeed contain a mixture of different phases. Such explanation, however, would imply that concentrations in the interfacial regions must be averaged as well, thus excluding the possibility of Ag enrichment everywhere in the profiles. Another possible explanation for the preferential phase separation near the back contact could be that the phase decomposition simply occurs faster than equilibration of the AAC profile. In this case, the system would stabilize by decomposing locally in the least stable Ga-rich region near Mo layer instead of taking a slower AAC equilibration route to reach the energy minimum. To test this hypothesis, we applied the same energy minimization algorithm to profiles with AAC pinned at constant level, thus only enabling local decomposition in each model cell. As shown in Fig. S3,† in such a case, the decomposition predictably occurs near the back contact. Furthermore, the local decomposition is also seen for the profiles with considerably lower integral AAC if Ag level is fixed. Comparing these values with AAC = 0.10 for the heat-treated sample and noting that the overall shape of the Ag profile does not change much after the annealing, one can conclude that the scenario of frozen Ag profile is indeed more consistent with the condition of post-deposition annealing at 350 °C. However, it cannot be excluded at this point that the preferential precipitation at the Mo surface is a product of other factors neglected in the model, such as off-stoichiometry, an interfacial energy contribution, or simply a higher density of nucleation sites at the ACIGS/Mo interface.
Since the described decomposition upon post-deposition heat treatment stems from a temperature-dependent equilibrium, it ought to be reversible. We studied the reversibility of the decomposition by performing two further anneals for the samples with the formed Ag peak, first at 550 °C for 30 minutes (second anneal) and then again at 350 °C for 25 hours (third anneal). Not only was the annealing at 550 °C sufficient to reset the Ag profile closer to its original shape, but also the following annealing at 350 °C caused the Ag peak to reappear near the back contact, as shown in Fig. 4b, confirming the expected reversibility. An attempt was made to characterise this peak region using STEM-EDS, but no Ag-rich grains were identified within the studied lamellas. Moreover, no distinct signature that could be attributed to an Ag-rich phase was found with Raman analysis on the back side of peeled-off absorbers (not shown here). These negative results can be rationalized by the fact that integral AAC value for this sample is relatively low (0.10), and thus, if the Ag-rich grains segregated near the back contact, their amount would be too small for these techniques to detect.
In order to promote phase separation to such a degree that it could be detected with STEM-EDS, we performed another annealing for 60 hours at 350 °C on Ga-rich ACIGS sample (target integral GGI = 0.85 without grading) with integral AAC = 0.50. This composition corresponds to the magenta marker in Fig. 2a and b, which shows that such an alloy should undergo an extensive decomposition. This film properties and the resulting solar cell performance are briefly summarised in Table S2† and characterised in detail as a part of our previous work.8 As can be seen in the cross-section STEM-EDS in Fig. 5 and S7,† the morphology of the ACIGS film changed drastically after the prolonged annealing. First and foremost, an extensive formation of small Ag-rich/Ga-depleted precipitates was found near the absorber surface. A detailed characterisation of these precipitations is yet to be performed but the very fact of their formation is a direct proof of the tendency for decomposition. Remarkably, not only the Ag-rich character but also the Ga-depletion are in accordance with the concentrations of stable phases indicated by yellow markers in Fig. 2b. Here, the surface localization of the Ag-rich precipitates can be attributed to the absence of GGI grading, which means that all regions of this film are equally unstable, whereas the surface of ACIGS layer serves as a nucleation site. It is not clear, however, why similar Ag-rich grains did not appear near the Mo layer in this case. Second, severe degradation of the film was observed due to the extensive formation of large Sn–In–Ga–O patches throughout the film (see Fig. S7†). These oxides are likely to be a product of decomposing Zn–Sn–O (ZTO) buffer and i-ZnO(70 nm)/ZnO:Al(210 nm) bi-layer that were present during annealing as a residual from the cell characterization.8 This degradation mechanism might be critical for life-span of the solar cells but it is unclear whether this phenomenon is also driven by the same thermodynamic force as the discussed phase separation effect. Further cross-checking by annealing comparable samples with CdS buffer layers instead of ZTO, as well as samples without a buffer layer would be beneficial to determine the role of the buffer layer on the kinetics of the underlying process (i.e. nucleation and growth rates). It can be safely concluded, however, that segregation of Ag-rich phase has no direct relation to the Sn–In–Ga–O patches, since the locations of the Ag-rich precipitates were not correlated to those of the oxide patches (see Fig. 5 and S7†).
