
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Choosing the right carbon additive is of vital importance for high-performance Sb-based Na-ion batteries†
Kristina
Pfeifer
a,
Stefanie
Arnold
 ab,
Öznil
Budak
bc,
Xianlin
Luo
a,
Volker
Presser
bc,
Helmut
Ehrenberg
ad and
Sonia
Dsoke
*ad
ab,
Öznil
Budak
bc,
Xianlin
Luo
a,
Volker
Presser
bc,
Helmut
Ehrenberg
ad and
Sonia
Dsoke
*ad
aInstitute for Applied Materials (IAM), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: sonia.dsoke@kit.edu
bINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany
cDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany
dHelmholtz-Institute Ulm for Electrochemical Energy Storage (HIU), P.O. Box 3640, 76021 Karlsruhe, Germany
First published on 7th March 2020
Abstract
Electrodes based on alloying reactions for sodium-ion batteries (NIB) offer high specific capacity but require bespoken electrode material design to enable high performance stability. This work addresses that issue by systematically exploring the impact of carbon properties on antimony/carbon composite electrodes for NIBs. Since the Sb surface is covered by an insulating oxide layer, carbon additives are crucial for the percolation and electrochemical activity of Sb based anodes. Instead of using complex hybridization strategies, the ability of mechanical mixing to yield stable high-performance Sb/C sodium-ion battery (NIB) electrodes is shown. This is only possible by considering the physical, chemical, and structural features of the carbon phase. A comparison of carbon nanohorns, onion-like carbon, carbon black, and graphite as conductive additives is given in this work. The best performance is not triggered by the highest or lowest surface area, and not by highest or lowest heteroatom content, but by the best ability to homogenously distribute within the Sb matrix. The latter provides an optimum interaction between carbon and Sb and is best enabled by onion-like carbon. A remarkable rate performance is attained, electrode cracking caused by volume expansion is successfully prevented, and the homogeneity of the solid/electrolyte interphase is significantly improved as a result of it. With this composite electrode, a reversible capacity of 490 mA h g−1 at 0.1 A g−1 and even 300 mA g−1 at 8 A g−1 is obtained. Additionally, high stability with a capacity retention of 73% over 100 cycles is achieved at charge/discharge rates of 0.2 A g−1.
1. Introduction
Energy storage technologies beyond lithium are of high interest since the demand for energy storage systems has highly increased throughout the past few years.1 When exploiting lithium sources, the price of the metal will rise as a consequence of the growing electrification.2 Sodium is the second-lightest alkali metal, and it is one of the Earth's most abundant elements. Therefore, sodium-ion batteries (NIBs) are a promising alternative combining low cost and similar intercalation chemistry to that of lithium.3,4 Tremendous efforts have been made to develop suitable materials for NIB electrodes.1,3,5,6 One of the main differences between sodium and lithium-ion batteries is the intercalation behavior into graphite. Graphite is successfully used as anode material for commercial lithium-ion batteries but cannot intercalate Na+-ions.7 Due to the chemical potential of sodium electroplating of the metal on the surface of graphite is energetically favorable.8 Hard carbons are alternatives to graphite, but have limited de-sodiation capacities of 100–300 mA h g−1.9 Therefore, a class of promising high capacity materials like tin, phosphorus, or antimony which can form Na-rich compounds through an alloying reaction was proposed in the literature.5,10–13 A key issue of alloy-based materials is the severe capacity fading, which prevents the commercialization of such anodes.10,14 The main reason for this fading is the large volume change during sodiation and de-sodiation, which can lead to aggregations and slower kinetics.12 If the volume expansion is not properly buffered, a massive pulverization can take place, causing severe cracking of the electrode.12,14 As a result, new surface areas are exposed, and the solid/electrolyte interphase (SEI) is continuously restructured. This is a crucial drawback as side reactions steadily proceed during cycling leading to a low coulombic efficiency (CE). Furthermore, an inhomogeneous thickness and composition of the SEI can partly block the charge transfer and cause a rapid capacity fading.15–17Besides the adjustments of binders and electrolytes, two main approaches are presented in state-of-the-art literature to counteract these problems: designing suitable nanostructures to improve the kinetics, and developing carbon matrices to buffer the volume changes and offer additional diffusion paths.12,18–23
Antimony (Sb) is a very common alloy material, which reacts with sodium to form Na3Sb with a theoretical capacity of 660 mA h g−1.24 Still, this material suffers from high volume change (∼300%)11,20,24 slow reaction kinetics during the Na+-ion insertion and extraction process, and the capacity is fading.20,25 The addition of carbon can counteract these drawbacks. Hence, the influence of carbon is widely discussed, and several different carbon materials in various concentrations have been proposed in the literature (Table 1).
| Potential vs. Na/Na+/V | C type | C amount in Sb/C/% | Preparation of Sb/C | Total electrode composition | Electrolyte | Normalization | Capacity/mA h g−1 at 100 mA g−1 | Reference |
|---|---|---|---|---|---|---|---|---|
| a Not available data from literature references are donated as “n.a.”. AB: acetylene black. MWCNTs: multi-walled carbon nanotubes. *: not mentioned if conductive additive is includes to the normalization. | ||||||||
| 0–2.5 | Cross-linked C | 56 | Wet synthesis of Sb/C (from Sb-acetate precursor) | 35% Sb, 45% C, 10% AB, 10% PVDF | 1 M NaPF6 in EC/DMC | Total mass of Sb and C* | ∼630 | Cui et al.21 |
| 0–2 | C fibers and carbon black | 15 | Mixing in a ball mill (all electrode components) | 70% Sb, 15% C, 15% CMC | 1 M NaClO4 in PC + 5% FEC | n.a. | ∼580 | Darwiche et al.24 |
| 0.01–2 | AB | 29 | Co-precipitation of Sb on C (SbCl3 and NaBH4) | 49% Sb, 21% C, 15% SuperP, 15% CMC | 1 M NaClO4 in PC + 5% FEC | Total mass of Sb and C* | ∼500 | Hou et al.27 |
| 0–2 | SuperP | 30 | Dry ball milling of Sb and C under Ar | 56% Sb, 24% C, 10% SuperP, 10% CMC | 1 M NaPF6 in EC/DEC + 5% FEC | Sb | ∼600 | Qian et al.26 |
| 0.01–2 | C-Nanofibers | 62 | Single-nozzle electrospinning (PAN and SbCl3) | 27% Sb, 43% C, 20% SuperP, 10% PAA | 1 M NaPF6 in EC/DEC + 5% FEC | Sb/C + SuperP | ∼500 | Wu et al.22 |
| 0–2 | MWCNTs | 22 | Wet ball milling of Sb and C in acetone | 62% Sb, 18% C, 10% SuperP, 10% CMC | 1 M NaClO4 in EC/PC + 5% FEC | Sb/C without SuperP | ∼500 | Zhou et al.46 |
| 0–2 | C-Fibers | 46 | Electrospinning (PAN and SbCl3) | 54% Sb, 46% C | 1 M NaClO4 in EC/DMC | Sb | ∼400 | Zhu et al.23 |
A key issue is the difficult comparison of electrode design operated in various electrochemical systems. For example, a discrepancy in the work of Qian et al.26 was highlighted by Darwiche et al.24 for bulk Sb. Qian et al.26 reported that bulk Sb (100 mesh = 149 μm) in an electrode composition of 80 mass% Sb, 10 mass% SuperP, and 10 mass% carboxymethyl cellulose (CMC) do not show comparable stability to their corresponding Sb/C composite. The bulk Sb electrode degraded during the initial cycles, whereas a nanocomposite of Sb and acetylene black obtained from a mechanical ball milling process could reach a capacity of 610 mA h g−1 normalized to the Sb amount. The work of Darwiche et al.24 revealed that bulk Sb (325 mesh = 44 μm) could exhibit similar stability as nanosized Sb. They presented a bulk Sb electrode combined with a conductive additive of vapor-grown carbon fibers (VGCF-S) and carbon black. An electrode composition of 70 mass% Sb, 15 mass% carbon black/carbon fibers, and 15 mass% CMC was able to deliver a stable capacity of about 550 mA h g−1 over 160 cycles.
