
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen exchange kinetics and nonstoichiometry of pristine La0.6Sr0.4CoO3−δ thin films unaltered by degradation
Matthäus
Siebenhofer
 *a,
Tobias Martin
Huber
ab,
Gernot
Friedbacher
a,
Werner
Artner
c,
Jürgen
Fleig
a and
Markus
Kubicek
*a
*a,
Tobias Martin
Huber
ab,
Gernot
Friedbacher
a,
Werner
Artner
c,
Jürgen
Fleig
a and
Markus
Kubicek
*a
aInstitute of Chemical Technologies and Analytics, Vienna University of Technology, Austria. E-mail: matthaeus.siebenhofer@tuwien.ac.at; markus.kubicek@tuwien.ac.at
bKyushu University, Next-Generation Fuel Cell Research Center (NEXT-FC), Japan
cX-Ray Center, Vienna University of Technology, Austria
First published on 25th March 2020
Abstract
La0.6Sr0.4CoO3−δ thin films grown on YSZ single crystals were investigated directly in the stage of deposition by means of in situ impedance spectroscopy during pulsed laser deposition (IPLD). This method allows the observation of dense thin films unaltered by degradation and provides information about the oxygen exchange kinetics as well as the defect chemistry of pristine LSC thin films. These measurements revealed remarkably low surface resistance values (1.3 Ω cm2 at 600 °C and 0.04 mbar O2) compared to films measured outside the PLD chamber (∼20 Ω cm2 at 600 °C and 0.04 mbar O2). Also the activation energy of the surface exchange resistance at 0.04 mbar p(O2) is significantly lower than at ambient conditions (∼1 eV vs. ∼1.3 eV) and degradation happens considerably slower. Furthermore, the grain size of the LSC thin film does not affect its initial surface resistance directly after deposition. The chemical capacitance of LSC thin films was linked to the concentration of oxygen vacancies and shows that LSC thin films exhibit lower oxygen vacancy concentrations than the corresponding bulk material.
1 Introduction
The optimization of cathode materials for solid oxide fuel cells (SOFCs) and electrolysis cells (SOECs) is an important step towards efficient and sustainable energy conversion and storage.1,2 Because of its high catalytic activity for the oxygen reduction reaction, La0.6Sr0.4CoO3−δ (LSC) is a promising candidate for this challenge.3,4 However, one of the remaining obstacles in the broader use of LSC is its susceptibility to changes of the surface structure e.g. due to Sr-segregation5–9 and chromium or sulfur poisoning.10,11To further deepen the understanding of the degradation mechanisms and possible countermeasures, it is necessary to retrace the properties and the structure of LSC to a point before degradation starts. When standard impedance spectroscopic measurements are used to analyze materials,12,13 every sample is exposed to several environmental factors such as degradation sources as well as thermal expansion stress between deposition and measurement.14 Consequently, data for the electrical properties of LSC after deposition calculated from measurements of different authors at different conditions vary within several orders of magnitude.9,15–17
In this study, a recently developed technique was employed to perform electrochemical impedance spectroscopy measurements on thin films during and directly after pulsed laser deposition18,19 on LSC thin films, grown on yttria stabilized zirconia (YSZ) single crystals. It was possible to track the electrochemical properties of the deposited material directly during growth and thus achieve an electrochemical characterization of pristine LSC thin films yet unaltered by degradation processes. This contribution reports the results of these measurements and compares them to ex situ experiments.
