
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transparent lignin-containing wood nanofiber films with UV-blocking, oxygen barrier, and anti-microbial properties†
Juho Antti
Sirviö
 *a,
Mostafa Y.
Ismail
a,
Kaitao
Zhang
a,
Mysore V.
Tejesvi
bc and
Ari
Ämmälä
a
*a,
Mostafa Y.
Ismail
a,
Kaitao
Zhang
a,
Mysore V.
Tejesvi
bc and
Ari
Ämmälä
a
aFibre and Particle Engineering Research Unit, University of Oulu, P. O. Box 4300, 90014 Oulu, Finland. E-mail: juho.sirvio@oulu.fi
bDepartment of Ecology and Genetics, University of Oulu, Oulu, Finland
cCHAIN Antimicrobials LLC, Teknologiantie 2, Oulu, 90590, Finland
First published on 2nd April 2020
Abstract
Lignocellulose is a potential raw material for film and membrane applications, such as packaging for every day consumables or a supporting barrier layer for flexible electronics. Here, lignin-containing cationic wood nanofiber (CWNF) films were produced using sawdust as the starting material. Sawdust was directly cationized using four different aqueous solvents containing tetraethylammonium hydroxide with different carbamides (urea, methylurea, ethylurea, or dimethylurea) and glycidyltrimethylammonium chloride as the cationization agent. Cationic wood was obtained with a high cationic group content (around 1.5 mmol g−1) and yield (89–100%), and CWNFs were easily obtained by mechanical disintegration as a water suspension. The films produced using solvent-casting exhibited excellent visible-light transparence (around 80% at 600 nm), and the presence of lignin allowed high UV-adsorption (below 380 nm, transmittance was under 1%). The films showed high oxygen barrier properties (below 400 and 4000 m3 μm/m2 day atm at a relative humidity of 50 and 92%, respectively) and good mechanical strength. An antimicrobial test conducted using the disk method showed that the CWNF films exhibited bacterial anti-adhesive properties with a small inhibition zone. CWNFs are therefore potential environmentally friendly packaging materials to prevent food spoilage, or useful as a UV-absorption layer for electronic devices such as solar cells.
Introduction
Films and membranes have become a part of everyday life as they are used in common items such as food packaging and electronics (e.g. in displays and flexible electronics). The majority of current films and membranes on the market are produced from oil-derived polymeric materials and plastics due to their inexpensive production and durability.1 However, there is a growing concern about the use of plastics in packaging due to their severe environmental impact. Significant amounts of oil-based plastics are non-biodegradable, that is, persist in nature for long periods, causing negative impacts on the environment.2 Although some of these plastics can be recycled, current recycling efforts are not sufficient to prevent the pollution.3 There is also concern about the contamination of recycled plastics, which reduces their safety in use for food packaging.4 In addition, some of the starting materials (i.e. monomers) of synthetic polymers are toxic, which can cause issues during material synthesis, and if those monomers remain in the product beyond synthesis they will also be in the final product.5Several strategies have been developed to minimize the environmental impact of plastics. Biodegradable polymers have been produced from oil-derived chemicals and use of biomass-derived monomers in polymer synthesis has gained a significant amount of interest due to concerns about the non-renewable nature of oil resources and the toxicity of oil-based monomers.6–8 Another means to replace current plastics is the use of natural polymers as film and membrane materials. Lignocellulosic biomass is a highly abundant potential polymer source.9 The main components of lignocellulose, cellulose and lignin are the most common polysaccharide and natural aromatic polymers, respectively.10 The third main component of lignocellulose, hemicelluloses, are a class of polysaccharides with useful properties such as water solubility.
Compared to polysaccharides, lignin has certain unique properties, namely low polarity (i.e. hydrophobic characteristics), the ability to absorb ultraviolet (UV) light, and antioxidant properties.11 Due to these properties, lignin has been studied as an active component to produce film materials with decreased hydrophilicity12 and UV blocking properties.13 Cellulose fibers have been investigated to produce films and membranes in the form of nanosized materials (e.g. cellulose nanofibers [CNFs] and cellulose nanocrystals [CNCs]). The films produced from nanosized cellulose can exhibit high transparency typical of plastics; however, the mechanical strength of nanocellulose films can highly surpass those of synthetic materials.14
Individualized lignin and nanocellulose have been studied to produce composite materials, such as films.15–17 However, the separation of lignin and cellulose (and hemicellulose) requires harsh reaction conditions and use of hazardous chemicals (e.g. chlorine-based bleaching chemicals). Therefore, to reduce the environmental burden and to obtain materials with the advantages of the individual components (e.g. hydrophobicity and UV-blocking traits of lignin together with the high strength of nanosized cellulose), there has been growing interest in the production of nanomaterials directly from biomass.18–22 However, due to the recalcitrant nature of the natural cellulose–hemicellulose–lignin-complex, the production of nanomaterials directly from biomass can be a more cumbersome process compared to nanoparticles obtained from individual components. Several methods, such as intensive mechanical refining,21 heat-intensified grinding,19 and enzymatic treatment23 have been studied to produce biomass-based nanomaterials, generally termed lignocellulose nanofibers or wood nanofibers (WNFs). Similar to the production of CNFs and CNCs, the introduction of charged chemical groups into biomass has been shown to improve the disintegration of biomass in its nanosized constituents20,24,25 due to the generation of steric repulsion and improved swelling (i.e. osmotic pressure). The additional chemical groups can provide functionality to biomass-based nanomaterials which can be used, for example, in water purification.26
In this study, we investigated the production of cationic WNFs (CWNFs) from sawdust without pre-removal of wood content, in order to obtain a film material with the sufficient mechanical properties and transparency of cellulose and the UV-blocking and the humidity–resistant oxygen barrier properties of lignin. The cationic group was introduced in order to improve disintegration efficiency and to facilitate anti-microbial functionality. Chemical modification was conducted using aqueous tetraalkylammonium hydroxide (recently demonstrated as an environmentally friendly solvent for biomass27) in the presence of four carbamides (urea, methylurea [MU], ethylurea [EU], and 1,3-dimethylurea [DMU]). Carbamates are ideal components for solvents and reaction media due to their low toxicity and biodegradability.28
Materials and methods
Materials
Unbleached spruce sawdust was obtained in a never-dried form. The sawdust was first oven-dried (24 h at 60 °C) and ground with an Ultra Centrifugal Mill ZM 200 (Retsch, Germany) using a sieve size of 250 μm. The mean particle size of the ground sawdust was 208 μm, as determined using a Beckman Coulter LS 13 320 laser diffraction particle size analyzer. The lignin (28.6 wt%), acetone-soluble extract (1.2 wt%), and alkaline-soluble hemicellulose and degraded cellulose (19.7 wt%) contents of the sawdust were analyzed using TAPPI T 222 om-02, TAPPI T 280 standard pm-99, and TAPPI T 212 om-02 standards, respectively.Tetraethylammonium hydroxide solution (TEAOH, 35 wt% in water), N-methylurea (MU), N-ethylurea (EU), and 1,3-dimethylurea (1,3-DMU) were obtained from Sigma Aldrich (Germany) and urea from Borealis Biuron (Austria). The cationization agent glycidyltrimethylammonium chloride (GTAC, 80 wt% in water) was obtained from TCI. Polyethylenesulfonate (PES-Na, BTG Mütek GmbH, Germany) was used as a polyelectrolyte titrant. Expect for cationization of wood, deionized water was used as the medium for all the steps. All chemicals were used as received.
Cationization of wood
Prior to cationization, carbamides (urea, 1,3-DMU, MU, or EU) were individually mixed with TEAOH at a carbamide:TEAOH molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at room temperature to obtain 18 g of clear, colorless solvent. Then, 2 g of sawdust was added followed by the addition of 4.42 ml of GTAC (the mass ratio of sawdust:GTAC was 1:2). The mixture was allowed to react at room temperature for 24 h while mixing. After the desired reaction, 10 ml of water was added and the product was transferred into centrifugation tubes. The reaction vessel was carefully rinsed with water and the product (with solvent and rinsing water) was transferred into a centrifugation tube and diluted with water up to a volume of 50 ml. The mixture was centrifuged for 10 min using a rotation speed of 10000 rpm at 25 °C. The supernatant was carefully discarded and the sample was repeatedly diluted with water and centrifuged until the pH of the supernatant water was neutral, indicating the removal of highly basic reaction solvent. After this, the samples were collected and stored at 4 °C as a water suspension having a consistency of about 10%.
:1 at room temperature to obtain 18 g of clear, colorless solvent. Then, 2 g of sawdust was added followed by the addition of 4.42 ml of GTAC (the mass ratio of sawdust:GTAC was 1:2). The mixture was allowed to react at room temperature for 24 h while mixing. After the desired reaction, 10 ml of water was added and the product was transferred into centrifugation tubes. The reaction vessel was carefully rinsed with water and the product (with solvent and rinsing water) was transferred into a centrifugation tube and diluted with water up to a volume of 50 ml. The mixture was centrifuged for 10 min using a rotation speed of 10000 rpm at 25 °C. The supernatant was carefully discarded and the sample was repeatedly diluted with water and centrifuged until the pH of the supernatant water was neutral, indicating the removal of highly basic reaction solvent. After this, the samples were collected and stored at 4 °C as a water suspension having a consistency of about 10%.