To complicate matters, it is often not clarified in literature if the added conductive additive is included in the normalization.21,24,27 Since the capacity of SuperP is reported to exceed 150 mA h g−1 for NIBs,22,26 one must include the carbon additive to the normalization. Especially the main properties of the carbon as a conductive additive, buffering matrix, or element with synergetic added-value cannot be evaluated by comparison of the current literature. Several different electrolytes, carbon types, Sb/C compositions, Sb sizes, and morphologies, etc. lead to an incomparability of these electrochemical systems and a lack of consistency.
Another study was performed by Ramireddy et al.,28 which focuses on the amount of carbon and Sb particle size. Different ratios of Sb and graphite were ball milled, and the resulting composites were combined in an electrode with SuperP and CMC with the overall composition of 80 mass% Sb/C, 10 mass% SuperP, and 10 mass% CMC. The authors concluded that a high amount of carbon with a small particle size (ca. 1 nm) leads to the highest cycling stability. However, a loss of energy density needs to be tolerated when enhancing the amount of graphite.28 The authors identified important correlations between the electrochemical performance and the (initial) Sb size, but yet a systematic investigation focusing on the properties of the applied carbon is in high demand.
We present a guideline for choosing the right carbon additive for Sb/C composite electrodes in NIBs. We focus on the examination of carbon characteristics and their influence on the electrochemical performance. Therefore, we ensure ideal conditions for comparability by first adjusting the electrode composition, electrolyte, and way of normalization. By using six different carbons with a fixed (optimized) amount of 20 mass% C, 70 mass% Sb, 10 mass% CMC, we demonstrate that the properties of the carbon are of essential importance. A facile mixing process for electrode preparation allows keeping the integrity of the carbons as well as feasible commercialization. Our data demonstrate that a conductive additive is enough to be the only carbonaceous component in an Sb/C electrode, but the choice of the type of carbon is of vital importance. Since many carbons have a significant intrinsic specific capacity when operated in sodium-ion containing electrolytes, we also closely investigate the influence of normalizing the electrode performance just to the Sb content versus normalization to the Sb + C mass. By variation of different carbon additives, we adjust the electrochemical performance and the SEI. When applying 20 mass% onion-like carbon (OLC) with unique characteristics, we can tune the SEI properties and prevent cracking caused by volume expansion.
2. Results and discussion
2.1. Sb synthesis and characterization
Antimony nanopowder was obtained by a co-precipitation method. Fig. 1a and b shows the refined X-ray diffraction pattern and scanning electron micrographs. All reflexes of the XRD pattern can be assigned to trigonal Sb with the space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (ICSD: 55402) comprised within agglomerated nanoparticles with a size of 10–40 nm (Fig. 1b). Fig. 1c displays the results obtained from transmission electron microscopy which verify the SEM findings. A lattice plane distance of 3.11 Å was determined (inset Fig. 1c). The crystallographic planes of selected-area electron diffraction measurement (Fig. 1d) confirm the results obtained from XRD. The resulting Sb powder was used to prepare an electrode without carbon (90 mass% synthesized Sb, 10 mass% CMC). In the O 1s and Sb 3d X-ray photoelectron spectra of the pristine electrode (Fig. 1e), peaks originating from metallic Sb and Sb3+ can be found. This means that the Sb particles are covered by a thin (<10 nm) oxide layer. Sb as pure metal is electronically conductive, but the metal oxide layer acts as an insulating layer between the Sb particles. The result is a high resistance of the electrode and a very poor electrochemical performance with a capacity close to zero (Fig. 1f). This is why the presence of a conductive additive is of vital importance to enable Sb-based NIB electrodes.
m (ICSD: 55402) comprised within agglomerated nanoparticles with a size of 10–40 nm (Fig. 1b). Fig. 1c displays the results obtained from transmission electron microscopy which verify the SEM findings. A lattice plane distance of 3.11 Å was determined (inset Fig. 1c). The crystallographic planes of selected-area electron diffraction measurement (Fig. 1d) confirm the results obtained from XRD. The resulting Sb powder was used to prepare an electrode without carbon (90 mass% synthesized Sb, 10 mass% CMC). In the O 1s and Sb 3d X-ray photoelectron spectra of the pristine electrode (Fig. 1e), peaks originating from metallic Sb and Sb3+ can be found. This means that the Sb particles are covered by a thin (<10 nm) oxide layer. Sb as pure metal is electronically conductive, but the metal oxide layer acts as an insulating layer between the Sb particles. The result is a high resistance of the electrode and a very poor electrochemical performance with a capacity close to zero (Fig. 1f). This is why the presence of a conductive additive is of vital importance to enable Sb-based NIB electrodes.
 | ||
| Fig. 1 (a) Rietveld-refined diffractogram, including the peak positions of antimony and its crystal structure. As-synthesized Sb powder: (b) scanning electron micrograph, (c) transmission electron micrograph, (d) selected-area electron diffraction including peak assignment. (e) O 1s and Sb 3d XPS spectrum of a pristine electrode with 90 mass% synthesized Sb and 10 mass% CMC composition; (f) electrochemical cycling stability of this electrode at 0.2 A g−1. | ||
2.2. Influence of the carbon amount and normalization
To understand the effect of a carbon additive on the Sb electrode performance, five electrodes with various ratios of Sb and carbon black (C65) were prepared. For all measurements, the amount of 10 mass% CMC was kept constant, and the ratios of Sb/C65 were set to 85/5, 80/10, 75/15, 70/20, and 40/50. The cycling stabilities of the composite electrodes are illustrated in Fig. 2a and b with normalization based on either Sb mass or combined Sb/C65 mass. As reported by Widmaier et al.,18 the amount and distribution of carbon can severely influence the electrochemical performance of battery electrodes. Therefore, an optimized electrical percolation of the electrode can significantly counteract side reactions with the electrolyte and enhance the electrochemical kinetics. In accordance, an increase of stability with higher amount of carbon is also observed in our case. Additionally, the coulombic efficiency (CE) is improved when using a larger amount of carbon (Fig. 2d). Stabilization of the CE reaching >97% is evident when increasing the amount of carbon from 15% to 20%. However, by further addition of carbon, this trend reverses, and the CE values remain under 95%. CEs higher than 100% for sample 85/5 and 80/10 are not physically meaningful since almost no charge (∼2 mA h g−1 for 85/5 and ∼40 mA h g−1 for 80/10 after 20 cycles) was stored, and a high error of CE measurement can be expected. Due to its significant outer surface area (ESI, Table S1†), C65 undergoes detrimental side reactions. As the amount of C65 is increased, we see a decline in the CE. This aligns with the report of Spahr et al.29 that a percolation threshold is reached by a certain amount of carbon and further addition of carbon does not further improve the electrode resistivity.29 Since the cycling performance of the 70/20 electrode does not drastically improve by further C65 addition, we expect that the percolation threshold is reached. For this reason, a ratio of 70 mass% Sb and 20 mass% C was selected for all follow-up experiments due to its superior performance. | ||
| Fig. 2 Electrochemical cycling stability at 0.2 A g−1 for different Sb/C65 ratios normalized to (a) the Sb active mass and (b) the Sb/C65 active mass; (c) corresponding coulombic efficiencies of all composites; (d) magnification of coulombic efficiencies for the Sb/C65 ratios of 75/15, 70/20, and 40/50. | ||
In Fig. 2a the capacity values are normalized to solely the Sb mass, whereas a normalization to the total Sb + C65 mass is given in Fig. 2b. With rising C65 amounts, the distinction between both normalizations becomes more relevant. After 20 cycles, the 40/50 electrode normalized to Sb reaches a capacity of 669 mA h g−1, which even exceeds the theoretical value of 660 mA h g−1. This is probably caused by the capacity contribution of C65. The corresponding capacity of 297 mA h g−1 for this electrode normalized to Sb/C65 is 44% lower as compared to normalization to Sb mass only.