2 Methods
2.1 Sample preparation
At first current collector grids (5 nm Ti/100 nm Pt, 30 μm square holes/5 μm stripes) were prepared by lift-off photolithography and metal sputtering (Bal Tec MED 020, Leica Microsystems GmbH, Germany) on both sides of (100) oriented yttria stabilized zirconia (YSZ, 9.5 mol% Y2O3, Crystec GmbH, Germany) single crystalline substrates (5 × 5 × 0.5 mm3). This grid served as the base for working electrode (WE) and counter electrode (CE) and ensured that the whole film was active (Fig. 1). After deposition each sample was examined with optical microscopy to calculate the exact area of the active LSC surface (=area of the LSC/YSZ interface), which is typically in the range of 35% to 42% of the entire sample area. | ||
| Fig. 1 Microscope photographs of grid details (a and c) as well as overviews of larger sample areas before and after deposition (b and d). | ||
The LSC targets for the pulsed laser deposition (PLD) of the thin films were synthesized from powders via a Pechini routine.20 La2O3, SrCO3 powders and Co granules (all Sigma-Aldrich, 99.995%) were each dissolved in nitric acid (Aldrich, 70% purified by redistillation, 99.999%), diluted with water and mixed. 1.1 mol citric acid (Aldrich 99.9998% metals basis) was added for each mol of cations for chelation. The mixture was heated on a hotplate and after sufficient evaporation of water a viscous foam developed which self-ignited after continuous heating. The product was calcined at 1000 °C for 2 h and pressed to a pellet by cold isostatic pressing (300–310 MPa) which was then sintered in air for 12 h at 1200 °C, yielding a La0.6Sr0.4CoO3−δ PLD target. The exact composition of the thin films slightly differed from the target so the preparation routine was modified until the thin film composition was of satisfying quality.21 The final thin film composition (La0.612±0.002Sr0.406±0.003Co0.983±0.005O3) was determined from a thin film deposited on YSZ by standard PLD, which was dissolved in hydrochloric acid and analyzed with inductively coupled plasma-mass spectroscopy (Thermo Scientific iCAP Q, USA).
LSC films were then deposited by PLD on top of the current collector grids. All depositions were done by means of a KrF (λ = 248 nm) excimer laser (Lambda Physics, COMPex Pro 201) with a laser fluence of approximately 2 J cm−2. For the counter electrode 9000 laser pulses at a frequency of 5 Hz were shot on the LSC target with the target substrate distance set to 5 cm, the atmosphere was set to 0.4 mbar p(O2) and the substrate was heated to a temperature of 450 °C, measured with a pyrometer (Heitronics). These deposition conditions yielded columnar and porous films which showed an especially low polarization resistance due to an increased inner surface.4,21 After the counter electrode deposition process was finished the sample was cooled to room temperature in the deposition atmosphere at a cooling rate of 15 °C min−1 to room temperature. Then the edge sides of the sample were treated by grinding to remove excess material, thus preventing short circuits. The resulting samples were then used for the following IPLD study in which dense LSC thin films were investigated.
2.2 In situ impedance spectroscopy during PLD (IPLD)