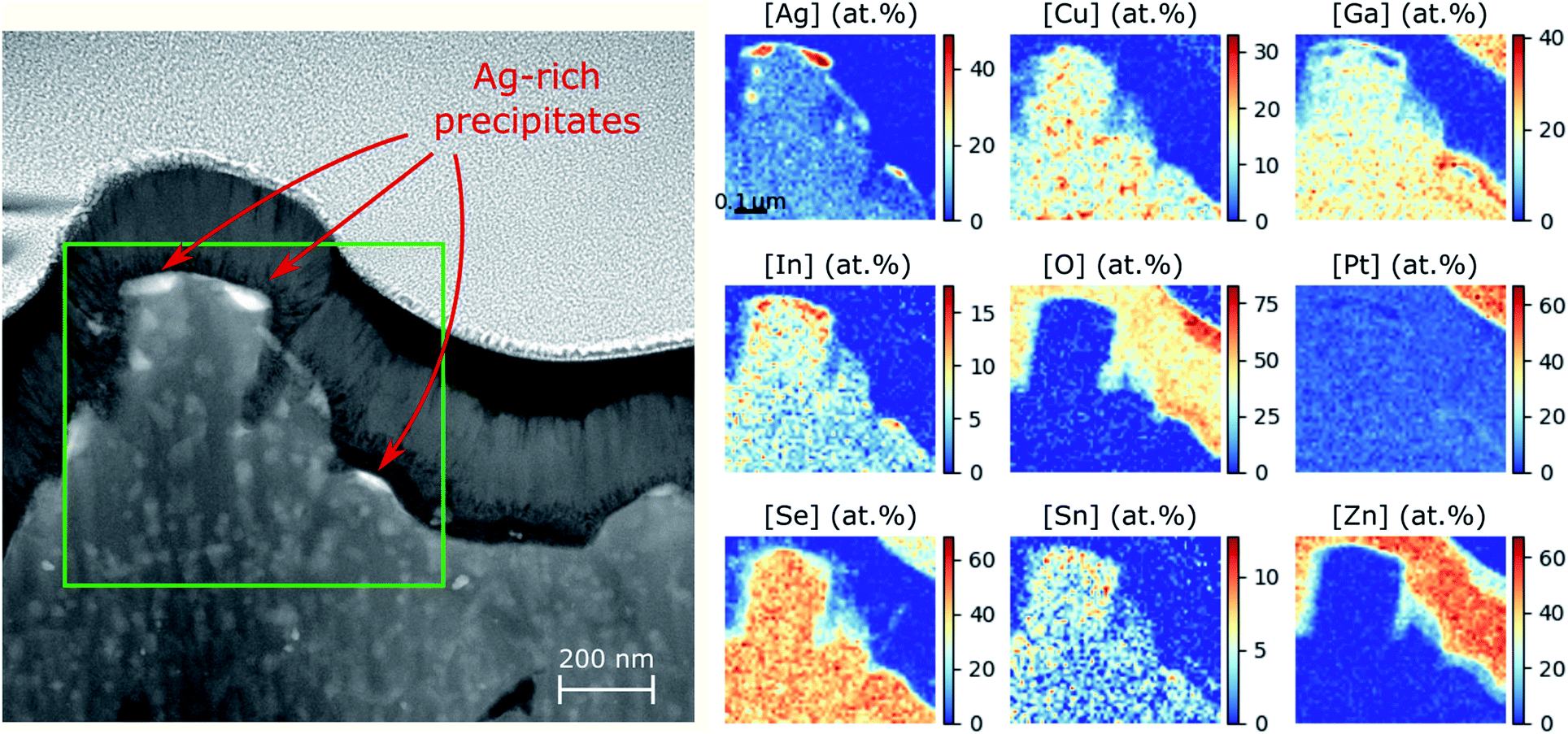 | ||
| Fig. 5 Cross-section STEM-EDS analysis of ACIGS film with integral AAC = 0.50 and GGI = 0.85 (sample AAC50@SLG in Table S2†) after heat treatment at 350 °C for 60 hours. The annealing and STEM-EDS analysis were performed with Zn–Sn–O (ZTO) buffer layer (more details on this device can be found in our previous work).8 Nucleation of Ag-rich precipitates is evident near the surface, indicating the phase decomposition. | ||
The decomposition-related phenomena in ACIGS have several important implications for designing thin-film solar cells. On the one hand, preventing the formation of Ag-rich regions within ACIGS absorbers is a desirable goal considering that impurity phases can be a source of trap-like regions. Indeed, previous calculations have shown that Ag-based chalcopyrites and kesterites have both band edges at lower energies compared to those of their Cu-based counterparts.7,28 This tendency has recently been extended to alloy systems and exploited to fabricate efficient wide-gap ACIGS cells.8 Hence, Ag-rich precipitates embedded in Ag-poor ACIGS would probably act as sinks for photogenerated electrons. In this work, decomposition was only obvious in GDOES profiles after 25 hours annealing at 350 °C, which is far from the typical deposition conditions. However, localised deviations in composition could have occurred earlier, for instance due to spinodal decomposition, which would be invisible to large-area measurements while still having a negative impact on the band profiles. Thus, it is of importance to analyse the compositional (in)homogeneity in ACIGS alloys at the nanometer scale by high-resolution characterisation methods. The phase separation may also interfere with the effects of post-deposition treatments, including alkali deposition that is commonly employed for optimizing CIGS performance.50 On the other hand, the unintentional AAC grading and the corresponding band profiling can facilitate separation of photogenerated carriers, akin to the carrier selectivity at the back surface achieved with GGI grading in CIGS. While a GGI gradient is realized by evaporating excess Ga at the first stage of deposition, which is preserved by relatively slow diffusion on the (III) sublattice33 and different formation kinetics of CuGaSe2 and CuInSe2,57 such a strategy fails for forming AAC gradients, due to fast Ag redistribution. Thus, alternative routes should be developed instead. With the possibility to tailor the AAC gradient by optimizing the deposition temperature, integral AAC level, and Ga grading, the corresponding band profiling could be yet another strategy to boost efficiency of chalcogenide solar cells.