Similar differences are found when applying different ways of normalization to various results presented in the literature (Table 1). For example, Cui et al.21 reached 630 mA h g−1 normalized to the Sb + C mass. According to our calculations, the maximum capacity with the electrode composition of Cui et al.21 (Table 1) is 396 mA h g−1 if Sb would reach its theoretical capacity, and the carbon could deliver 300 mA h g−1 (approximately the maximum value reported in literature).9,30 Different from that, Qian et al.26 reported a similar capacity of 610 mA h g−1 by normalizing to solely the Sb mass. The necessity of correct referencing to which mass the normalization is applied but also the inclusion of all active components is highlighted by these examples. Consequently, comparisons to literature need to be considered carefully, and the normalization should be based on Sb and carbon content. Accordingly, all values are normalized to the combined mass of Sb and C for the remainder of our work.
2.3. Properties of the conductive carbon materials
We selected six different carbon materials with different characteristics as conductive additives in Sb-based NIB electrodes, namely, two types of carbon blacks (SuperP and C65), graphite (KS6L), carbon nanohorns (nanostars), and two types of onion-like carbons (OLCs). We selected these specific carbon materials to cover an array of properties, including different size, shape, surface area, degree of ordering (graphitization), and heteroatom concentration (Fig. 4).Regarding our carbon materials, the following aspects should be noted. SuperP and C65 are widely used commercial conductive additives for LIBs and NIBs.18,29 These carbons are reported to impact the electrical resistivity, ionic resistivity, and electrode density.29 SuperP was reported as an anode material for NIBs due to its ability to insert sodium-ions.26,31 Nevertheless, their effects as conductive additives were almost solely studied for LIBs.29,32–35 KS6L graphite has been applied as a conductive additive,35,36 as well as the active material for LIBs.33,34 Carbon nanohorns consist of graphene tubes with horn-shaped tips.37,38 Several thousand linked nanohorns form nanostructured spherical aggregates, so-called graphene nanostars.38 This carbon type has been used as conductive component in LIBs or supercapacitors.39 To the best of our knowledge, such carbon nanohorns have solely been applied in carbon/phosphorous composites for NIBs.37 Onion-like carbons are composed of multiple concentric fullerene-like carbon shells.40 A large scale production via annealing of nanodiamonds as precursor was established by Vladimir Kuznetsov.41 Various structural properties can be obtained depends on the synthesis conditions.42 We synthesized two types of OLCs to evaluate the influence of their different characteristics on the electrochemical performance of the Sb/C electrode. Nanodiamonds were annealed under vacuum at 1300 °C to obtain the OLC1300V sample, whereas the annealing of the OLC1700A sample was done at 1700 °C under argon atmosphere which is known to yield, besides carbon onions, a secondary phase of multi-layer graphene nanoribbons.43
Transmission electron micrographs in Fig. 3a–f show the morphology of all six carbon samples. No morphological difference can be found between C65 and SuperP (Fig. 3a and b). Both carbons form chain-like networks of round shaped particles with 20–30 nm diameter. Different from that, KS6L is composed of micrometer-sized edgy graphite flakes (Fig. 3c). In Fig. 3d, we see aggregated nanohorns are arranged to 50–100 nm nanostars with sharp tips forming their exterior shape. The conical spikes of the nanohorns can be spotted in the high magnification image of Fig. 3d. An agglomerated network of carbon onions is seen for both OLC samples on a sub-micrometer length scale, while the individual OLC particles exhibit a multi-shell fullerene-like architecture with a diameter of 5–10 nm (Fig. 3e and f). In contrast to OLC1300V, OLC1700A shows nanometer-sized carbon onions alongside few-layer graphene nanoribbons.40 The latter originate from carbon redistribution when carrying out the OLC synthesis in an inert atmosphere at ambient pressure and not in vacuum.40
 | ||
| Fig. 3 Transmission electron micrographs of (a) C65, (b) SuperP, (c) KS6L, (d) nanostars, (e) OLC1300V, and (f) OLC1700A. | ||
Chemical analysis (Table 2) reveals that all samples are mainly composed of carbon with small amounts of oxygen- and nitrogen-containing surface functional groups. SuperP carbon has the lowest amount of surface functional groups with 1 mass%, whereas OLC1300V contains 3.83 mass%.
| Sample | C/mass% | N/mass% | O/mass% |
|---|---|---|---|
| C65 | 98.20 ± 1.03 | — | 0.88 ± 0.14 |
| KS6L | 98.36 ± 0.67 | 0.21 ± 0.01 | 0.99 ± 0.26 |
| Nanostars | 96.38 ± 0.49 | 0.57 ± 0.06 | 3.13 ± 1.00 |
| OLC1300V | 96.17 ± 1.32 | 1.55 ± 0.02 | 1.14 ± 0.31 |
| OLC1700A | 98.79 ± 0.78 | 0.36 ± 0.09 | 0.83 ± 0.22 |
| SuperP | 99.00 ± 0.90 | 0.35 ± 0.08 | 0.60 ± 0.04 |
Raman spectra of the carbon samples were recorded and are shown in Fig. 5a. The D- and G-modes in the area of 1200–1700 cm−1 appear for each sample at different positions and with different intensities depending on their structural features. The degree of carbon ordering (graphitization) can be obtained from the D- and G-mode FWHM (full-width at half-maximum) values and the intensity ratio ID/IG.44 A high ID/IG ratio together with low FWHM values indicate a high degree of structural order within the carbon.45 A detailed analysis of the D- and G-modes and their intensities is given in the ESI, Fig. S3, and Table S2.† A high graphitization is found for KS6L and OLC1700A due to low ID/IG values of 0.2 and 1.2, respectively. High D- and G-mode FWHM values of 43 cm−1 and 19 cm−1 for KS6L and both 60 cm−1 for OLC1700A correspond to that finding. The OLC1300V carbon possesses the lowest degree of graphitization with an ID/IG ratio of 3.1 and D- and G-mode FWHM values of 152 cm−1 and 80 cm−1, respectively.