Elemental analysis of sulfated cellulose
The nitrogen content of the oven-dried samples was analyzed using a PerkinElmer CHNS/O 2400 Series II elemental analyzer. As GTAC introduces one nitrogen per cationic group, the cationic group content is directly related to the nitrogen content.Diffuse reflectance infrared Fourier transform spectroscopy
The chemical characterization of the original and cationic wood was performed by diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy using a Bruker Vertex 80v spectrometer (USA). Spectra of the dried samples were obtained in the 600–4000 cm−1 range and 40 scans at a resolution of 2 cm−1 were taken.Disintegration of cationic wood into nanofibers
The disintegration of cationic wood was performed with a microfluidizer (Microfluidics M-110EH-30, USA). The cationic wood suspension in water (a consistency of 0.5 wt%) was passed twice at a pressure of 1000 bar through the 400 and 200 μm chambers and separately once at a pressure of 1500 bar through the 400 and 100 μm chambers of the microfluidizer (Microfluidics M-110EH-30, USA).Charge density
The surface charge densities of the CWNFs were determined using the polyelectrolyte titration method through a particle charge detector (BTG Mütek PCD-03, Germany). 10 ml of nanofiber suspension (at 0.01 wt% in water) was titrated with PES-Na (1 meq. l−1). The charge density was calculated based on the PES-Na consumption.Transmission electron microscopy
The analysis of the size and the morphology of the CWNFs was performed by transmission electron microscopy (TEM, JEOL JEM-2200FS, Japan). The samples were first diluted to around 0.001 wt% and then applied on poly-L-lysine treated carbon coated copper grids.29 2 wt% uranyl acetate was used as the staining agent. The average diameter of the nanofibers was measured using ImageJ software 1.50i.Fabrication of films
CWNF films were fabricated using the solvent-casting method as follows: the desired amount of CWNF suspension was diluted with water to a total mass of 60 g and sonicated for 10 min in order to remove any air bubbles trapped in the suspension. After sonication, the suspension was poured on a polystyrene tray (area of 0.0147 m2) and the water content was allowed to evaporate at room temperature for 5 days to produce films with a grammage of 30 g m−2. The films were then stored at 50% relative humidity (RH) for two days and then peeled off from the tray.Tensile test
Tensile tests were conducted with a universal testing device (Instron 5544, USA). Prior to measurement, samples were conditioned at 23 °C and a humidity of 50% for 2 days. The samples were prepared by cutting films into strips at a length of 70 mm and a width of 5 mm. The average thickness of the samples was measured at 3 random positions using a thickness gauge (Precision Thickness Gauge FT3, Hanatek Instrument, UK). The tensile test was conducted using a 2 kN force sensor, a gauge length of 40 mm, and a strain speed of 5 mm min−1 until breaking. Five samples of each film were measured.UV-Vis spectroscopy
The transmittance of the CWNF films was measured in the wavelength range of 200–800 nm using a UV-Vis spectrometer (Shimadzu, Japan). In order to ensure that the films were perpendicularly aligned against the incoming beam and to avoid wrinkling, the films were put between two quartz glass slides before being set up in a cuvette stand.The absorption coefficient was calculated using the Beer–Lambert law:
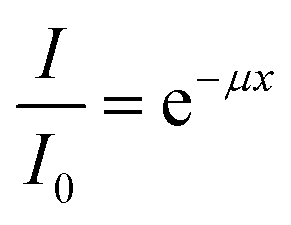 | (1) |
Oxygen barrier test
The oxygen barrier properties of the CWNF films were investigated by measuring the oxygen transmission rate (OTR) values using a MOCON OxTran 2/20 (Minneapolis, MN). The oxygen permeability (OP) was calculated by multiplying the OTR values with the thickness of the film and dividing it by the difference in the partial pressure of the oxygen gas between the two sides of the film. During measurement, the films were exposed to 100% oxygen on one side and to oxygen-free nitrogen gas (98% nitrogen and 2% hydrogen) on the other side. The measurements were conducted at 23 °C, with a relative humidity (RH) of 50 and 92%, and normal atmospheric pressure. Two individual samples from each of the films were measured.Antimicrobial test
The antibacterial activity of the fabricated films was examined against Staphylococcus aureus (S.aureus, Gram-positive) and Escherichia coli (E.coli, Gram-negative). A mixture of Mueller–Hinton (MH) broth and agar in 500 ml distilled water was autoclaved at 120 °C for 15 min. After autoclaving, the mixture was cooled down to 46 °C, and then an appropriate amount of E.coli and S.aureus suspension was added into the MH agar and mixed, respectively, resulting in a bacterial concentration of 6 × 105 CFU ml−1. Subsequently MH agar containing bacteria was poured into Petri dishes. Square 15 × 15 mm film samples were placed on the surface of MH agar plates and incubated at 37 °C for 24 h. Afterwards, the halo sizes in the plates were visually observed. All the samples were analyzed in triplicate.Thermogravimetric analysis
The thermal properties of the original sawdust and CWNF films were investigated using a thermal analyzer (Netzsch STA 449F3, Germany) under an air flow (dynamic air) at a constant rate of 60 ml min−1. Approximately 5 mg of dry sample was weighed in an aluminum oxide pan and heated from 30 to 950 °C at a rate of 10 °C min−1. The first derivate curves of TGA (DTG) were recorded using OriginPro 2019 software.Statistical analysis
One-way analysis of variance (ANOVA) was conducted using OriginPro 2019 to determine the statistical significance (p < 0.05) of the tensile barrier test results.Results and discussion
Cationization of wood
Previously, cationization of cellulose30 and lignin31 with GTAC (and its parent component, 3-chloro-2-hydroxypropyltrimethylammonium chloride) has been conducted using alkaline aqueous solutions (Scheme 1). Alkaline conditions allow the deprotonation of the hydroxyl groups of cellulose, lignin, and hemicelluloses, making them more nucleophilic towards the epoxide ring opening reaction. In the current study, aqueous TEAOH solutions with four different carbamides (urea, MU, EU, and 1,3-DMU) were used as reaction media for wood. TEAOH together with carbamides, described as aqueous deep eutectic solvent (DES), has been previously used for room temperature dissolution of low-molecular weight microcrystalline cellulose.32 Although the dissolution of high-molecular weight cellulose, such as that present in wood, is limited, it was assumed that TEAOH with carbamides could serve as an efficient reaction medium for wood. The wood material used here was dry sawdust, which was mildly ground to a smaller size, leaving the majority of the wood components still intact. It was assumed that a TEAOH–carbamide solution would function as a swelling agent, allowing the efficient chemical modification of the wood material, as well as providing sufficient alkalinity to enhance the reactivity of the hydroxyl groups. In addition, the TEAOH—carbamide solutions exhibit low viscosity (even as 5 wt% microcrystalline cellulose solutions) allowing the use of a low-weight ratio between sawdust and solvent (here 1:9), thus decreasing the consumption of the solvent.
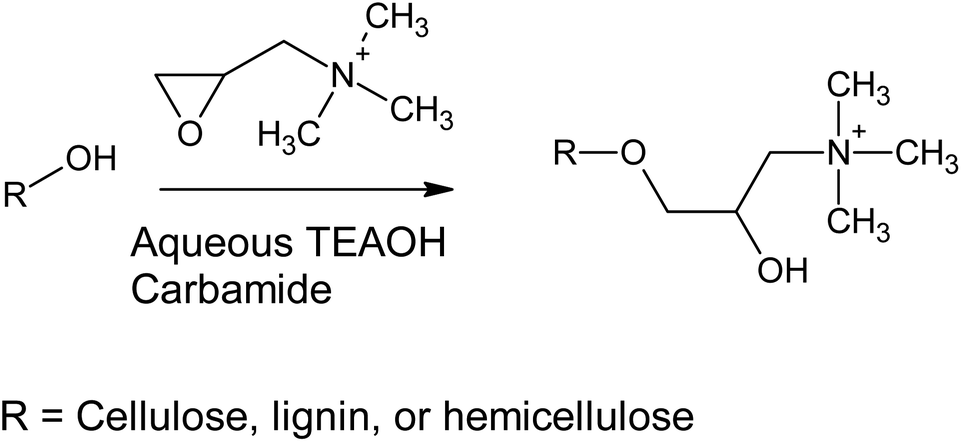 | ||
| Scheme 1 A simplified reaction scheme of the cationization of wood. It should be noted that the hydroxyl group generated by the epoxide ring opening might also react, possibly forming a cationic side-chain in the wood structure. | ||
The TEAOH–carbamide solutions were prepared using 35 wt% aqueous TEAOH solution and carbamides with a TEAOH–carbamide molar ratio of 1:2. Table 1 shows that the solvents gave similar results: the cationic group content and charge densities were 1.4–1.6 mmol g−1 and 1.5–1.9 meq. g−1, respectively. The small variation in the cationic group content and charge density originates from the use of different analysis methods, as they are measured using elemental analysis and polyelectrolyte titration, respectively. The most apparent differences between samples was observed in the yield, as the solution containing MU or 1,3-DMU showed quantitative yields, indicating that most of the wood components are preserved after cationization. On the other hand, the yields obtained with urea and EU were around 90%. These variations in the yield may originate from the different solubility of the wood components and the product in the solvents used. In addition, it is possible that the water used in the washing stage could dissolve part of the cationic wood.