The IPLD setup is shown in Fig. 2. Details of the method are described elsewhere.19 At the beginning a corundum plate is placed directly on the platinum heating wires. To contact the counter electrode, a platinum sheet is placed on top and fixed by a corundum frame with an opening. Then the sample with both current collectors and the counter electrode already deposited is placed into this opening such that the counter electrode is in contact with the platinum sheet. Afterwards it is covered with another corundum plate with a smaller opening which serves as a mask during PLD to avoid coating along the sample faces. The top current collector is then contacted by a Pt tip attached to a Cu arm.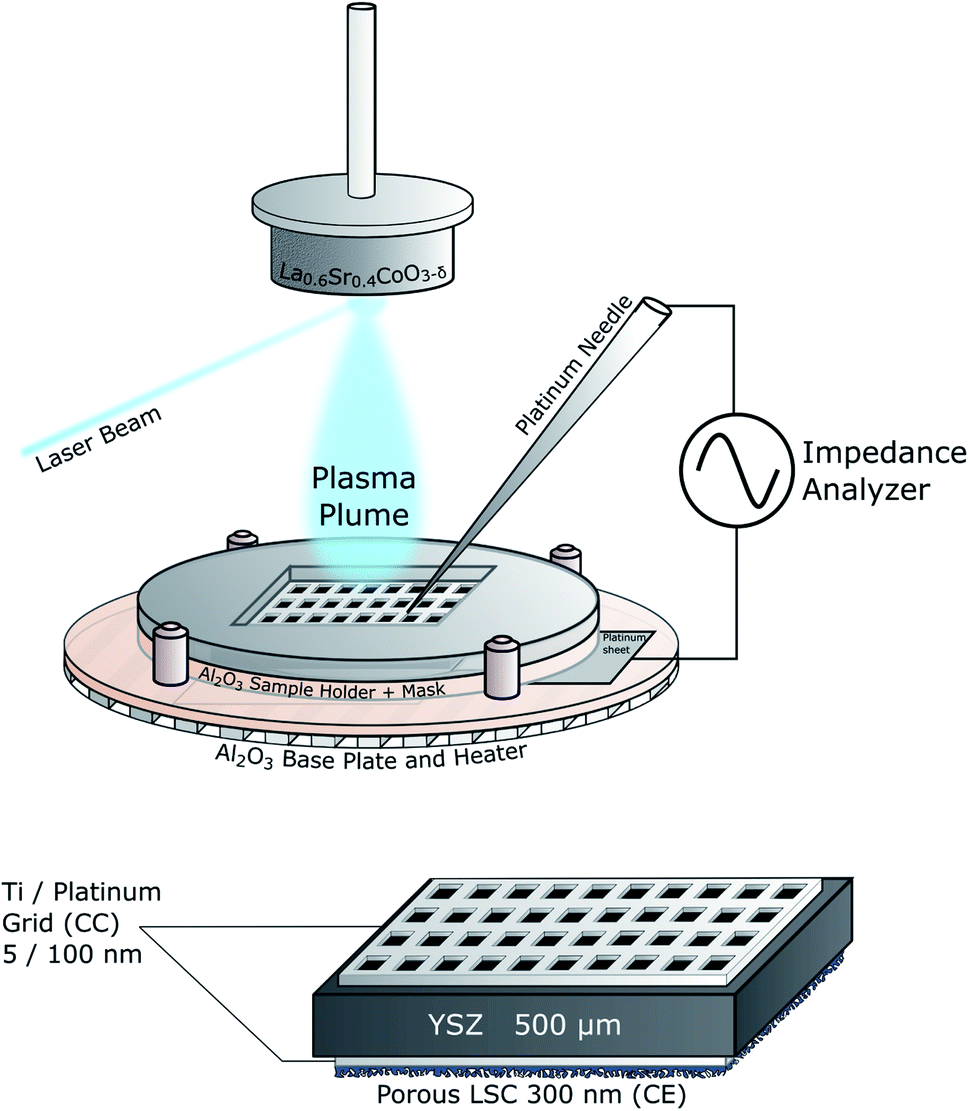 | ||
| Fig. 2 Sketch of the IPLD setup and of the standard sample structure. | ||
Before the actual IPLD measurements started the LSC target was ground and cleaned, mounted in the PLD chamber and preablated for 60 s at 5 Hz. The atmosphere was then set to 0.04 mbar p(O2) and the sample was heated to the desired temperature which was controlled by measuring the high frequency minimum or real axis intercept of impedance measurements. This resistance is largely caused by the ionic conduction of YSZ but also includes an internal resistance of the setup which was previously measured and subtracted. Combined with the known temperature-conductivity relation of YSZ single crystals13 the resulting resistance was used to determine the actual sample temperature. During the deposition of the working electrode the laser was operated with a frequency of 2 Hz for a predefined pulse number.
Then impedance measurements were performed with an Alpha-A High Performance Frequency Analyzer and Electrochemical Test Station POT/GAL 30V/2A setup by Novocontrol Technologies in the frequency regime from 106 to 10−1 Hz. The resolution was set to five points per decade and an alternating voltage of 10 mV rms was used. Two impedance spectra were recorded for each film thickness. Those generally agreed very well, ensuring that the defect chemistry of the sample was equilibrated and that degradation processes inside the PLD chamber were negligibly slow on this timescale. The LSC growth rate was about 0.025 nm/pulse which was confirmed with profilometer (DekTakXT, Bruker, USA) measurements of the deposited films.
2.3 Structural characterization
Samples with thin films deposited at different temperatures were examined by means of grazing incidence X-ray diffraction (XRD) with an incidence angle of 2° in an Empyrean X-ray diffractometer (Malvern Panalytical) equipped with a parallel beam mirror on the incident beam side and a parallel plate collimator and a scintillation detector on the diffracted beam side. All scans were done with a measuring time of ∼1 h per sample. The diffractograms were analyzed with Panalytical Highscore.22Atomic force microscopy (AFM) was performed on samples deposited at different temperatures to analyze differences in surface microstructures. The measurements were done in tapping mode with a Nanoscope V multimode setup (Bruker) over a scan area of 1 × 1 μm2. The graphs of the experimental data were treated with ImageJ to gain information about grain size and grain density.