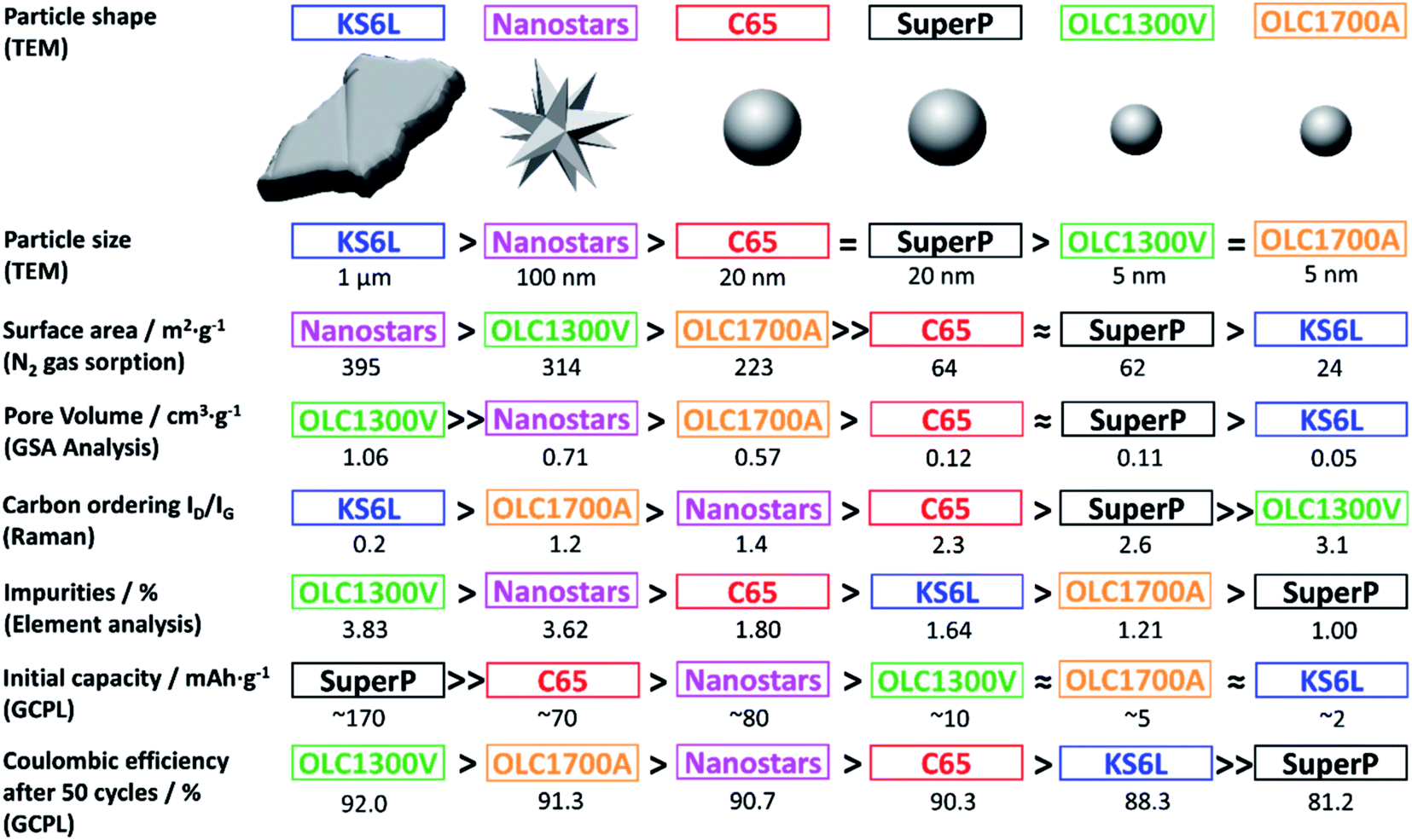 | ||
| Fig. 4 Overview of parameters obtained from GSA, TEM, Raman, and electrochemical analysis (galvanostatic charge/discharge cycling with potential limitation). The carbons are arranged depending on their properties towards the related characteristic with the corresponding value below. | ||
 | ||
| Fig. 5 (a) Raman spectra of the carbon samples, (b) electrochemical cycling stability at 0.2 A g−1 for electrodes composed of 90 mass% of the respective carbon and 10 mass% CMC; (c and d) the related coulombic efficiencies. | ||
The specific surface area, pore-volume, and average pore width (volume-weighted) were determined by nitrogen gas sorption analysis (GSA), and the data are found in ESI, Fig. S2, and Table S1.† OLC1300V and the nanostars possess the highest BET surface area (314 m2 g−1 and 395 m2 g−1) as well as the highest total pore volume (1.06 cm3 g−1 and 0.71 cm3 g−1). KS6L shows the lowest porosity with a total pore volume of 0.05 cm3 g−1 and a BET surface area of 24 m2 g−1. C65 and SuperP present similar BET surface areas of 64 m2 g−1 and 62 m2 g−1 together with similar total pore volumes of 0.12 cm3 g−1 and 0.11 cm3 g−1.
Electrodes composed of the respective carbons and 10 mass% CMC were prepared to evaluate the sodium-ion storage capacities. The de-sodiation capacity for each carbon sample over 100 cycles is depicted in Fig. 5b. Two groups of carbons do not show any significant intrinsic de-sodiation capacity: graphite and carbon onions. KS6L is typical graphite incapable of sodium ion insertion within its structure in our system.8 Moreover, OLCs are composed of closed carbon shells that do not offer any possibility for sodium-ion insertion. The capacities delivered by both OLC samples as well as KS6L are attributed to physical electrosorption of Na+ at the outer surface; accordingly, we see a higher capacity for the onions (4–8 mA h g−1) compared to KS6L (ca. 2 mA h g−1) considering the higher surface area of the former (205–286 m2 g−1). All other carbons exhibit the ability to insert considerable amounts of Na+, and SuperP delivers the highest de-sodiation capacity of 170 mA h g−1. The Na+ insertion mechanism in SuperP has already been studied elsewhere.31 Atomic thick interlayer spaces of graphite and voids around defects of graphite can insert sodium-ions. Due to its similarities, we attribute the measured capacity of around 70 mA h g−1 provided by the C65 electrode to a similar sodium insertion as described for SuperP. Besides that, the carbon nanostars can insert Na-ions with a resulting initial capacity of about 80 mA h g−1.
The CEs for all carbons (Fig. 5c and d) remain under a value of 90%, which indicates a high amount of side reactions. For carbons that do not insert sodium-ions but have a large surface area (i.e., both OLCs), the deviation to 100% CE is mainly attributed to side reactions located at the material surface. Especially SuperP suffers from side reactions. The CE of SuperP after 50 cycles persist at ∼80%. This irreversibility is attributed to enclosures of metallic sodium in the pores between the particles.31 Comparable processes are conceivable for the other carbons which insert Na+. For comparability, an overview of all carbon characteristics is displayed in Fig. 4. These properties will be correlated to the electrochemical performance of the Sb/C composite electrodes in the following.
2.4. Influence of carbon characteristics on Sb/C electrodes
The galvanostatic profiles (ESI, Fig. S4†) of the first three cycles are very similar for all cells. It can be confirmed for all electrodes, that the sodiation takes place through several steps. Three pronounced plateaus are in-line with previous findings forming amorphous NaxSb, crystalline NaSb, and crystalline Na3Sb during sodiation.24,46 This indicates that the fundamental electrochemical process is not significantly varied by varying the type of carbon. The coulombic efficiencies of all electrodes are presented in Fig. 6c and d. All coulombic efficiency values are comparable and do not exceed 98%.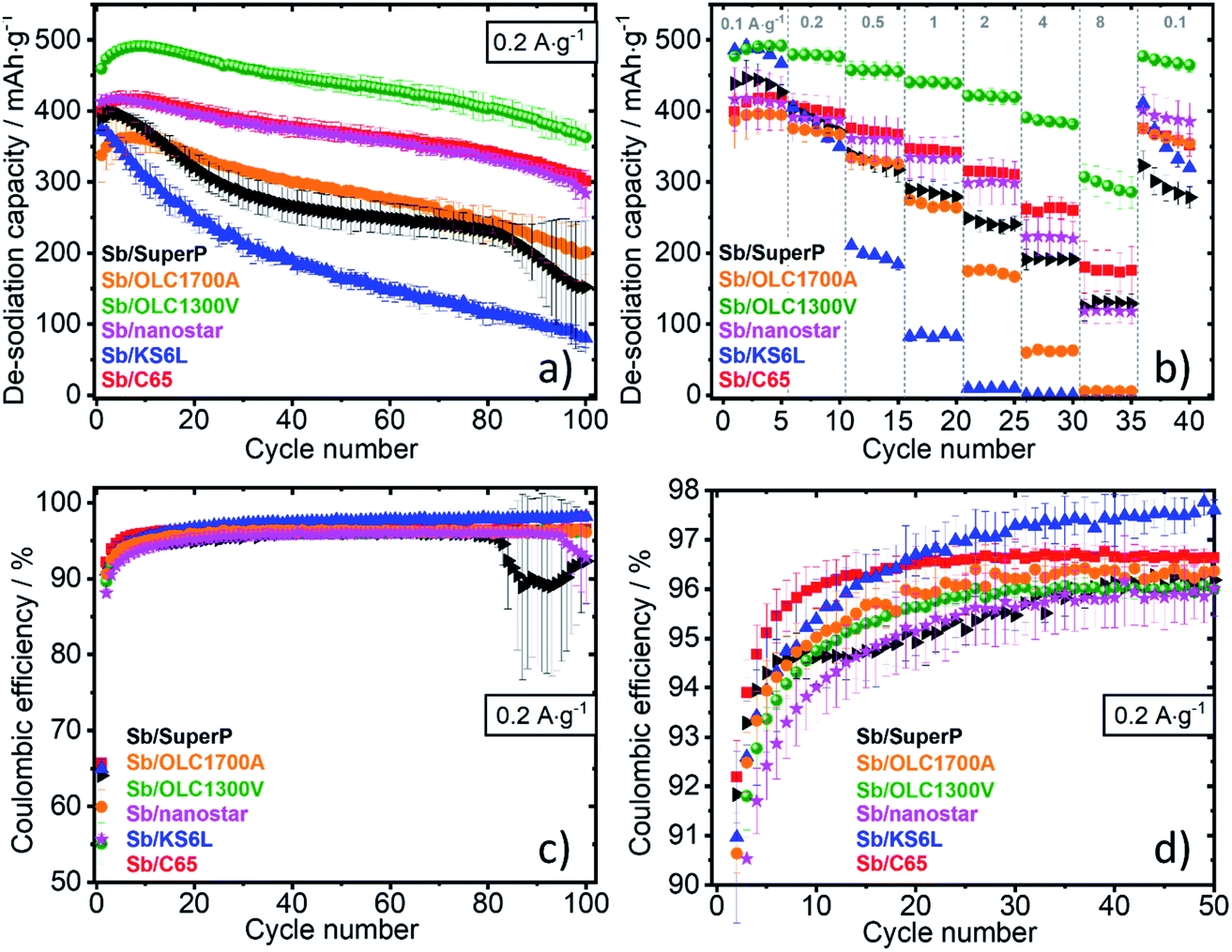 | ||
| Fig. 6 (a) Electrochemical stability at 0.2 A g−1 and (b) rate performance of the respective Sb/C electrodes; (c and d) coulombic efficiencies. | ||
The lowest electrochemical performance is delivered by the Sb/KS6L electrode, especially at higher specific currents of 0.5 A g−1 (Fig. 6b). Because of the flaked shape of KS6L and its considerably large carbon particles, the contact resistance between the active material and current collector is high. This hypothesis is confirmed by the highest resistance of 1.907 Ω cm2 calculated from the IR drop (Table 3). The large flakes of KS6L lead to inhomogeneous charge and voltage distribution with local degradation spots causing electrolyte decomposition.18 Even if the graphite itself does not suffer from volume expansion, with its large and rigid particles, it cannot buffer the volume expansion of Sb. KS6L is not suitable as a conductive additive in NIBs, and this conclusion aligns with the literature.47
| Electrodes | U/V | I/A g−1 | R/Ω cm2 |
|---|---|---|---|
| Sb/C65 | 0.456 | 4 | 0.515 |
| Sb/SuperP | 0.464 | 4 | 0.524 |
| Sb/KS6L | 1.686 | 4 | 1.907 |
| Sb/OLC1300V | 0.313 | 4 | 0.352 |
| Sb/OLC1700A | 0.370 | 4 | 0.420 |
| Sb/nanostars | 0.505 | 4 | 0.569 |