Conclusions
By combining computational and experimental techniques, we investigated thermodynamic stability of ACIGS alloy and discussed its implications for fabrication of thin-film solar cells. First, our calculations revealed that Ag alloying into Ga-rich CIGS makes it unstable and can trigger decomposition during post-deposition treatment and/or storage. On the device level, we predicted that the composition-dependent instability can manifest itself in a form of anticorrelation between [Ag]/([Ag] + [Cu]) (AAC) and [Ga]/([Ga] + [In]) (GGI). Then, we showed experimentally by both GDOES and STEM-EDS that the anticorrelation leads to unintentional AAC grading within ACIGS films with a steep GGI profile. This result is directly related to the application, considering that GGI grading is a common strategy for promoting separation of photogenerated carriers in solar cells. Next, we demonstrated local phase decomposition of ACIGS in regions with high Ga content occurring during annealing at 350 °C for 25 hours. By comparing elemental distributions after a series of heat treatments, we showed the reversibility of the effect, thus proving its thermodynamic origin. Finally, using STEM-EDS imaging, we revealed segregation of Ag-rich/Ga-depleted precipitates in a Ga-rich ACIGS film after a prolonged annealing at 350 °C. The obtained results are expected to serve as a thermodynamic roadmap for engineering ACIGS solar cells and to provide a fundamental insight into stability issues in semiconductor alloys in general.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Swedish Foundation for Strategic Research (project number: RMA15-0030). We acknowledge the access to high-performance computing resources provided by Swedish National Infrastructure for Computing (SNIC). Many thanks to O. I. Malyi for constructive discussions on thermodynamic analysis and modelling of semiconductor alloys. We also acknowledge L. Riekehr for carrying out STEM-EDS analysis, as well as all other members of the thin-film solar cell group at Ångstrom laboratory for their help and valuable contributions.References
- S. Siebentritt, M. Igalson, C. Persson and S. Lany, Prog. Photovoltaics Res. Appl., 2010, 18, 390–410 CrossRef CAS.
- M. Nakamura, K. Yamaguchi, Y. Kimoto, Y. Yasaki, T. Kato and H. Sugimoto, IEEE Journal of Photovoltaics, 2019, 9, 1863–1867 Search PubMed.
- P. Jackson, R. Wuerz, D. Hariskos, E. Lotter, W. Witte and M. Powalla, Phys. Status Solidi RRL, 2016, 10, 583–586 CrossRef CAS.