| Sample | Carbamide | N (%) | Cationic group content (mmol g−1) | Charge density (meq. g−1) | Yield (%) | Width of CWNFs (nm) |
|---|---|---|---|---|---|---|
| 1 | Urea | 2.1 ± 0.08 | 1.5 ± 0.06 | 1.6 ± 0.07 | 89 | 4.8 ± 1.3 |
| 2 | 1,3-DMU | 2.2 ± 0.08 | 1.6 ± 0.06 | 1.7 ± 0.06 | 100 | 3.7 ± 0.8 |
| 3 | MU | 2.1 ± 0.02 | 1.5 ± 0.02 | 1.9 ± 0.01 | 100 | 3.9 ± 1.0 |
| 4 | EU | 2.0 ± 0.04 | 1.4 ± 0.03 | 1.5 ± 0.03 | 91 | 4.8 ± 1.6 |
The high yield shown by cationized wood is in line with previous studies, where chemically modified wood showed yields of over 90%.25,33 Previous studies have shown that the yield of chemically modified wood can be higher when compared to the corresponding cellulose samples. The higher yield of wood compared to pure cellulose may be due to the composition of the wood. In addition to cellulose, lignin and hemicellulose have reactive hydroxyl groups that most likely take part in chemical reactions, lowering the overall charge density of the individual components and making them less soluble, for example, in water during washing. Therefore, in addition to the use of a starting material with a higher production yield (e.g. wood vs. cellulose), the use of wood can give a material with an excellent yield.
In the literature, cationic CNFs have been produced using GTAC in a mixture of cellulose, water, isopropanol and alkaline salt (e.g. NaOH).34 Other solvents have also been studied to minimize the epoxide ring opening reaction with water, which consumes the reagent and decreases the reaction efficiency.35,36 Depending on the reaction conditions (e.g. solvent content, time, and temperature), CNFs with charge densities of 0.354 (ref. 37) and 0.403 (ref. 38) meq. g−1 or a cationic group content of 0.21–1.140,36 0.59–2.31,39 and 1.91 (ref. 40) mmol g−1 have been reported. Chlorocholine chloride together with DMSO as a solvent was used to produce CNFs with a cationic group content of 0.09–0.19 mmol g−1,41 whereas Girard's reagent (after periodate oxidation) was used to obtain nanofibers with a charge density of 1.10–2.13 meq. g−1.42 In addition, WNFs with a cationic group content of 0.70 mmol g−1 were obtained using betaine hydrochloride as a reagent and Tosyl chloride as a coupling agent in DES.33 Therefore, the current method can be seen as an efficient way to produce highly charged wood-based materials with a charge density on par with that of CNFs with the highest reported charge densities. The high cationic charge of the wood produced in this study could allow it to be used in water purification applications, for example as a flocculant,42,43 or in the removal of toxic components, such as dyes, from water.39,44
Chemical characterization of cationic wood
The most distinguished chemical differences in sawdust before and after cationization are the disappearance of the ester bond (at 1738 cm−1) that naturally occurs in the wood (the acetyl group of hemicellulose and the ester linkage of the carboxylic groups in the ferulic and p-coumaric acids of lignin/hemicellulose)45 and the appearance of a carbon-nitrogen stretching band at 1483 cm−1 (ref. 46) (Fig. 1a and b; spectra of other samples are presented in ESI, Fig. S1†). The ester bond is cleaved due to the highly alkaline conditions during cationization, resulting in a hydrolysis reaction in the presence of water.47 On the other hand, the carbon–nitrogen bond originated from a tertiary amine group of GTAC. In addition, broadening of the peak in the OH-region (around 3300 cm−1) and the water peak around 1650 cm−1 were observed, indicating that the chemical modification altered the hydrogen bonding pattern and the water uptake of the wood. The increased water uptake was later confirmed by TG analysis.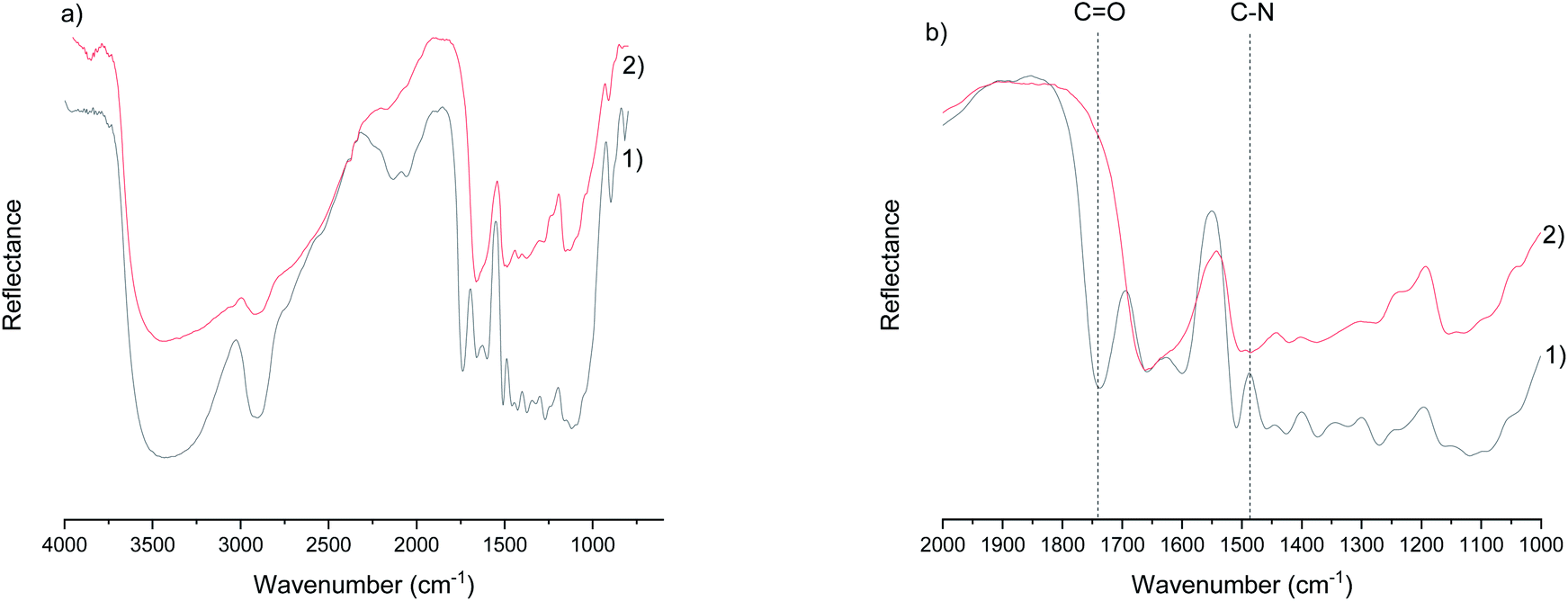 | ||
Fig. 1 (a) the DRIFT spectra of wood (1) and cationic wood obtained using urea as carbamide (2), and (b) the fingerprint region of the DRIFT spectra showing the most distinguished differences between the two spectra: the disappearance of the carbonyl peak (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the ester bond and the appearance of a carbon–nitrogen peak (C–N) after chemical modification. O) of the ester bond and the appearance of a carbon–nitrogen peak (C–N) after chemical modification. | ||
Nanofibrillation of cationic wood
The production of CWNFs was achieved by the mechanical disintegration of the water suspension of cationic wood with a microfluidizer. All of the samples passed through the microfluidizer without blocking its narrow chambers to produce very low viscosity, yellow suspensions (Fig. S2†). Previously, WNFs with a gel-like appearance have been obtained by sulfation with reactive DESs.48,49 Sulfate groups are more hydrophilic compared to tertiary amine groups introduced into wood in the current study50 and the sulfated WNFs exhibited significantly higher charge density compared to CWNFs produced here, both contributing to their high viscosity values.Generally, the viscosities of the nanofibers produced from high lignin-containing starting materials are lower compared to those obtained from bleached (lignin-free) cellulose.19 Low viscosities can originate from the lower degree of hydrophilicity of lignin, decreasing the interaction between WNFs and water (i.e. swelling) when compared to the corresponding CNFs. It should be noted that although CNFs with high viscosity can be highly desirable, for example when used as a rheology modifier,51 the low viscosity of WNFs can also be beneficial in the preparation of film-forming solution, as the entrapped air bubbles are more easily removed from low viscosity samples. In addition, low viscosity might enable the production of CWNFs with a high solid content, for example, using a twin-screw extruder.52
TEM analysis showed that cationic wood disintegrated into individual nanofibers with a width of approximately 5 nm (Table 1 and Fig. 2). This measurement is close to that of elementary fibrils (width of 3.5 nm) and similar to the values reported for the CNFs produced using bleached cellulose pulp.36,53–56 CWNFs varied from micrometer-long nanofibers to shorter, 200–300 nanometer long CNC-like particles. Some larger particles with a width of a few hundred nanometers were observed (Fig. 2b). All the samples also contained high amounts of coarse nanosized particles, which can easily be distinguished from nanofibers that exhibited a sharp, needle-like appearance. The coarse nanoparticles are associated with the amorphous materials (lignin and hemicellulose) present in wood.33,48 Due to the high yield of CWNFs, it can be assumed that around half of the materials consist of these amorphous nanoparticles. As cellulose and lignin/hemicellulose are present as separated components, CWNFs can be regarded as a composite between these two individual nanoparticles.