C65 and SuperP are the most similar carbons concerning their appearance and physicochemical properties resumed in Fig. 4. The main difference is the higher initial de-sodiation capacity of 170 mA h g−1 and the lower CE of SuperP respect to C65. These factors cause inferior electrochemical stability of Sb/SuperP as compared to Sb/C65 since high storage capacity is usually connected to high volume changes. This leads to a rapid contact loss and explains the observed capacity fading for Sb/SuperP electrode (Fig. 6a).
Carbon nanostars are also capable of sodium-insertion and have an intrinsic capacity of about 80 mA h g−1, which is similar to C65 (∼70 mA h g−1). Fig. 6a and b illustrates that the electrochemical behavior (stability, and de-sodiation capacity) of Sb/C65 and Sb/nanostars is also similar. Due to its higher resistance of 0.569 Ω cm2, the rate handling ability of Sb/nanostars is inferior to Sb/C65 (0.515 Ω cm2, Table 3). This poorer conductivity is likely attributed to the shape of the carbon nanohorns with cone-like structures extending beyond the surface to crates a rough morphology. The latter will reduce the effective contact area between Sb and carbon particles and decrease the electric conductivity.
An explanation for the exceptionally high and stable performance of Sb/OLC1300V can be deduced by comparing the main differences between OLC1300V and OLC1700A. The OLCs are both 5–10 nm sized particles, which are expected to provide excellent contact between carbon and Sb due to their round shape and smooth surface. However, the pristine electrodes differ from each other. Sb/OLC1300V consists of sub-micrometer-sized Sb agglomerates covered by the carbon particles (Fig. 7c). Thereby, the Sb particles are well distributed within a carbon matrix. Contrary, in the case of Sb/OLC1700A, Sb agglomerates are located on top of a carbon/Sb network (ESI, Fig. S5c†). We explain the lower performance of the latter by the presence of few-layer graphene nanoribbons physically attached to individual carbon onions and clusters thereof. These OLC1700A carbon particles can form sintered clusters due to a higher pressure and temperature applied during annealing as compared to the synthesis of OLC1300V. This difference reduces the ability of OLC1700A to disperse within the Sb-matrix with the same homogeneity as OLC1300V. Accordingly, the sample with the best distribution of the current around the active material particles results in the highest conductivity (Table 3) and the best rate performance of OLC1300V (Fig. 6b). Optimized conductive paths allow a homogenous charge transfer at the surface of the Sb particles. A larger volume of Sb becomes, active causing the highest de-sodiation capacity (Fig. 6a). The inhomogeneity of Sb/OLC1700A leads to poor buffering performance and electrode cracking (ESI, Fig. S5d†). Besides the morphology, OLC1700A is highly graphitic with a lower degree of heteroatoms and defects compared to OLC1300V. Consequently, OLC1700A is presumably more inert towards reactions with the electrolyte or the active material. Disordered parts and impurities could catalyze the chemical reactions leading to an improved SEI, better-buffering effects, and better interaction between the active material and carbon.
 | ||
| Fig. 7 Scanning electron micrographs of (a) pristine Sb/C65, (b) Sb/C65 after 50 cycles, (c) pristine Sb/OLC1300V and (d) Sb/OLC1300V after 50 cycles. | ||
All electrodes suffer from cracking after cycling (Fig. 7b and ESI, Fig. S5b, d, f, h†) caused by the Sb volume expansion. Sb/OLC1300V electrode (Fig. 7d) is the only exception and has also the best rate capability, highest conductivity, and highest capacity. For comparison, scanning electron micrographs of Sb/C65 and Sb/OLC1300V before and after cycling are reported in Fig. 7. The pristine electrodes (Fig. 7a and c) do not differ significantly. Still, the particles are unevenly dispersed, and several cracks are apparent. A massive change of the electrodes after 50 cycles can be observed. Cracking occurs for all electrodes across the entire area (Fig. 7b and ESI, Fig. S5b, d, f, h†) with the noticeable exception of Sb/OLC1300V (Fig. 7d). Crack formation leads to the disruption of conductive pathways. No cracks are apparent for Sb/OLC1300V, and conductive paths were not affected for Sb/OLC1300V during cycling. The Sb/OLC1300V electrode was able to attain 300 mA h g−1 even at 8 A g−1, which is remarkable when considering the slow reaction kinetics usually associated with alloy materials.12,20,25
We then recorded the X-ray photoelectron emission spectra of the cycled Sb/C composite electrodes for a better understanding of the electrode properties. The XPS data can be seen in Fig. 8 and ESI, Fig. S7,† and the data are in agreement with the works of Darwiche et al.16 and Bodenes et al.15 The SEI layers of all electrodes are composed of the same carbonaceous species in different amounts. C 1s, O 1s, and Sb 3d spectra can be found in ESI, Fig. S7.†Fig. 8 shows the amount of metallic Sb in the cycled and de-sodiated electrodes. Metallic Sb is found for each sample except from OLC1300V. XPS is a surface-sensitive technique that only probes about 10 nm of the upper sample layer. Hence, the absence of metallic Sb means that the entire Sb/OLC1300V electrode is covered by a homogenous SEI during cycling, whereas the other samples exhibit SEI free parts. Such an observation is in accordance with the SEM illustrations in Fig. 7, ESI, Fig. S5 and S6.† It is conceivable that the SEI-free parts are in the cracked areas. Most probably, the volume expansion and the resultant electrode cracking leads to bare areas of the electrodes where side reactions take place primarily due to a missing SEI. These effects favor a faster capacity fading of the electrodes.