2.4 Ex situ electrochemical characterization
Ex situ impedance spectroscopy was performed with an Alpha-A High Performance Frequency Analyzer and Electrochemical Test Station POT/GAL 30V/2A setup by Novocontrol Technologies in the frequency regime from 106 to 10−2 Hz. The samples were heated in a tube furnace and the temperature was measured with a type S thermocouple positioned about 1 cm next to the sample. For low oxygen partial pressure experiments the ex situ setup was evacuated to a base pressure of 10−5 mbar and afterwards the desired p(O2) was set with a controlled oxygen gas flow. Ex situ measurements at ambient conditions were conducted in the same setup filled with air.3 Results and discussion
3.1 Structure and surface of La0.6Sr0.4CoO3−δ thin films
LSC thin films were found to grow in a columnar way on the YSZ substrates. Thin films deposited at different temperatures all showed the same crystal structure and phase purity. Additional peaks from the platinum grid (visible in Fig. 3) could not be avoided as the grid must be prepared before the deposition.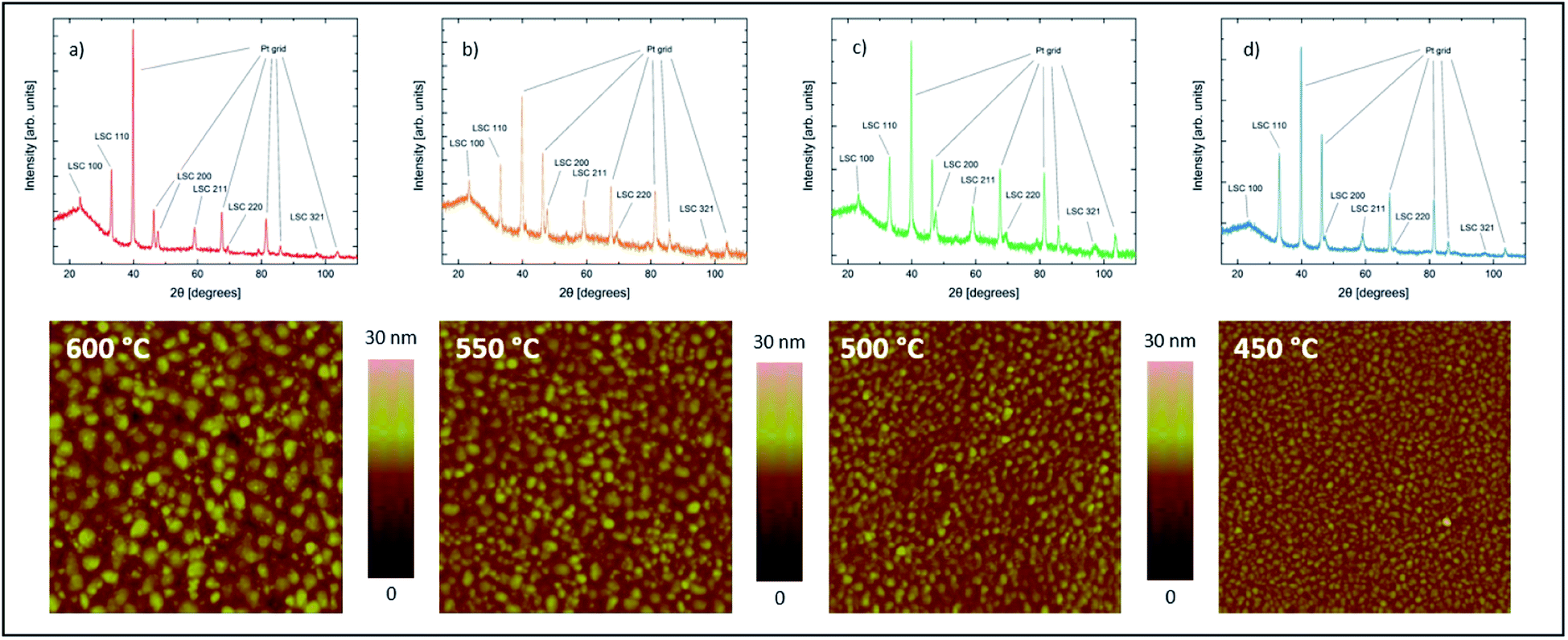 | ||
| Fig. 3 Diffractograms and AFM scans of samples with LSC films (100 nm thickness) deposited on YSZ at different temperatures of 600 °C (a), 550 °C (b), 500 °C (c) and 450 °C (d). | ||
As expected from earlier studies6 and from nucleation theory23 the AFM scans confirmed different surface morphologies for different deposition temperatures. All four samples show clearly distinguishable and homogeneously distributed grains with different grain size and different grain boundary groove depth (Fig. 4). The average grain diameter increases from 16.6 nm for the sample deposited at 450 °C to 35.5 nm for the sample deposited at 600 °C, the highest deposition temperature. Similarly the maximum grain boundary groove depth increases from 7.0 to 12.6 nm. The average roughness of the dense films increases from 1.11 nm at 450 °C to 2.33 nm at 600 °C.