 | ||
| Fig. 2 TEM images of CWNFs obtained using (a) urea, (b) 1,3-DMU, (c) MU, and (d) EU, demonstrating the presence of both short and long nanofibers (a), a small number of bigger, a few hundred nanometer sized aggregates (b) and network like structure (c and d) with a large number of coarse nanoparticles originating from the amorphous components of wood (lignin and hemicelluloses) (see the ESI for high-resolution TEM images, Fig. S3–S6†). | ||
Although the average width of all the samples was within the error range, it appears that CWNFs produced using MU and 1,3-DMU had a slightly more narrow size distribution (see the ESI for histograms of the width of the samples, Fig. S7†) and thus more uniform nanofibers are obtained using either of these two components in a solvent system. The higher charge density of the CWNFs produced with MU and 1,3-DMU might be explained by the slightly smaller side of the nanofibers, which in turn allows for more efficient interaction between the polymeric titrant used in polyelectrolyte titration when compared to samples produced using urea and EU.
The widths of the CWNFs were in line with previous results obtained from sulfated WNFs produced from sawdust48 and groundwood pulp49 and CWNFs produced from groundwood pulp using DES.33 However, compared to CWNFs obtained previously, in the current study, the large particles were present to a lesser extent. Compared to the anionic WNFs produced by succinylation in DES,25 the CWNFs produced here have a significantly smaller size distribution.
Cationic wood nanofiber films
CWNF films were produced using the solvent-casting method, and all the samples produced transparent, yellowish films, with good mechanical robustness (Fig. 3 and S8†). The average thickness of CWNF1, CWNF2, CWNF3, and CWNF4 films was 44, 39, 57, and 63 μm, respectively. When placed directly on top of the background, the underlying image was readily visible (Fig. 3a), whereas when the film was held a few centimeters above the background, haziness could be observed (Fig. 3b), although the background could still be recognized. All the films were also easy to handle and tolerated bending (Fig. 3c).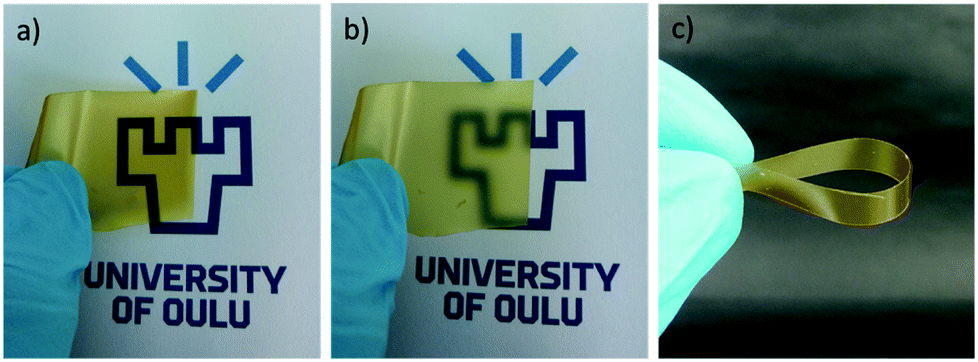 | ||
| Fig. 3 CWNF film (a) directly on top and (b) held a few centimeters above the background showing good transparence and a slight haziness of the films, and (c) bending of the film to demonstrate good mechanical robustness. | ||
UV blocking and visible light transparency of the cationic wood nanofiber films
Transparency is desirable in food packaging due to possibility to see the product, and blocking UV radiation helps to prevent the spoilage of food. UV light can generate singled oxygen that accelerates the oxidation of lipids and degrades antioxidants and vitamins.57 High visible light transparency and UV-blocking capabilities are also desirable traits in electronic applications such as solar cells, as many of these devices harvest visible light, and UV light can cause damage to the components, decreasing their lifespan.58 Hence, the UV and visible light absorption and transmittance of the CWNF films were investigated.For all the films tested, strong absorption can be observed in the range of 250–350 nm (Fig. 4a), associated with the presence of lignin (characteristic bands in the UV spectrum of lignin appear at 200–230 nm and 260–280 nm (ref. 59)). Due to the strong absorption, the transmittance of UV light (<400 nm) was practically zero in all the films (Fig. 4b), showing that CWNF films are excellent UV barrier materials. Moderate absorption at the low end of the visible light spectrum explains the intense yellow color, which is visible at 565–590 nm, of the CWNF suspensions and films. Nevertheless, at wavelengths higher than 500 nm, the transmittance values of all the films were above 60% and around 80% at 600 nm. The transparency of the CWNF films was higher compared to the previous WNF films obtained from sulfated sawdust, having a transmittance of 41–68% at a wavelength of 600 nm.48 UV blocking films (almost zero transparency below 400 nm) similar to the CWNF films have previously been produced by mixing CNCs with isolated lignin (10 wt% lignin content); however, their visible light transmittance was around 20% and 60% at wavelengths of 500 and 600 nm, respectively.15 Slightly higher visible light transparency compared to the current study has been reported with a coating of CNCs and organosolv lignin, although complete UV blocking was not achieved.16 Also, the visible light transmittance of CWNF films is higher compared to the cationic CNF film studied previously.60
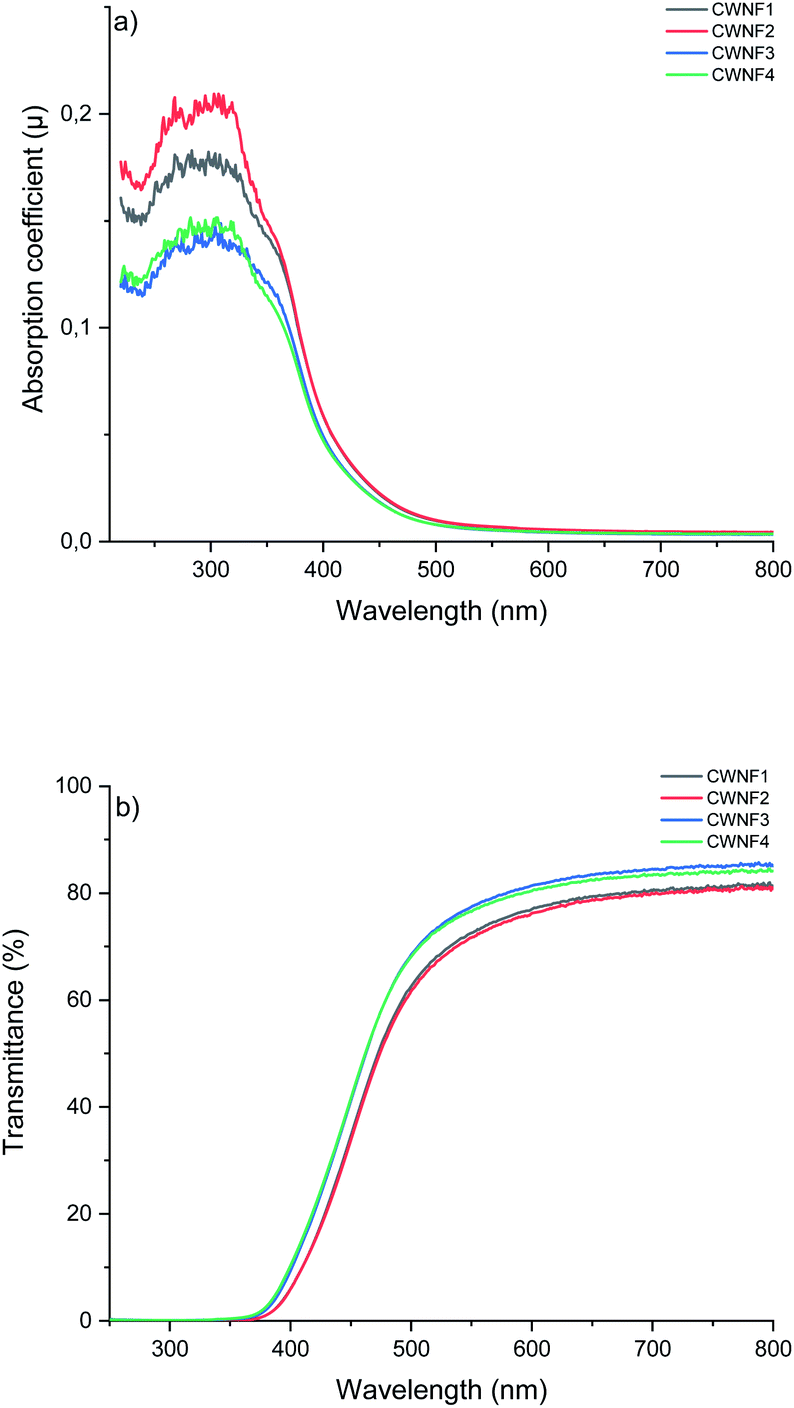 | ||
| Fig. 4 UV and visible light (a) absorption coefficient and (b) transmittance of CWNF films, showing strong absorption behavior in the UV region (<400 nm) and high transparency in the visible light region (380–760 nm (ref. 61)). | ||
Oxygen barrier properties of the cationic wood nanofiber films
As stated before, oxygen is one of the major causes of food spoilage due to its high reactivity.62 The results presented in Table 2 indicate that at an RH of 50%, the oxygen barrier properties (OP values) of CWNF films can be described either as high (≤400 cm3 μm (m2 d atm)−1) or medium (400–4000 cm3 μm (m2 d atm)−1).63 The results indicate that CWNFs are significantly better oxygen barriers than the hydrophobic synthetic polymers used in packaging materials.63 Nevertheless, their OP values are higher compared to efficient oxygen barriers, such as polyvinylidene chloride (10–300 cm3 μm (m2 d atm)−1)64 and films produced from CNFs at an RH of 50%. For example, OP values of 12,55 35,65 and 37 (ref. 66) cm3 μm (m2 d atm)−1 have been reported with anionic CNF films. On the other hand, the OP values of CWNF films at 50% RH are lower compared to other polysaccharides such as amylose and amylopectin (709 and 1418 cm3 μm (m2 d atm)−1, respectively),67 and close to the OP values of cellophane, measured at 0% RH.63| Sample | Densitya (g m−3) | Mechanical properties | Oxygen barrier properties at different RHs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tensile strengtha (MPa) | Tensile modulusa (GPa) | Yield strengtha (MPa) | Strain at breaka (%) | Specific tensile strengtha (kN m kg−1) | Specific tensile modulusa (MN m kg−1) | Specific Yield strengtha (MPa) | 50% | 92% | ||||
| OTRa (cm3 (m2 d)−1 | OPa (cm3 μm (m2 d atm)−1) | OTRa (cm3 (m2 d)−1 | OPa (cm3 μm (m2 d atm)−1) | |||||||||
| a Means that are marked by different superscript letters within the same column are significantly different at the 0.05 level based on one-way ANOVA. | ||||||||||||
| 1 | 0.682 | 70.3 ± 9.4a | 4.0 ± 0.5a | 60.4 ± 8.2a | 5.3 ± 1.0a | 103.1 ± 13.8a | 5.9 ± 0.7a | 88.6 ± 12.0a | 10.4 ± 0.5a | 498 ± 9a | 43.5 ± 5.1a | 2047 ± 372a |
| 2 | 0.774 | 60.9 ± 5.7a | 3.6 ± 0.2a | 55.2 ± 5.0a | 4.4 ± 0.6a | 78.7 ± 7.4b | 4.6 ± 0.2a | 71.2 ± 6.5b | 8.7 ± 1.7a | 355 ± 29a | 54 ± 21a | 2170 ± 601a |
| 3 | 0.527 | 70.4 ± 2.1a | 3.9 ± 0.1a | 61.6 ± 0.3a | 5.7 ± 1.2a | 133.6 ± 3.9c | 7.5 ± 0.2c | 116.9 ± 0.6c | 7.9 ± 0.9a | 433 ± 47a | 42 ± 12a | 2299 ± 655a |
| 4 | 0.555 | 70.7 ± 1.4a | 3.7 ± 1.7a | 62.5 ± 0.6a | 4.0 ± 0.4a | 127.6 ± 2.6c | 6.6 ± 3.1a | 112.8 ± 3.2c | 6.6 ± 0.0a | 351 ± 0a | 50. 7 ± 23a | 2762 ± 1209a |