- Solibro Research AB - News, http://solibro-research.com/en/news/, accessed October 31, 2019.
- M. Edoff, T. Jarmar, N. S. Nilsson, E. Wallin, D. Högström, O. Stolt, O. Lundberg, W. Shafarman and L. Stolt, IEEE Journal of Photovoltaics, 2017, 7, 1789–1794 Search PubMed.
- K. Kim, S. K. Ahn, J. H. Choi, J. Yoo, Y.-J. Eo, J.-S. Cho, A. Cho, J. Gwak, S. Song, D.-H. Cho, Y.-D. Chung and J. H. Yun, Nano Energy, 2018, 48, 345–352 CrossRef CAS.
- Z.-K. Yuan, S. Chen, H. Xiang, X.-G. Gong, A. Walsh, J.-S. Park, I. Repins and S.-H. Wei, Adv. Funct. Mater., 2015, 25, 6733–6743 CrossRef CAS.
- J. Keller, K. V. Sopiha, O. Stolt, L. Stolt, C. Persson, J. J. S. Scragg, T. Törndahl and M. Edoff, Prog. Photovoltaics Res. Appl., 2020, 28, 237–250 CrossRef CAS.
- T. Kato, Jpn. J. Appl. Phys., 2017, 56, 04CA02 CrossRef.
- M. Powalla, S. Paetel, E. Ahlswede, R. Wuerz, C. D. Wessendorf and T. Magorian Friedlmeier, Appl. Phys. Rev., 2018, 5, 041602 Search PubMed.
- S. Wei and A. Zunger, J. Appl. Phys., 1995, 78, 3846–3856 CrossRef CAS.
- J. Lindahl, J. Keller, O. Donzel-Gargand, P. Szaniawski, M. Edoff and T. Törndahl, Sol. Energy Mater. Sol. Cells, 2016, 144, 684–690 CrossRef CAS.
- T. Kato, J. Wu, Y. Hirai, H. Sugimoto and V. Bermudez, IEEE Journal of Photovoltaics, 2019, 9, 325–330 Search PubMed.
- T. Kobayashi, H. Yamaguchi, Z. Jehl Li Kao, H. Sugimoto, T. Kato, H. Hakuma and T. Nakada, Prog. Photovoltaics Res. Appl., 2015, 23, 1367–1374 CrossRef CAS.
- U. P. Singh, W. N. Shafarman and R. W. Birkmire, Sol. Energy Mater. Sol. Cells, 2006, 90, 623–630 CrossRef CAS.
- S. Kim, J. Nishinaga, Y. Kamikawa, S. Ishizuka, T. Nagai, T. Koida, H. Tampo, H. Shibata, K. Matsubara and S. Niki, Jpn. J. Appl. Phys., 2018, 57, 055701 CrossRef.
- J. K. Larsen, J. Keller, O. Lundberg, T. Jarmar, L. Riekehr, J. J. S. Scragg and C. Platzer-Björkman, IEEE Journal of Photovoltaics, 2018, 8, 604–610 Search PubMed.
- H. Aboulfadl, J. Keller, J. Larsen, M. Thuvander, L. Riekehr, M. Edoff and C. Platzer-Björkman, Microsc. Microanal., 2019, 25, 532–538 CrossRef PubMed.
- J. Keller, O. V. Bilousov, E. Wallin, O. Lundberg, J. Neerken, S. Heise, L. Riekehr, M. Edoff and C. Platzer-Björkman, Phys. Status Solidi A, 2019, 216, 1900472 CrossRef CAS.
- J. J. Scragg, T. Ericson, T. Kubart, M. Edoff and C. Platzer-Björkman, Chem. Mater., 2011, 23, 4625–4633 CrossRef CAS.
- C. P. Thompson, L. Chen, W. N. Shafarman, J. Lee, S. Fields and R. W. Birkmire, IEEE 42nd Photovoltaic Specialist Conference (PVSC), 2015, pp. 1–6 Search PubMed.
- G. M. Hanket, J. H. Boyle and W. N. Shafarman, 34th IEEE Photovoltaic Specialists Conference (PVSC), 2009, pp. 001240–001245 Search PubMed.
- J. L. Shay and J. H. Wernick, Ternary Chalcopyrite Semiconductors: Growth, Electronic Properties, and Applications: International Series of Monographs in The Science of The Solid State, Elsevier, 2017 Search PubMed.
