
Controlling the C2+ product selectivity of electrochemical CO2 reduction on an electrosprayed Cu catalyst†
 ab
Chan Woo
Lee,
ac
Dang Le Tri
Nguyen,
abd
Hyung-Suk
Oh,
ab
Byoung Koun
Min
aef
and
Yun Jeong
Hwang
*abg
ab
Chan Woo
Lee,
ac
Dang Le Tri
Nguyen,
abd
Hyung-Suk
Oh,
ab
Byoung Koun
Min
aef
and
Yun Jeong
Hwang
*abg
Abstract
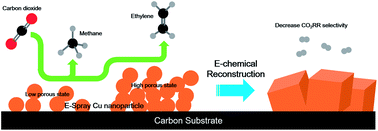
Cu catalysts prepared by modifying bulk Cu foils have achieved high performance for value-added C2+ compounds from electrochemical CO2 reduction (CO2RR) but the transformation of active sites can be affected by the bulk substrate, which make it complex to design the catalyst. Herein, we newly introduce a simple electrospray pyrolysis method to take advantage of a facile wet-chemical synthesis applicable on non-copper substrates, such as a porous carbon paper, and demonstrate highly enhanced selectivity for C2H4 production from CO2RR. The electrosprayed copper oxide on the carbon paper showed uniquely improved C2 selectivity compared with that on the copper substrate. The improved performance is proposed to be related to the presence of Cu mixed state and retention of morphology of the electrosprayed catalyst on the carbon paper, showing the importance of the substrate. In addition, the C2 product selectivity can be tuned by the electrospray synthesis time as it affects the size of the surface nanostructure as well as the porosity of the catalyst, which can provide an effective way to regulate the C2/C1 ratio.
- This article is part of the themed collection: Journal of Materials Chemistry A Lunar New Year collection 2021
 Please wait while we load your content...
Please wait while we load your content...