The powerful oxygen barrier properties of CNFs and many synthetic films originate from their strong polar characteristics as well as their crystallinity.63 Due to polarity, films have reduced interaction between non-polar oxygen atoms, decreasing the oxygen solubility, and the crystalline structure creates a torturous path, hindering the diffusion. Polarity increases the water adsorption, resulting in the swelling of the film structure, thus allowing oxygen to permeate through films more readily when humidity is increased. The most dramatic increase is usually observed at an RH of 65% and higher. For example, in the case of carboxymethylated CNF films, the OP values increased from 0.061 cm3 μm (m2 d atm)−1 measured at 0% to 37–86 and 3617 cm3 μm (m2 d atm)−1 when the RH was increased to 50 and 80%, respectively.66 In the case of the film produced from bacterial CNCs, the OP values increased from 6.12 to 52–264 cm3 μm (m2 d atm)−1, when the RH increased from 0 to 80%.
The OP values of CWNF films at an RH of 92% were around 2000–3000 cm3 μm (m2 d atm)−1, thus still being a medium oxygen barrier.63 The barrier properties of CWNF films at elevated humidity are in the range of polyethylene terephthalate (1000–5000 cm3 μm (m2 d atm)−1) and over 20 times lower compared to polypropylene and polyethylene. At an RH of 96%, around two times higher OP values compared to CWNFs were obtained for CNF films produced using a two hour hot-pressing method, whereas by wax dip coating, similar barrier properties compared to CWNFs could be obtained (OP value of 1723 cm3 μm (m2 d atm)−1 at 97%).68 For cellophane (RH of 95%) and chitosan (RH of 93%), an OP value of 25470 and 92477 cm3 μm (m2 d atm)−1, respectively, has been reported,63 being notably higher when compared to CWNF films. Therefore, at an elevated RH, CWNF films exhibit oxygen barrier properties on par or significantly better when compared with many other biomass-based films and synthetic packaging polymers.
Lignin is a less polar component in comparison to cellulose and hemicellulose and is thus proposed as a valuable component to improve the hydrophobicity of WNF films. The effects of lignin have previously been shown in contact angle measurement19 and water absorption18 and in improved strength properties at elevated humidity and in a wetted state.69 Contradictory results have also been reported, highlighting the effect of its morphology, and other possible factors, on the hydrophobicity of WNF films. In addition, the presence of lignin has also been shown to improve the oxygen barrier properties of WNFs at an RH of 50%, whereas the opposite effect was observed at higher humidity.18 Although the exact lignin content of CWNFs was not determined due to the possible chemical modification of lignin, high yield from the cationic wood indicates that most of the lignin was preserved, and the lignin content in CWNF films is around 24 wt%. The TEM images indicated that both lignin and hemicellulose nanoparticles and nanofibrillated cellulose fibers were relatively homogenously distributed in CWNFs. The homogenous distribution is further supported by the high transmittance of the films, as the presence of large aggregates can decrease light transmittance. Therefore, as previously stated, CWNFs can be described as a composite material, in which oxygen barrier properties are the result of a combination of the individual components. Cellulose and hemicellulose are hydrophilic components which produce effective oxygen barrier properties at low humidity, whereas the presence of lignin reduces swelling of the film structure at elevated humidity. However, less polar lignin slightly decreased the overall oxygen barrier properties due to the higher interaction between oxygen and lignin compared to cellulose. Due to the effective oxygen barrier properties, CWNFs could be potentially used successfully for food packaging. For applications that require extremely high oxygen barriers, such as solar cells, the oxygen barrier properties of CWNFs are too low to be effective. Nevertheless, CWNFs could be used as a UV-blocking layer in laminated structures, where they could also provide a supportive oxygen barrier after the hydrophobic moisture barrier and before the actual oxygen barrier (e.g. poly(vinyl alcohol)70).
Mechanical properties of the cationic wood nanofiber films
The mechanical properties of the CWNF films are presented in Table 2. The mean tensile strength of the films varied in between 60 and 71 MPa. The differences of means, however, were statistically insignificant at the 0.05 level, as determined by ANOVA analysis. The tensile modulus values were also in the same range for all films (around 4 GPa). The results obtained here are similar to the ones previously reported for sulfated WNFs produced from sawdust. However, it should be noted that in a previous study, sulfated WNF films exhibited non-yielding characteristics, whereas in the case of CWNF films, yielding was observed and the yield strength of the films was close to 60 MPa (see the ESI† for an example of stress–strain curves). Yielding is a typical property of cellulosic nanofiber films, indicating the point at which nanofibers begin to slip, causing permanent damage to the film structure (i.e. irreversible straining). In the case of CNF films, the yield point is followed by a strong strain-hardening tendency, that is, the ultimate tensile strength is generally significantly higher compared to the yield strength. For example, CNF films with similar strain compared to these CWNFs showed a yield strength of 84 MPa, approximately 20 MPa higher in comparison.71 On the other hand, the ultimate tensile strength of CNFs was over two times higher compared to CWNF films. The strain-hardening effect is likely due to the straightening and alignment of the long and curved nanofibers.72 The CWNF contains somewhat round and coarsely shaped nanoparticles, originating from lignin and hemicellulose, that do not align (or are poorly aligned); therefore the strain-hardening of the CWNFs is minimal compared to CNF films. A similar behaviour is observed in CNF composites.73,74 The strain-hardening of CNWFs is also strongly associated with the reorganization of the hydrogen bonding network under the strain.75 In the case of WNFs, the presence of lignin might partially block the formation of the hydrogen bonding network, thus decreasing the overall tensile strength and the strain-hardening.The density of the CWNF films was lower when compared to the WNF films produced previously, which typically have reported densities of 1–1.5 g m−3.18,19,76 In a previous study, films are produced using filtration methods that are usually followed by vacuum or hot press-drying at an elevated temperature, which can result in the production of denser films when compared to the solvent-casting method used in this study. Previously, similar densities to the CWNF films (0.5 and 0.8 g m−3) were obtained by the solvent-casting of WNFs produced using homogenization.21 To examine the differences in the density, the specific tensile strengths, modulus, and yield were calculated. Except for CWNF 2, the specific tensile strengths were over 100 kN m kg−1, the highest being 134 kN m kg−1 for the film produced using MU. The specific tensile strength of CNWs is close to the average tensile strength of CNF films reported in the literature. For example, values of 127 kN m kg−1 and 160 kN m kg−1 have been reported for films produced with Masuko ground77 and TEMPO-oxidized CNFs.78 Nevertheless, it should be noted that a specific tensile strength of 268 kN m kg−1 has previously been obtained using TEMPO-oxidation.79 For WNFs with a high lignin content (>20%), specific tensile strengths of 79,19 83,25 and 90 (ref. 76) kN m kg−1 have been obtained.