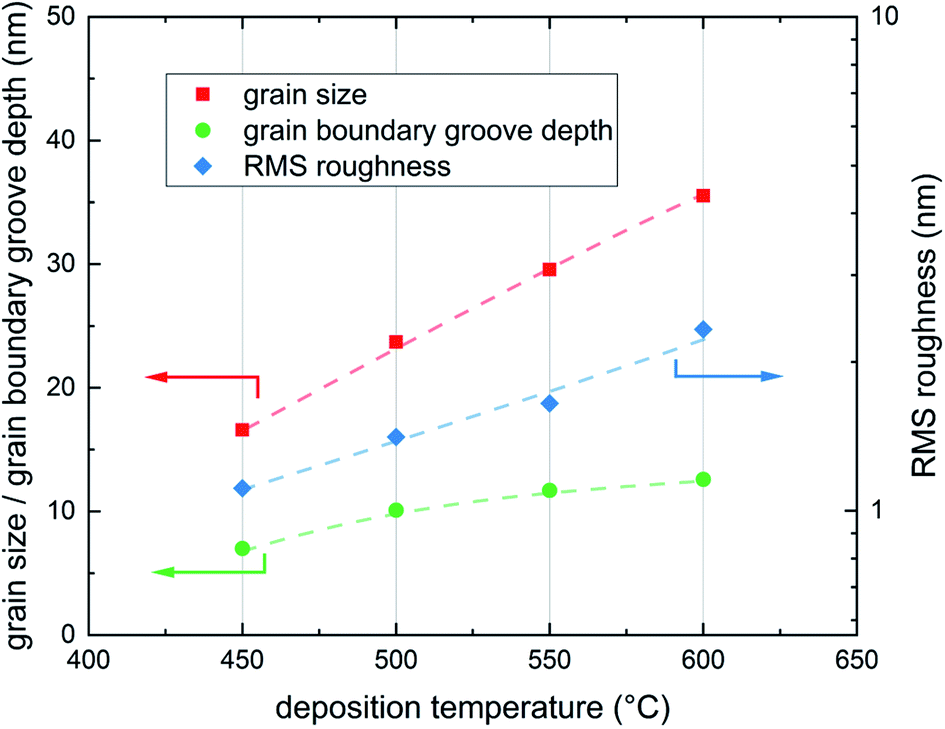 | ||
| Fig. 4 Grain size, grain boundary groove depth and film roughness for the four different deposition temperatures 600, 550, 500 and 450 °C; all three properties, grain size, grain boundary groove depth and RMS roughness increase with deposition temperature, lines are a guide to the eye. | ||
3.2 Impedance spectroscopy on LSC thin films during growth
In an earlier study using IPLD it was already discussed that about 2–3 nm LSC are required to cover the Pt current collectors to an extent that true LSC properties are measured.19 In accordance with this study, the first measured spectrum after 2 nm (100 pulses) includes three clearly distinguishable features (Fig. 5). The following interpretation of these arcs is based on the current paths discussed in ref. 19 and 24. The first feature in the high frequency regime (100 kHz - 1 kHz) is attributed to a current path which is limited by the sheet resistance of the LSC thin film RWE, sheet and the small double layer capacitance at the interface between the current collector and the electrolyte CCC, dl. This impedance feature becomes smaller with increasing film thickness. Some samples show a second high frequency feature which was attributed to the resistive and capacitative properties of the LSC/YSZ interface.12 At lower frequencies the common current path of such thin film cells becomes dominant, i.e. current flow with rate limiting oxygen surface exchange at either the working or the counter electrode and chemical capacitors of the electrodes. This results in the mid frequency arc (1 kHz to 1 Hz), corresponding to the growing LSC top electrode, and a low frequency arc (<1 Hz), reflecting the LSC counter electrode. The chemical capacitance of the counter electrode is much higher due to its larger thickness and the corresponding impedance feature remains constant over the course of the experiment.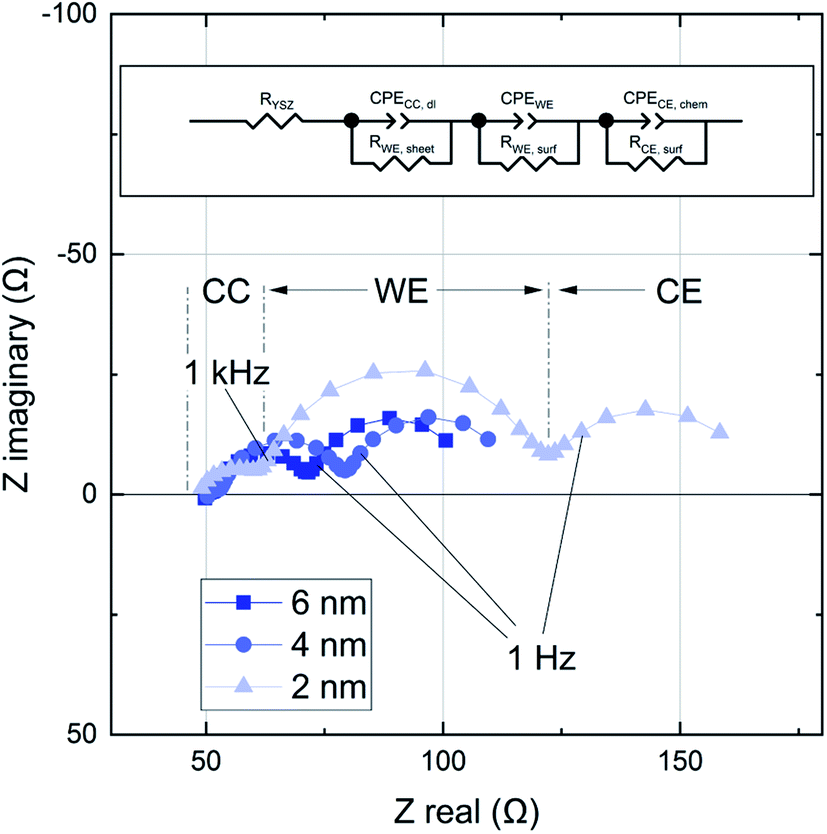 | ||