- G. M. Hanket, C. P. Thompson, J. K. Larsen, E. Eser and W. N. Shafarman, 38th IEEE Photovoltaic Specialists Conference, 2012, pp. 000662–000667 Search PubMed.
- J. H. Werner, J. Mattheis and U. Rau, Thin Solid Films, 2005, 480–481, 399–409 CrossRef CAS.
- L. Gütay and G. H. Bauer, Thin Solid Films, 2007, 515, 6212–6216 CrossRef.
- B. Huang, S. Chen, H. Deng, L. Wang, M. A. Contreras, R. Noufi and S. Wei, IEEE Journal of Photovoltaics, 2014, 4, 477–482 Search PubMed.
- S. Chen, X. G. Gong and S.-H. Wei, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205209 CrossRef.
- J. E. Avon, K. Yoodee and J. C. Woolley, J. Appl. Phys., 1984, 55, 524–535 CrossRef CAS.
- J. H. Boyle, B. E. McCandless, W. N. Shafarman and R. W. Birkmire, J. Appl. Phys., 2014, 115, 223504 CrossRef.
- J. H. Boyle, B. E. McCandless, G. M. Hanket and W. N. Shafarman, Thin Solid Films, 2011, 519, 7292–7295 CrossRef CAS.
- K. Kim, J. W. Park, J. S. Yoo, J. Cho, H.-D. Lee and J. H. Yun, Sol. Energy Mater. Sol. Cells, 2016, 146, 114–120 CrossRef CAS.
- S. Nakamura, T. Maeda and T. Wada, Jpn. J. Appl. Phys., 2013, 52, 04CR01 CrossRef.
- I. Ho and G. B. Stringfellow, Appl. Phys. Lett., 1996, 69, 2701–2703 CrossRef CAS.
- J. B. Varley, X. He, A. Rockett and V. Lordi, ACS Appl. Mater. Interfaces, 2017, 9, 5673–5677 CrossRef CAS PubMed.
- E. Petrishcheva and R. Abart, Acta Mater., 2012, 60, 5481–5493 CrossRef CAS PubMed.
- A. Zunger, S.-H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS PubMed.
- A. van de Walle, P. Tiwary, M. de Jong, D. L. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L.-Q. Chen and Z.-K. Liu, Calphad, 2013, 42, 13–18 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- C. K. Gan, Y. P. Feng and D. J. Srolovitz, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235214 CrossRef.
- A. Sher, M. van Schilfgaarde, A.-B. Chen and W. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 4279–4295 CrossRef PubMed.
- F. Soisson and G. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 203–214 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- O. Donzel-Gargand, F. Larsson, T. Törndahl, L. Stolt and M. Edoff, Prog. Photovoltaics Res. Appl., 2019, 27, 220–228 CrossRef CAS.
- H. T. Xue, W. J. Lu, F. L. Tang, X. K. Li, Y. Zhang and Y. D. Feng, J. Appl. Phys., 2014, 116, 053512 CrossRef.
- A. M. Holder, S. Siol, P. F. Ndione, H. Peng, A. M. Deml, B. E. Matthews, L. T. Schelhas, M. F. Toney, R. G. Gordon, W. Tumas, J. D. Perkins, D. S. Ginley, B. P. Gorman, J. Tate, A. Zakutayev and S. Lany, Sci. Adv., 2017, 3, e1700270 CrossRef PubMed.
- T. Kobayashi, H. Yamaguchi and T. Nakada, Prog. Photovoltaics Res. Appl., 2014, 22, 115–121 CrossRef CAS.
- N. V. Storozhuk, K. V. Sopiga and A. M. Gusak, Philos. Mag., 2013, 93, 1999–2012 CrossRef CAS.
- G. M. Hanket, J. H. Boyle, W. N. Shafarman and G. Teeter, 35th IEEE Photovoltaic Specialists Conference, 2010, pp. 003425–003429 Search PubMed.
- L. Chen, J. Lee and W. N. Shafarman, IEEE Journal of Photovoltaics, 2014, 4, 447–451 Search PubMed.
- H. Rodriguez-Alvarez, A. Weber, J. Lauche, C. A. Kaufmann, T. Rissom, D. Greiner, M. Klaus, T. Unold, C. Genzel, H.-W. Schock and R. Mainz, Adv. Energy Mater., 2013, 3, 1381–1387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta00363h |
| This journal is © The Royal Society of Chemistry 2020 |