Therefore, it can be concluded that the mechanical properties of the CWNF films are on par with those of the state-of-the-art lignocellulose nanomaterials. In addition, the CWNF films also outperform many synthetic packaging materials, such as polyethylene, propylene and poly(vinyl chloride) having tensile strength in the range of 22.1–31.0, 31.0–41.4 and 40.7–51.7 MPa, respectively.80 As stated above, filtration-pressing methods can be used to produce CNF and WNF films with higher density, thus improving the tensile properties. In addition, it has been shown that by using a high temperature during drying, lignin can be softened, resulting in the filling of voids in the nanofiber network.19,20 Consequently, hot-pressing can increase the mechanical properties of the films. However, filtration is usually done using a polyvinylidene fluoride membrane with a small anionic surface charge, making the peeling of the cationically charged films from the membrane cumbersome, due to their electrostatic attraction to the membrane. Therefore, alternative membrane materials are recommended for this purpose. However, the strong electrostatic adhesion of CWNFs to surfaces could be used in coatings or in the fabrication of materials with interfacial nanoparticle complexation.81
Antimicrobial properties of the cationic wood nanofiber films
Food spoilage due to microbial growth causes serious health and safety issues due to toxicity. To improve the quality, safety, and shelf life of food products, developing active packaging materials capable of reducing the growth of food pathogens is a possible solution.82 In addition to food packaging, antimicrobial properties are also desirable for medical packaging to avoid microbial infections and promote sterilization.83 Here, the film produced using 1,3-DMU as the carbamide component of the solvent was investigated as a model film for anti-microbial properties. A narrow inhibition zone around the sample was observed in E. coli and S. aureus, indicating that the CWNF film has certain antimicrobial properties (Fig. 5). In addition, no bacteria were observed to grow on the film, demonstrating strong bacterial anti-adhesive properties.84 Previous studies of PVA-composite films containing cationic CNFs showed no inhibition zone in disk testing, but the bacteria did not grow on top nor under the film.36 The lack of an inhibition zone was associated with the immobilization of cationic CNFs (as an antimicrobial agent) in films, resulting in the death of the bacteria in direct contact with the film. Our control test with the sulfated WNFs also showed no inhibition zone (Fig. S10 and S11†). Therefore, it seems that a small portion of the CWNF film leaches out of the film structure, causing the anti-bacterial effect around the film. It is possible that amorphous nanoparticles originating from lignin and hemicellulose might not be tightly bound to the film. This may be due to the cationization of lignin and hemicellulose, which then act as antimicrobial agents. However, the inhibition zone is significantly smaller compared to, for example, bacterial nanocellulose films loaded with silver nanoparticles85 and curcumin,86 where silver ions or small molecular curcumin are easily released from the film. This minimal leaching effect of the CWNF films can be desirable in packaging applications, as leaching of the chemicals (here chemically modified wood components) has to be limited. Therefore, further studies should be conducted to identify which components, if any, are actually leaching and whether they have any toxic effects on animals. | ||
| Fig. 5 Photograph of the antimicrobial performance of the CWNF film against (a) E. coli and (b) S. aureus. Narrow inhibition zones can be observed in the light area around the film. | ||
Thermal properties of the cationic wood nanofiber films
TGA and DTG curves presented in Fig. 6a and b indicate that CWNFs exhibit a different thermal degradation behavior compared to sawdust. Under dynamic air (i.e. presence of oxygen), sawdust showed a bimodal mass loss curve. The onset temperature for the first degradation step was around 270 °C and the highest mass-loss rate was at 314 °C. The second peak of the mass-loss rate was observed at a temperature of 349 °C. The first degradation phase is associated with the degradation of aliphatic components, that is, hemicellulose and cellulose and the first thermal degradation step of lignin.87 The second mass-loss peak, starting around 400 °C, is due to the final degradation state of lignin. Due to the presence of different oxygen-bearing chemical groups, lignin has a wider degradation range than hemicellulose and cellulose.88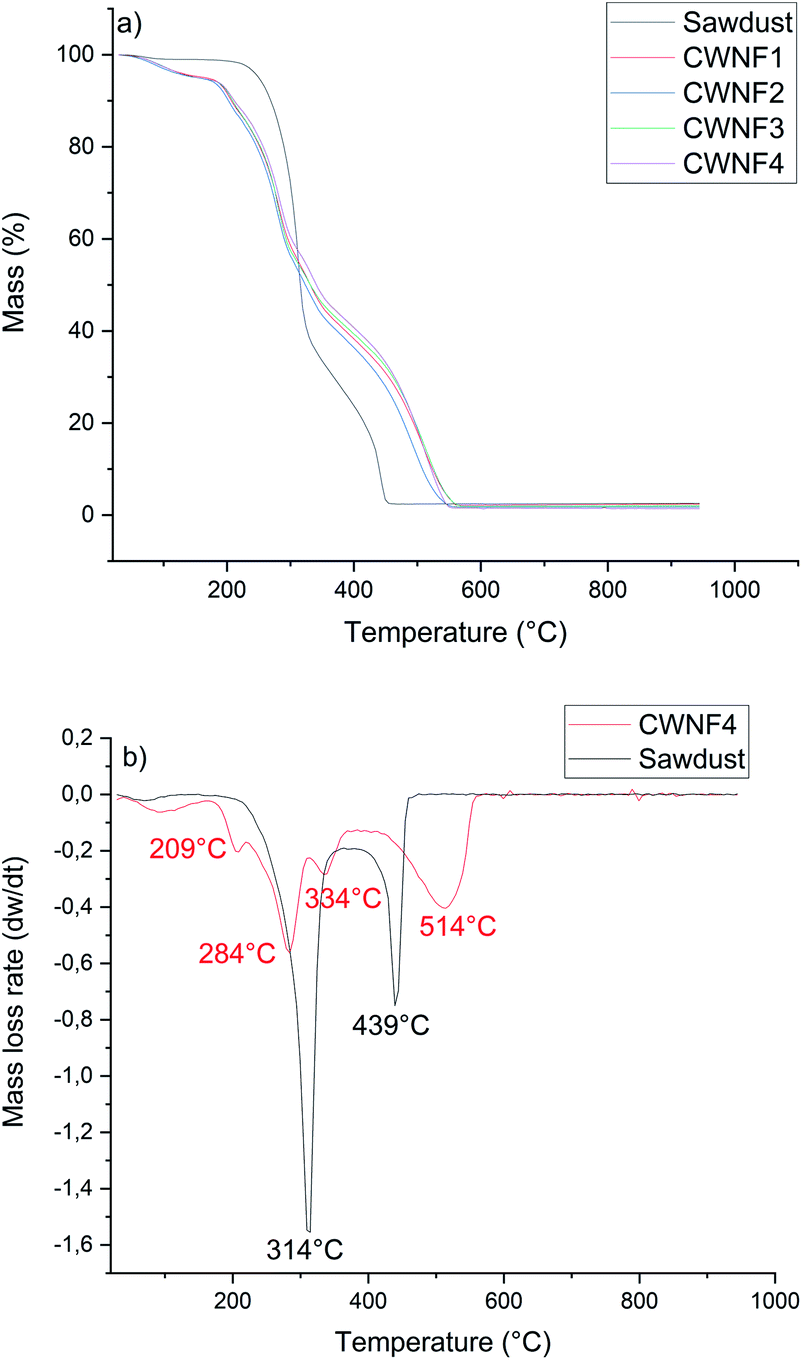 | ||
| Fig. 6 (a) TGA curves of original sawdust and CWNF films and (b) DTG curve of the CWNF4 film with original sawdust. | ||
In contrast to the original sample of sawdust, several degradation steps were observed for CWNFs. The first significant difference between the TGA curves of CWNFs and sawdust can be seen at temperatures between 30 and 170 °C. The mass loss in the low temperature range can be attributed to the loss of adsorbed water.89,90 For the sawdust, only about 1% of mass was lost before the first degradation step, whereas for the CWNFs, around 5% of mass was lost as water. Differing mass loss at low temperatures indicates that the hydrophilicity of sawdust increased during the CWNF production. The onset temperature of degradation for the CWNFs was observed to be around 200 °C, which is around 70 °C lower in comparison to pure sawdust. The first degradation step, associated with the degradation of the labile quaternary ammonium groups91,92 of CWNFs, shows a maximum mass-loss rate at 209 °C. The decomposition of quaternary ammonium groups is related to the formation of volatile components, such as (NCH2(CH3)2),93 either by reverse Menschutkin decomposition or Hofmann elimination.94
After the degradation peak of quaternary ammonium groups, two separated mass-loss rate peaks were observed at 284 and 334 °C, and the first degradation step of sawdust was observed in between these peaks. The final degradation step of CWNFs begins around 410 °C, which is slightly higher than that of the sawdust (400 °C). The highest mass-loss rate of the final degradation step was observed at significantly higher temperature for CWNFs (514 °C) than that of the sawdust (439 °C).