| Fig. 5 Impedance spectra for a growing LSC thin film after 80, 160 and 400 pulses (2, 4 and 10 nm). The film was grown at 600 °C and 0.04 mbar p(O2). All impedance data were fitted using the equivalent circuit shown on top. | ||
This study has a focus on the mid frequency arc which represents the oxygen exchange resistance RWE, surf and the largely chemical capacitance CWE of the growing LSC film.18 These results can be interpreted with regard to the kinetic and thermodynamic properties of the LSC film. The impedance contribution was quantified by an equivalent circuit fit of the spectra to three serial R-CPE elements, see Fig. 5 (CPE = constant phase element). The capacitance of the LSC working electrode can be calculated from the corresponding CPE element, which describes a non ideal capacitor, using the following relations:25
| ZCPE = Q−1(iω)−n and C = (R1−nQ)1/n, | (1) |
The LSC oxygen exchange resistance RWE, surf was then normalized to the film area in direct contact with YSZ, excluding the inactive area above the Pt current collectors.19 The area specific surface exchange resistance of LSC thin films is shown in Fig. 6. The plot shows that the resistance drops significantly over the first 20 nm and levels off at about 30 to 50 nm. However, this decay is not due to improved surface exchange properties but is a consequence of the LSC sheet resistance which reduces the active LSC surface area for very thin LSC films.19 Thus, for very early stages of film growth only a part of the free LSC area between the current collector grid is active and the resistance normalized to the whole LSC area is increased. Therefore, the first 50 nm are not taken into account when averaging the actual surface exchange resistance of the films. The average values resulting above 50 nm are given in Fig. 6 and plotted in Fig. 7versus the deposition (=measurement) temperature. These resistances are remarkably low, particularly when taking into account the low oxygen partial pressure of 0.04 mbar (e.g. 1.3 Ω cm2 at 600 °C).