 | ||
| Fig. 8 Amounts of metallic Sb in Sb/C electrodes obtained from X-ray photoelectron emission spectroscopy. | ||
Our data demonstrate that the carbon properties are of vital importance for the performance of Sb/C electrodes in a NIB. We also can derive important design guidelines for high-performance Sb/C electrodes. Based on the carbon characteristics (Fig. 4), electrochemical studies (Fig. 6, and ESI, Fig. S4†), and the subsequent analytics (Fig. 7, 8 and ESI, Fig. S5–S7†), we were able to elaborate crucial properties of the carbon additives to enable high rate and high stability performance of Sb/C electrodes. The ability to insert sodium counteracts the buffering feature of the carbon, as can be seen for SuperP containing electrodes. The particle morphology should allow good contact between the carbon and the active material. Thus, a round shape as provided by OLCs or SuperP and C65 is preferable to the spikey surface of nanostars or the large graphite flakes of KS6L. A high pore volume and surface area lead to an enlarged electrode/electrolyte interface. This interface is of high importance as it controls the charge transfer, SEI formation, CE, and stability of the battery.48 Further, nanosized carbon can enable high distribution and percolation of the electrode, since it can fill inter-particle voids between the active material particles. We further observe that low graphitization, together with a high amount of impurities is beneficial for improved electrochemical performance. A possible explanation could be that OLC1300V, with its partially disordered structure, can act as a crystallization seed to obtain the special electrode and SEI morphology after cycling (Fig. 7d). It is well known that a high number of defects can lead to a homogenous and mechanically more stable SEI.49 The Sb/OLC1300V electrode after cycling is covered by a very uniform SEI layer, which supports this hypothesis. Additionally, the absence of cracks indicates that either the volume expansion is effectively buffered or the mechanical stability of the SEI prevents crack formation.
3. Conclusions
Due to the inconsistencies and lacking knowledge of the role of the carbon in composite electrodes, a systematic study of six different carbon materials was performed. We first highlight the importance of normalization for comparability for carbon-containing composite electrodes. All carbonaceous components in an electrode could store sodium-ions, as we have demonstrated by probing different pure carbon electrodes. Therefore, the normalization of Sb-based electrodes must include the mass of the entire Sb/C composite.Further, we demonstrated that a minimum amount of carbon is necessary for an Sb based electrode to store sodium-ions. Sb particles are covered by a thin insulating oxide layer, which enhances the electrode resistance and hinders the electrochemical charge storage. Nanosized carbon offers a conductive matrix with embedded Sb particles. We found that an amount of 20 mass% carbon is sufficient to enable a well-percolated electrode with a stable electrochemical performance. A simple and inexpensive mechanical mixing of the active Sb material, conductive carbon, and the binder solution leads to comparable electrochemical performance in contrast to complex Sb/C nanocomposite synthesis found elsewhere in the literature.23,27,46
Finally, we provide a basis on which the influence of carbon characteristics was systematically evaluated. By consideration of different carbon morphologies, sizes, amounts of impurities, graphitization, surface areas, pore volumes, and the electrochemical activity of the carbons, we identified the aspects which are beneficial and improve the electrochemistry of Sb/C electrodes. If the carbon can insert Na+-ions, the connected volume expansion will disrupt the buffering feature and lead to electrode cracking. High purity of the carbon and well-developed graphitization are found to be disadvantageous properties of carbon additives. Carbons with such properties are more inert towards reactions with the electrolyte or the active material. Disordered parts and heteroatoms could catalyze the chemical reactions leading to an improved SEI. In addition, nanosized carbon with round-shaped particles is estimated to offer the best percolation of the electrode as it homogeneously covers the agglomerated Sb particles. This leads to an improved distribution of the current around the active material particles resulting in high conductivity and superior rate performance. Such characteristics can be achieved with onion-like carbon synthesized at 1300 °C under vacuum by nanodiamond deposition. Sb/OLC1300V electrodes show virtually no cracks and a very uniform SEI after electrochemical cycling. An Sb/OLC1300V electrode can attain 490 mA h g−1 at 0.1 A g−1 and 300 mA h g−1 at 8 A g−1, which is remarkable when considering the slow reaction kinetics usually associated with alloy materials.12,20,25 The optimized conductive paths allow a homogenous charge percolation at the surface of the Sb particle. A large volume of the active particle is sodiated, causing a high de-sodiation capacity and high stability at 0.2 A g−1 over 100 cycles. Compared to the literature given in Table 1 our electrodes exceed most of the stated values if normalized to the Sb mass (630 mA h g−1 at 0.1 A g−1 and 386 mA h g−1 at 8 A g−1). Additionally, we use a lower amount of carbon (20%) compared to other studies. Therefore, we are able to compete with the results stated in state-of-the-art-literature, providing simpler processing and less carbon amounts in our study. These findings are expected to apply also to other alloy anodes for NIBs and should serve as important guidelines for the design of high-performance electrodes.
4. Experimental section
4.1. Antimony nanopowder synthesis
The synthesis of Sb nanopowder was previously described elsewhere.27 For our work, the parameters given in literature were optimized by using an excess of sodium borohydride (NaBH4, Sigma Aldrich) as a reducing agent. 1.216 g NaBH4 was dissolved in 400 mL ethanol in a beaker. 2.244 g antimony chloride (SbCl3, Sigma Aldrich) dissolved in 100 mL ethanol was added dropwise to the NaBH4 solution under stirring at room temperature. The resultant black mixture was reacted for another hour and afterward sonicated for ten minutes. The Sb particles were filtered under vacuum and washed with ethanol and water. Thereafter, the black powder was dried at 80 °C for 4 h.4.2. Carbon additives
Six different carbon compounds were compared as conductive additives for Sb based anodes. Carbon black C-NERGY C65, SuperP carbon black, and KS6L graphite were obtained from Imerys Graphite & Carbon. Graphene nanostars were received from Graphene Supermarket. The OLCs were synthesized by annealing high-purity detonation nanodiamond powder (NaBond) either in an inert atmosphere (argon) at 1700 °C (OLC1700A) or in vacuum at 1300 °C (OLC1300V).43 The carbonaceous additives will be labeled C65, SuperP, KS6L, nanostars, OLC1700A, and OLC1300V.4.3. Electrolytes
The preparation and handling of the electrolyte solvents and salts were conducted in an argon-filled glovebox (MBraun, O2, H2O < 0.5 ppm). The sodium salt was dried under vacuum at 80 °C for 48 h. Two different organic electrolytes were prepared: 1 M sodium perchlorate (NaClO4, Alfa Aesar, >99% purity) was dissolved in a mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (by mass) ethylene carbonate (EC, Sigma Aldrich, ≥99% purity) and dimethyl carbonate (DMC, Sigma Aldrich, ≥99% purity), respectively. As an additional electrolyte, 5 mass% fluoroethylene carbonate (FEC, Sigma Aldrich, 99% purity) was added to the previous mixture. The addition of FEC was proposed to be beneficial for the stability of Sb/C electrodes due to a better composition of the SEI layer.24,26,50 We demonstrate that not only the SEI of the Sb/C is stabilized but also the sodium metal reference and counter electrodes are protected towards side reactions with the electrolyte (ESI, Fig. S1†).51 For all other electrochemical measurements in this work, the FEC containing electrolyte is used. Both prepared electrolytes were examined via Karl-Fischer titration and were found to contain less than 25 ppm water.
:1 (by mass) ethylene carbonate (EC, Sigma Aldrich, ≥99% purity) and dimethyl carbonate (DMC, Sigma Aldrich, ≥99% purity), respectively. As an additional electrolyte, 5 mass% fluoroethylene carbonate (FEC, Sigma Aldrich, 99% purity) was added to the previous mixture. The addition of FEC was proposed to be beneficial for the stability of Sb/C electrodes due to a better composition of the SEI layer.24,26,50 We demonstrate that not only the SEI of the Sb/C is stabilized but also the sodium metal reference and counter electrodes are protected towards side reactions with the electrolyte (ESI, Fig. S1†).51 For all other electrochemical measurements in this work, the FEC containing electrolyte is used. Both prepared electrolytes were examined via Karl-Fischer titration and were found to contain less than 25 ppm water.