The multiple degradation steps in the TGA curve of CWNFs could indicate the separation of wood constituents as more individualized components from the original lignin–hemicellulose–cellulose complex. Hemicellulose is described as a short-chain polymer with the lowest thermal stability among the main wood components.95 Therefore, it can be assumed that the degradation of hemicelluloses is observed during the second step, after the degradation of quaternary ammonium groups, in CWNFs. The second step might also be the degradation point for the low crystalline parts of cellulose96 generated during the CWNF production. The third degradation step could be the degradation point of the individualized cellulose nanofibers,97 which were observed in TEM images. It is possible that the final degradation step in the breakdown of lignin requires a higher temperature due to the formation of a protective char layer around the lignin particles, decreasing the heat transfer. The remaining mass at the onset of the final degradation step of CWNFs at 410 °C was over 40%, whereas in the case of sawdust, the remaining mass was only 22% at this point.
Conclusions
TEAOH with carbamide was successfully used to produce high charge density cationic wood with an excellent yield from sawdust without any chemical pre-treatment to remove wood components. CWNFs were then easily obtained from cationic wood by mechanical disintegration. Due to the presence of lignin, the films produced from CWNFs exhibited strong UV-blocking properties, and the narrow side-distribution of cellulosic nanofibers facilitated high optical transparency. The effective oxygen barrier properties of CWNF films even at elevated humidity were also attributed to the presence of individualized cellulosic and lignin nanomaterials working synergistically by decreasing oxygen transmission and the swelling of the films, respectively. Furthermore, the films exhibited anti-bacterial properties due to their high cationic group content. Therefore, it is proposed that CWNF films could present an environmentally friendly alternative to oil-based plastics currently used in food packaging and flexible electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Elisa Wirkkala and Dr. Mika Kaakinen for their assistance in the elemental analysis and TEM measurements, respectively. B.Sc. Kalle Kälkäjä is gratefully acknowledged for their contributions to the experiment. J. A. S. would like to thank the Kone Foundation for its financial support. This research was conducted as part of the ARVOPURU project granted by the European Regional Development Fund of the European Union and funded by the Council of Oulu Region, Ha-Sa Oy, Junnikkala Oy, Keitele Forest Oy, Kuhmo Oy, Pölkky Oy and Westas Group Oy.Notes and references
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- B. Geueke, K. Groh and J. Muncke, J. Cleaner Prod., 2018, 193, 491–505 CrossRef CAS.
- K. J. Groh, T. Backhaus, B. Carney-Almroth, B. Geueke, P. A. Inostroza, A. Lennquist, H. A. Leslie, M. Maffini, D. Slunge, L. Trasande, A. M. Warhurst and J. Muncke, Sci. Total Environ., 2019, 651, 3253–3268 CrossRef CAS PubMed.
- V. Siracusa, P. Rocculi, S. Romani and M. D. Rosa, Trends Food Sci. Technol., 2008, 19, 634–643 CrossRef CAS.
- T. P. Kainulainen, J. A. Sirviö, J. Sethi, T. I. Hukka and J. P. Heiskanen, Macromolecules, 2018, 51, 1822–1829 CrossRef CAS PubMed.
- S.-A. Park, Y. Eom, H. Jeon, J. M. Koo, E. S. Lee, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Green Chem., 2019, 21, 5212–5221 RSC.
- J. Baruah, B. K. Nath, R. Sharma, S. Kumar, R. C. Deka, D. C. Baruah and E. Kalita, Front. Energy Res., 2018, 6, 1–19 CrossRef.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- E. M. Zadeh, S. F. O'Keefe and Y.-T. Kim, ACS Omega, 2018, 3, 7388–7398 CrossRef CAS PubMed.
- H. Sadeghifar, R. Venditti, J. Jur, R. E. Gorga and J. J. Pawlak, ACS Sustain. Chem. Eng., 2017, 5, 625–631 CrossRef CAS.
- A. Brhoum, A. Dufresne, T. Öhlund and P. Samyn, Nanoscale, 2017, 9, 15181–15205 RSC.
- M. Parit, P. Saha, V. A. Davis and Z. Jiang, ACS Omega, 2018, 3, 10679–10691 CrossRef CAS PubMed.
- A. Hambardzumyan, L. Foulon, B. Chabbert and V. Aguié-Béghin, Biomacromolecules, 2012, 13, 4081–4088 CrossRef CAS PubMed.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- E. Rojo, M. S. Peresin, W. W. Sampson, I. C. Hoeger, J. Vartiainen, J. Laine and O. J. Rojas, Green Chem., 2015, 17, 1853–1866 RSC.
- M. Visanko, J. A. Sirviö, P. Piltonen, R. Sliz, H. Liimatainen and M. Illikainen, Cellulose, 2017, 24, 4173–4187 CrossRef CAS.
- M. Herrera, K. Thitiwutthisakul, X. Yang, P. Rujitanaroj, R. Rojas and L. Berglund, Cellulose, 2018, 25, 3121–3133 CrossRef CAS.
- K. L. Spence, R. A. Venditti, Y. Habibi, O. J. Rojas and J. J. Pawlak, Bioresour. Technol., 2010, 101, 5961–5968 CrossRef CAS PubMed.
- I. Solala, A. Volperts, A. Andersone, T. Dizhbite, N. Mironova-Ulmane, A. Vehniäinen, J. Pere and T. Vuorinen, Holzforschung, 2012, 66, 477–483 CAS.
- M. Hassan, L. Berglund, E. Hassan, R. Abou-Zeid and K. Oksman, Cellulose, 2018, 25, 2939–2953 CrossRef CAS.
- S. Iwamoto and T. Endo, ACS Macro Lett., 2015, 4, 80–83 CrossRef CAS.
- J. A. Sirviö and M. Visanko, J. Mater. Chem. A, 2017, 5, 21828–21835 RSC.
- J. A. Sirviö and M. Visanko, J. Hazard. Mater., 2020, 383, 121174 CrossRef PubMed.
- N. Singh and K. Prasad, Green Chem., 2019, 21, 3328–3333 RSC.
- W. H. Evans, E. J. David and S. J. Patterson, Water Res., 1973, 7, 975–985 CrossRef CAS.
- M. Roman and W. T. Winter, Biomacromolecules, 2004, 5, 1671–1677 CrossRef CAS.
- H. J. Prado and M. C. Matulewicz, Eur. Polym. J., 2014, 52, 53–75 CrossRef CAS.
- R. Wahlström, A. Kalliola, J. Heikkinen, H. Kyllönen and T. Tamminen, Ind. Crops Prod., 2017, 104, 188–194 CrossRef.
- J. A. Sirviö and J. P. Heiskanen, Cellulose, 2020, 27, 1933–1950 CrossRef.
- J. A. Sirviö, Carbohydr. Polym., 2018, 198, 34–40 CrossRef PubMed.
- F. Rol, M. N. Belgacem, A. Gandini and J. Bras, Prog. Polym. Sci., 2019, 88, 241–264 CrossRef CAS.
- N. Odabas, H. Amer, M. Bacher, U. Henniges, A. Potthast and T. Rosenau, ACS Sustain. Chem. Eng., 2016, 4, 2295–2301 CrossRef CAS.
- A. Chaker and S. Boufi, Carbohydr. Polym., 2015, 131, 224–232 CrossRef CAS PubMed.
- A. Olszewska, P. Eronen, L.-S. Johansson, J.-M. Malho, M. Ankerfors, T. Lindström, J. Ruokolainen, J. Laine and M. Österberg, Cellulose, 2011, 18, 1213 CrossRef CAS.
- K. Littunen, J. Snoei de Castro, A. Samoylenko, Q. Xu, S. Quaggin, S. Vainio and J. Seppälä, Eur. Polym. J., 2016, 75, 116–124 CrossRef CAS.
- A. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC.
- F. Rol, S. Saini, V. Meyer, M. Petit-Conil and J. Bras, Ind. Crops Prod., 2019, 137, 81–88 CrossRef CAS.
- T. T. T. Ho, T. Zimmermann, R. Hauert and W. Caseri, Cellulose, 2011, 18, 1391–1406 CrossRef CAS.