4.4. Optical characterization
For visual analysis of the Na reactivity, 0.25 g Na metal pieces (Alfa Aesar, 99.95% purity) were thoroughly cleaned by scraping the surface layer of the metal with a scalpel in the argon-filled glove box to avoid any impurities of the metal. These shiny metal pieces were added to 5 mL of both electrolytes. Pictures were recorded immediately after the addition of the metal and three days later (ESI, Fig. S1†).4.5. Electrode materials and preparation
Pure carbon (C) working electrodes were obtained with a ratio of 90 mass% C, and 10 mass% carboxymethyl cellulose (CMC, DS = 0.7, Mw = 250000, Sigma Aldrich) dissolved in water and ethanol (1:1 mass ratio) following the subsequently described mixing steps.
Sb/C working electrodes were prepared by mixing Sb powders with the conductive carbon additive and 10 mass% CMC in a DAC150.1 FVZ speed-mixer from Hauschild. First, the amount of C65 (5 mass%, 10 mass%, 15 mass%, 20 mass%, and 50 mass%) was varied to determine the appropriate ratio for stable cycling of Sb and C. Subsequently, the type of carbon was varied keeping a composition of 70 mass% Sb, 20 mass% C, and 10 mass% CMC. The electrodes with the corresponding carbon will be assigned to as Sb/C65, Sb/SuperP, Sb/KS6L, Sb/nanostars, Sb/OLC1300V, and Sb/OLC1700A.
The Sb powder and carbons were first dry-mixed at 1000 rpm for 5 min. Then, the water/ethanol solution was added to obtain a viscous paste. This paste was again mixed at 1500 rpm for 10 min following 2500 rpm for 10 min. After 10 min of sonication, the paste was again mixed at 2500 rpm for 10 min before adding the binder solution (3 mass% CMC in water). The last mixing step was conducted at 800 rpm for 10 min. The slurries were doctor bladed on an aluminum foil and dried for three days at ambient conditions. Subsequently, the electrodes were punched out with a 12 mm diameter and transferred into a vacuum oven inside an Ar-filled glovebox. Finally, a vacuum drying step at 120 °C for 12 h was conducted. The thickness of the dried electrodes was typically 30–40 μm with a mass loading of 3 ± 1 mg cm−2 (the full electrode mass, including the current collector, was 7 ± 1 mg cm−2).
4.6. Cell preparation and electrochemical characterization
Custom-built polyether ether ketone (PEEK) cells with spring-loaded titanium pistons as a three-electrode system were used for electrochemical testing as described in ref. 52. After drying all cell parts at 120 °C, the cells were assembled inside an Ar-filled glovebox. When using sodium metal as a counter and a reference electrode, great importance was paid to the preparation as proposed in ref. 53. The oxide layer was removed thoroughly with particular care to obtain a smooth Na metal surface and to avoid inhomogeneity and impurities. The counter electrodes were pressed to a uniform thickness of 1 mm. 12 mm diameter sodium disc counter electrodes, and the working electrodes were separated by a 13 mm diameter vacuum dried glass-fiber disc (Whatman GF/D). Cells that were cycled for XPS and SEM analysis were additionally separated by a cellulose separator (Nippon Kodoshi). This separator was placed on top of the working electrode to avoid the adhesion of glass fibers on the surface. A copper foil current collector was placed on the backside of each counter electrode. The Na-reference was placed on a compressed glass fiber separator (GF/D, from Whatman) in a cavity close to the working electrode/counter electrode stack and contacted by a titanium wire. The cells were vacuum filled with the electrolyte.Electrochemical measurements were carried out in a climate chamber at 25 °C using a VMP3 multi-channel potentiostat/galvanostat (Bio-logic Science Instrument), equipped with the EC-Lab software. Galvanostatic charge/discharge cycling with potential limitation (GCPL) experiments was performed in a voltage window of 0.1–2.0 V vs. Na/Na+ with a charge/discharge current of 200 mA g−1. The cycling stability measurements were stopped after 100 cycles. Cells that were prepared for XPS and SEM analysis were stopped after 50 cycles in the de-sodiated state. Half-cell rate capability measurements were conducted at currents between 100 mA g−1 and 8 A g−1 (same rates for charge and discharge).
4.7. Electrode and materials characterization
X-ray diffraction (XRD) measurements of the Sb powder were performed with an STOE STADI P diffractometer (Mo-Kα-radiation, λ = 0.7093 Å) in rotating capillary transmission mode. For phase identification, the ICSD database was utilized.Raman spectra were recorded with a Renishaw inVia Raman Microscope equipped with an Nd–YAG laser with an excitation wavelength of 532 nm and a power of 0.05 mW at the surface of the sample, using an objective lens with a numeric aperture of 0.75. Ten different spots from each of the samples were recorded with 5 accumulations and 30 s acquisition time. All spectra were normalized to 100%, and fitting of the carbon signal was achieved assuming Voigt peak profiles for the D-mode, G-mode.
Nitrogen gas sorption analysis (GSA) was conducted to obtain a specific surface area (SSA), and pore volume of the samples. We carried out the measurements with a Quantachrome iQ system using nitrogen gas at −196 °C. Prior to the measurements, the powder samples were degassed at 250 °C and 102 Pa for 12 h. The SSA was calculated using the Brunauer–Emmett–Teller (BET) equation in the linear pressure range and the quenched solid density functional theory (QSDFT) by assuming slit-shaped pores with the ASiQwin-software. The values for the total pore volume were obtained at a relative pressure of P/P0 = 0.95.
The carbon morphology was characterized by scanning electron microscopy (SEM) using a JEOL JSM 7500F at an acceleration voltage of 3 kV. SEM measurements of the electrodes were conducted by a thermal field emission scanning electron microscope (FESEM, Carl Zeiss SMT AG) at an acceleration voltage of 5 kV. The samples were fixed on a steel sample holder by using carbon sticky tape.
Transmission electron microscopy (TEM) and selected area diffraction (SAED) investigations were carried out by using a JEOL JEM-2100F instrument operated at 200 kV. The sample was dispersed in ethanol through sonication for 5 min and drop-casted onto a copper grid with a lacy carbon film. Scanning TEM (STEM) was performed on a probe-corrected FEI Titan Themis 60-300 X-FEG S/TEM instrument operated at 300 kV equipped with an FEI Super-X windowless EDX system with 4 synchronized silicon drift detectors from Bruker. For all these measurements, the samples were dispersed in isopropanol or ethanol through sonication for 2 min and drop-casted onto a copper grid with a lacey carbon film.
For the XPS analysis, the Sb/C composite electrodes were charged–discharged 50 times and stopped in the de-sodiated state at 2.0 V vs. Na/Na+. The potential was held for 2 h. The cells were then transferred to an Ar-filled glovebox for disassembly. The electrodes were removed and rinsed with DMC prior to XPS measurements. X-ray photoemission measurements were performed using a K-Alpha XPS spectrometer from Thermo Fisher Scientific (East Grinstead). The samples were illuminated with monochromatic Al-Kα X-rays with a spot size of about 400 μm. The photoelectrons were detected with a hemispherical 180 dual focus analyzer with 128 channel detectors. To prevent any localized charge buildup, the K-Alpha charge compensation system was employed during analysis, using electrons of 8 eV energy and low-energy argon ions. The Thermo Avantage software was used for data acquisition and processing.54 The spectra were fitted with one or more Voigt profiles (binding energy uncertainty: ±0.2 eV). All spectra were referenced to the C 1s peak of hydrocarbon at 285.0 eV binding energy controlled by means of the well-known photoelectron peaks of metallic Cu, Ag, and Au.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr M. Widmaier, M. Bauer, and C. Li for support and helpful discussions. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe) and was funded by the German Research Foundation (DFG) under Project ID 390874152 (POLiS Cluster of Excellence). ÖB and VP thank Eduard Arzt (INM) for his continuing support.References
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 RSC.
- (a) A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacín, Energy Environ. Sci., 2012, 5, 8572 RSC; (b) J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, A1–A4 CrossRef CAS; (c) P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007–15028 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, A2476–A2482 CrossRef CAS.
- M.-S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- E. Edison, S. Sreejith, C. T. Lim and S. Madhavi, Sustainable Energy Fuels, 2018, 2, 2567–2582 RSC.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef PubMed.
- W. Brehm, J. R. Buchheim and P. Adelhelm, Energy Technol., 2019, 7, 1900389 CrossRef CAS.
- V. L. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, A1011–A1014 CrossRef CAS.
- L. Bodenes, A. Darwiche, L. Monconduit and H. Martinez, J. Power Sources, 2015, 273, 14–24 CrossRef CAS.
- A. Darwiche, L. Bodenes, L. Madec, L. Monconduit and H. Martinez, Electrochim. Acta, 2016, 207, 284–292 CrossRef CAS.
- (a) L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed; (b) S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CrossRef CAS PubMed; (c) V. Winkler, G. Kilibarda, S. Schlabach, D. V. Szabó, T. Hanemann and M. Bruns, J. Phys. Chem. C, 2016, 120, 24706–24714 CrossRef CAS.
- M. Widmaier, N. Jäckel, M. Zeiger, M. Abuzarli, C. Engel, L. Bommer and V. Presser, Electrochim. Acta, 2017, 247, 1006–1018 CrossRef CAS.
- (a) M. Widmaier, K. Pfeifer, L. Bommer and V. Presser, Batteries Supercaps, 2018, 1, 11–26 CrossRef CAS; (b) C. Li, A. Sarapulova, K. Pfeifer and S. Dsoke, ChemSusChem, 2020, 986–995 CrossRef CAS PubMed.
- H. Hou, M. Jing, Y. Yang, Y. Zhang, W. Song, X. Yang, J. Chen, Q. Chen and X. Ji, J. Power Sources, 2015, 284, 227–235 CrossRef CAS.
- C. Cui, J. Xu, Y. Zhang, Z. Wei, M. Mao, X. Lian, S. Wang, C. Yang, X. Fan, J. Ma and C. Wang, Nano Lett., 2019, 19, 538–544 CrossRef CAS PubMed.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 RSC.
- Y. Zhu, X. Han, Y. Xu, Y. Liu, S. Zheng, K. Xu, L. Hu and C. Wang, ACS Nano, 2013, 7, 6378–6386 CrossRef CAS PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- L. Wu, H. Lu, L. Xiao, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2015, 3, 5708–5713 RSC.
- J. Qian, Y. Chen, L. Wu, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2012, 48, 7070–7072 RSC.
- H. Hou, Y. Yang, Y. Zhu, M. Jing, C. Pan, L. Fang, W. Song, X. Yang and X. Ji, Electrochim. Acta, 2014, 146, 328–334 CrossRef CAS.
- T. Ramireddy, N. Sharma, T. Xing, Y. Chen, J. Leforestier and A. M. Glushenkov, ACS Appl. Mater. Interfaces, 2016, 8, 30152–30164 CrossRef CAS PubMed.
- M. E. Spahr, D. Goers, A. Leone, S. Stallone and E. Grivei, J. Power Sources, 2011, 196, 3404–3413 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- C. Wu, P. I. Pan, Y. Cheng, C. Liu, C. Chang, M. Avdeev and S. Lin, J. Power Sources, 2017, 14–21 CrossRef CAS.
- (a) J. Syzdek, M. Marcinek and R. Kostecki, J. Power Sources, 2014, 245, 739–744 CrossRef CAS; (b) A. Rezqita, R. Hamid, S. Schwarz, H. Kronberger and A. Trifonova, ECS Trans., 2015, 66, 17–27 CrossRef CAS; (c) J. Xiao, W. Xu, D. Wang, D. Choi, W. Wang, X. Li, G. L. Graff, J. Liu and J. G. Zhang, J. Electrochem. Soc., 2010, A1047–A1051 CrossRef CAS.
- M. Nádherná, J. Reiter, J. Moškon and R. Dominko, J. Power Sources, 2011, 196, 7700–7706 CrossRef.
- S. Rothermel, P. Meister, G. Schmuelling, O. Fromm, H.-W. Meyer, S. Nowak, M. Winter and T. Placke, Energy Environ. Sci., 2014, 7, 3412–3423 RSC.
- M. Indrikova, S. Grunwald, F. Golks, A. Netz and B. Westphal, J. Electrochem. Soc., 2015, A2021–A2025 CrossRef CAS.
- (a) D. Weingarth, D. Cericola, F. C. F. Mornaghini, T. Hucke and R. Kötz, J. Power Sources, 2014, 266, 475–480 CrossRef CAS; (b) W. Wu, J. Yan, A. Wise, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2014, A561–A567 CrossRef CAS.
- M. Li, R. Carter, L. Oakes, A. Douglas, N. Muralidharan and C. L. Pint, J. Mater. Chem. A, 2017, 5, 5266–5272 RSC.
- T. Murakami, K. Ajima, J. Miyawaki, M. Yudasaka, S. Iijima and K. Shiba, Mol. Pharmaceutics, 2004, 1, 399–405 CrossRef CAS PubMed.
- (a) H. Lai, J. Li, Z. Chen and Z. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2325–2328 CrossRef CAS PubMed; (b) W. Xu, Z. Wang, Z. Guo, Y. Liu, N. Zhou, B. Niu, Z. Shi and H. Zhang, J. Power Sources, 2013, 232, 193–198 CrossRef CAS; (c) C.-M. Yang, Y.-J. Kim, M. Endo, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 20–21 CrossRef CAS PubMed; (d) R. Yuge, N. Tamura, T. Manako, K. Nakano and K. Nakahara, J. Power Sources, 2014, 266, 471–474 CrossRef CAS; (e) Y. Zhao, J. Li, Y. Ding and L. Guan, RSC Adv., 2011, 1, 852 RSC; (f) Y. Zhao, J. Li, Y. Ding and L. Guan, Chem. Commun., 2011, 47, 7416–7418 RSC.
- M. Zeiger, N. Jäckel, V. N. Mochalin and V. Presser, J. Mater. Chem. A, 2016, 4, 3172–3196 RSC.
- V. L. Kuznetsov, A. L. Chuvilin, Y. V. Butenko, I. Y. Malkov and V. M. Titov, Chem. Phys. Lett., 1994, 343–348 CrossRef CAS.
- J. K. McDonough and Y. Gogotsi, Electrochem. Soc. Interface, 2013, 61–66 CrossRef CAS.
- M. Zeiger, N. Jäckel, D. Weingarth and V. Presser, Carbon, 2015, 94, 507–517 CrossRef CAS.
- (a) A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS; (b) A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 14095–14107 CrossRef CAS.
- L. Borchardt, M. Oschatz, M. Lohe, V. Presser, Y. Gogotsi and S. Kaskel, Carbon, 2012, 50, 3987–3994 CrossRef CAS.
- X. Zhou, Z. Dai, J. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727 RSC.
- C. W. Wang, K. A. Cook and A. M. Sastry, J. Electrochem. Soc., 2003, A385–A397 CrossRef CAS.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- R. Dugas, A. Ponrouch, G. Gachot, R. David, M. R. Palacin and J. M. Tarascon, J. Electrochem. Soc., 2016, A2333–A3339 CrossRef CAS.
- K. Pfeifer, S. Arnold, J. Becherer, C. Das, J. Maibach, H. Ehrenberg and S. Dsoke, ChemSusChem, 2019, 12, 3312–3319 CrossRef CAS PubMed.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- J. Conder and C. Villevieille, Chem. Commun., 2019, 55, 1275–1278 RSC.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta00254b |
| This journal is © The Royal Society of Chemistry 2020 |