- H. Liimatainen, T. Suopajärvi, J. Sirviö, O. Hormi and J. Niinimäki, Carbohydr. Polym., 2014, 103, 187–192 CrossRef CAS PubMed.
- T. Suopajärvi, J. A. Sirviö and H. Liimatainen, J. Environ. Chem. Eng., 2017, 5, 86–92 CrossRef.
- T. Selkälä, T. Suopajärvi, J. A. Sirviö, T. Luukkonen, P. Kinnunen, K. I. Kling, J. B. Wagner and H. Liimatainen, Chem. Eng. J., 2019, 374, 1013–1024 CrossRef.
- M. Jonoobi, K. O. Niska, J. Harun and M. Misra, BioResources, 2009, 4, 626–639 CAS.
- Y. Wang and W. Xie, Carbohydr. Polym., 2010, 80, 1172–1177 CrossRef CAS.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- P. Li, J. A. Sirviö, S. Hong, A. Ämmälä and H. Liimatainen, Chem. Eng. J., 2019, 375, 122050 CrossRef CAS.
- J. A. Sirviö and M. Visanko, Biomacromolecules, 2019, 20, 2413–2420 CrossRef PubMed.
- J. T. Davies, Proceedings of the Second International Congress on Surface Activity, Butterworth, London, 1957, vol. 1, p. 426 Search PubMed.
- E. Jiang, N. Amiralian, M. Maghe, B. Laycock, E. McFarland, B. Fox, D. J. Martin and P. K. Annamalai, ACS Sustain. Chem. Eng., 2017, 5, 3296–3304 CrossRef CAS.
- F. Rol, B. Karakashov, O. Nechyporchuk, M. Terrien, V. Meyer, A. Dufresne, M. N. Belgacem and J. Bras, ACS Sustain. Chem. Eng., 2017, 5, 6524–6531 CrossRef CAS.
- P. Li, J. A. Sirviö, B. Asante and H. Liimatainen, Carbohydr. Polym., 2018, 199, 219–227 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- J. A. Sirviö, A. Kolehmainen, M. Visanko, H. Liimatainen, J. Niinimäki and O. E. O. Hormi, ACS Appl. Mater. Interfaces, 2014, 6, 14384–14390 CrossRef PubMed.
- A. Naderi, A. Koschella, T. Heinze, K.-C. Shih, M.-P. Nieh, A. Pfeifer, C.-C. Chang and J. Erlandsson, Carbohydr. Polym., 2017, 169, 515–523 CrossRef CAS PubMed.
- V. Goudarzi, I. Shahabi-Ghahfarrokhi and A. Babaei-Ghazvini, Int. J. Biol. Macromol., 2017, 95, 306–313 CrossRef CAS.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- R. A. Lee, C. Bédard, V. Berberi, R. Beauchet and J.-M. Lavoie, Bioresour. Technol., 2013, 144, 658–663 CrossRef CAS PubMed.
- P. Li, J. A. Sirviö, A. Haapala, A. Khakalo and H. Liimatainen, Food Hydrocoll, 2019, 92, 208–217 CrossRef CAS.
- C. Starr, C. A. Evers and L. Starr, Biology: Concepts and Applications, Thomson, Brooks/Cole, 2006 Search PubMed.
- S.-I. Hong and J. M. Krochta, J. Food Sci., 2003, 68, 224–228 CrossRef CAS.
- J. Wang, D. J. Gardner, N. M. Stark, D. W. Bousfield, M. Tajvidi and Z. Cai, ACS Sustain. Chem. Eng., 2018, 6, 49–70 CrossRef CAS.
- J. Lange and Y. Wyser, Packag. Technol. Sci., 2003, 16, 149–158 CrossRef CAS.
- V. Kumar, R. Bollström, A. Yang, Q. Chen, G. Chen, P. Salminen, D. Bousfield and M. Toivakka, Cellulose, 2014, 21, 3443–3456 CrossRef CAS.
- C. Aulin, M. Gällstedt and T. Lindström, Cellulose, 2010, 17, 559–574 CrossRef CAS.
- A. Rindlav-Westling, M. Stading, A.-M. Hermansson and P. Gatenholm, Carbohydr. Polym., 1998, 36, 217–224 CrossRef CAS.
- M. Österberg, J. Vartiainen, J. Lucenius, U. Hippi, J. Seppälä, R. Serimaa and J. Laine, ACS Appl. Mater. Interfaces, 2013, 5, 4640–4647 CrossRef PubMed.
- J. Sethi, M. Visanko, M. Österberg and J. A. Sirviö, Carbohydr. Polym., 2019, 203, 148–156 CrossRef CAS.
- J. Gaume, P. Wong-Wah-Chung, A. Rivaton, S. Thérias and J.-L. Gardette, RSC Adv., 2011, 1, 1471–1481 RSC.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS.
- H. Sehaqui, S. Morimune, T. Nishino and L. A. Berglund, Biomacromolecules, 2012, 13, 3661–3667 CrossRef CAS PubMed.
- F. Ansari, S. Galland, M. Johansson, C. J. G. Plummer and L. A. Berglund, Compos. Part Appl. Sci. Manuf., 2014, 63, 35–44 CrossRef CAS.
- M. Y. Ismail, M. Patanen, J. A. Sirviö, M. Visanko, T. Ohigashi, N. Kosugi, M. Huttula and H. Liimatainen, Carbohydr. Polym., 2019, 218, 87–94 CrossRef CAS PubMed.
- Q. Meng and T. J. Wang, Appl. Mech. Rev., 2019, 71, 040801 CrossRef.
- A. Ämmälä, O. Laitinen, J. A. Sirviö and H. Liimatainen, Ind. Crops Prod., 2019, 140, 111664 CrossRef.
- J. Sethi, M. Farooq, S. Sain, M. Sain, J. A. Sirviö, M. Illikainen and K. Oksman, Cellulose, 2018, 25, 259–268 CrossRef CAS.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- M. Salajkova, L. Valentini, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2013, 87, 103–110 CrossRef CAS.
- W. D. C. Jr and D. G. Rethwisch, Fundamentals of Materials Science and Engineering: an Integrated Approach, John Wiley & Sons, 2012 Search PubMed.
- K. Zhang and H. Liimatainen, Small, 2018, 14, 1801937 CrossRef PubMed.
- B. Malhotra, A. Keshwani and H. Kharkwal, Front. Microbiol., 2015, 6, 1–9 Search PubMed.
- D. Sun, M. B. Shahzad, M. Li, G. Wang and D. Xu, Mater. Technol., 2015, 30, B90–B95 CrossRef.
- J. A. Sirviö, A.-K. Anttila, A. M. Pirttilä, H. Liimatainen, I. Kilpeläinen, J. Niinimäki and O. Hormi, Cellulose, 2014, 1–11 Search PubMed.
- S. Pal, R. Nisi, M. Stoppa and A. Licciulli, ACS Omega, 2017, 2, 3632–3639 CrossRef CAS.
- M. Khamrai, S. L. Banerjee and P. P. Kundu, Carbohydr. Polym., 2017, 174, 580–590 CrossRef CAS.
- K. Slopiecka, P. Bartocci and F. Fantozzi, Appl. Energy, 2012, 97, 491–497 CrossRef CAS.
- Q. Liu, S. Wang, Y. Zheng, Z. Luo and K. Cen, J. Anal. Appl. Pyrolysis, 2008, 82, 170–177 CrossRef CAS.
- F. Zhang, Z. Pang, C. Dong and Z. Liu, Carbohydr. Polym., 2015, 132, 214–220 CrossRef CAS PubMed.
- J. A. Sirviö, T. Hasa, J. Ahola, H. Liimatainen, J. Niinimäki and O. Hormi, Carbohydr. Polym., 2015, 133, 524–532 CrossRef PubMed.
- G. Li, Y. Fu, Z. Shao, F. Zhang and M. Qin, BioResources, 2015, 10, 7782–7794 CAS.
- P. P. Danilovas, R. Rutkaite and A. Zemaitaitis, Carbohydr. Polym., 2014, 112, 721–728 CrossRef CAS PubMed.
- W. Xie, Z. Gao, W.-P. Pan, D. Hunter, A. Singh and R. Vaia, Chem. Mater., 2001, 13, 2979–2990 CrossRef CAS.
- S. Sowmiah, V. Srinivasadesikan, M.-C. Tseng and Y.-H. Chu, Molecules, 2009, 14, 3780–3813 CrossRef CAS PubMed.
- T. Sebio-Puñal, S. Naya, J. López-Beceiro, J. Tarrío-Saavedra and R. Artiaga, J. Therm. Anal. Calorim., 2012, 109, 1163–1167 CrossRef.
- M. Poletto, H. L. Ornaghi and A. J. Zattera, Materials, 2014, 7, 6105–6119 CrossRef CAS.
- C. Borsoi, M. V. G. Zimmernnam, A. J. Zattera, R. M. C. Santana and C. A. Ferreira, J. Therm. Anal. Calorim., 2016, 126, 1867–1878 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DRIFT spectra and TEM images; photographs of CWNF solutions and films, and antimicrobial performance of the reference film; histograms; stress–strain curves. See DOI: 10.1039/c9ta13182e |
| This journal is © The Royal Society of Chemistry 2020 |