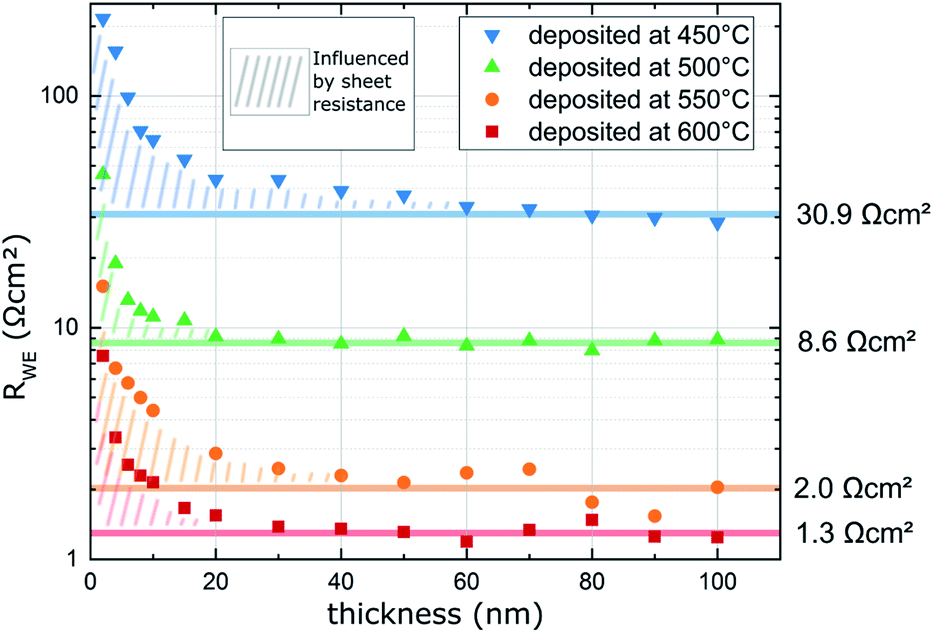 | ||
| Fig. 6 Surface exchange resistance (RWE, surf) of LSC thin films normalized to the grid-free area measured in situ during PLD at different deposition (=measurement) temperatures and 0.04 mbar p(O2). | ||
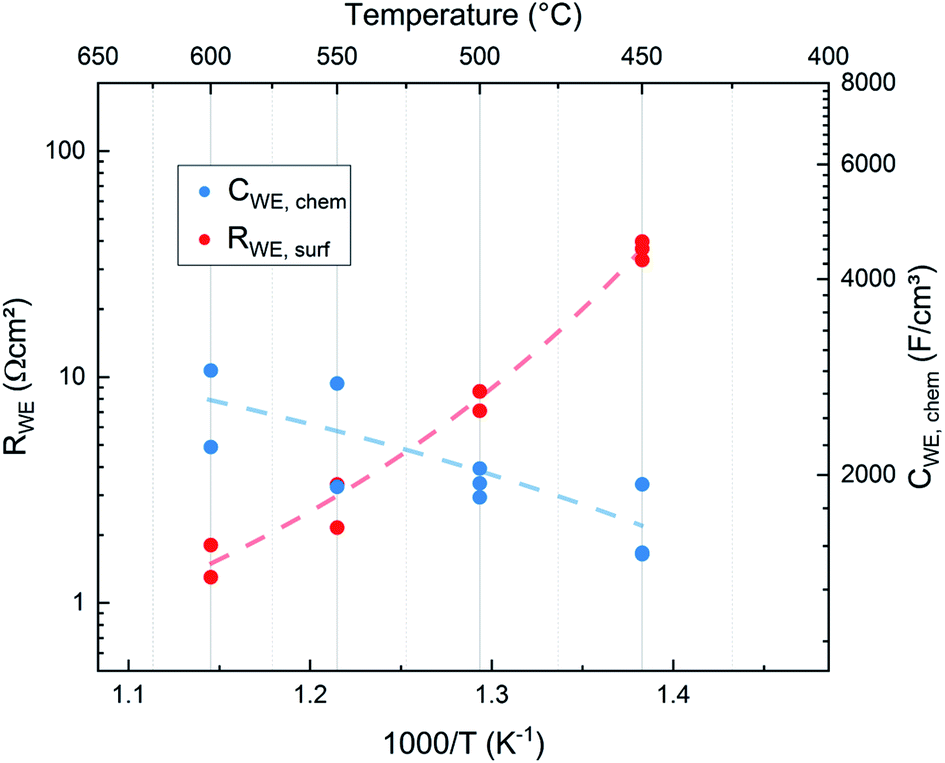 | ||
| Fig. 7 Surface exchange resistance and chemical capacitances of films directly measured in situ (IPLD) at the respective deposition temperature and a deposition pressure of 0.04 mbar p(O2). (Ten different films were investigated and thus an activation energy analysis is not performed here, lines are a guide to the eye.) | ||
The capacitances of the thin films are shown in Fig. 8. Those are largely due to the chemical bulk capacitance of LSC and increase during growth.26 Chemical capacitances are defined according to
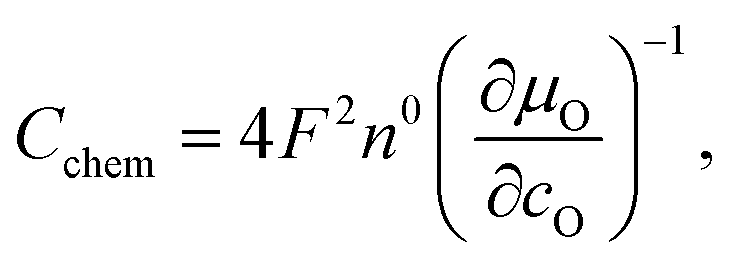 | (2) |
| CWE = CWE, int + CWE, chemx. | (3) |
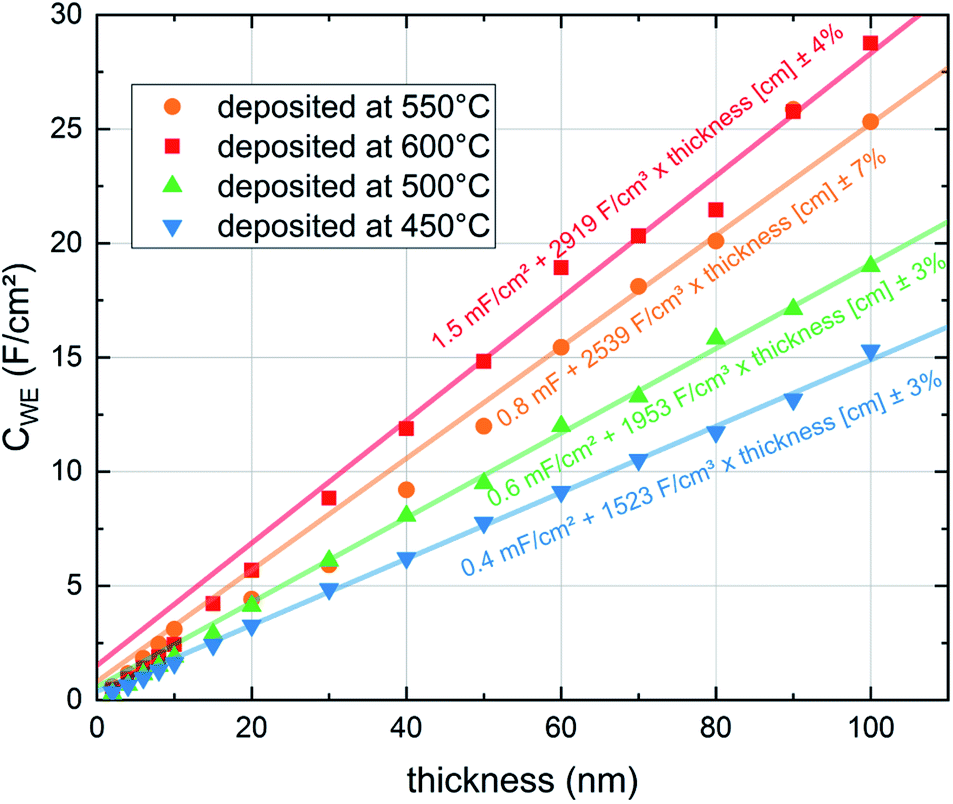 | ||
| Fig. 8 Capacitances (CWE) of LSC thin films normalized to the grid-free area during exemplary in situ PLD measurements at different deposition and measurement temperatures and 0.04 mbar p(O2). The values of the last 50 nm were used to extrapolate the capacitance to lower thicknesses. | ||
These normalized chemical bulk capacitances CWE, chem are given in Fig. 8 and are plotted in Fig. 7versus the deposition temperature.
As expected for a chemical capacitance, CWE, chem increases with temperature due to the accompanying rise of the oxygen vacancy concentration. It is higher than values reported in previous studies which themselves scatter, partly due to different measurement conditions4,15–17,19,27 (see Table 1). Our high capacitance is largely attributed to the low oxygen partial pressure in the PLD chamber which results in a high concentration of oxygen vacancies.
| Source | T [°C] | p(O2) | C WE, chem [F cm−3] |
|---|---|---|---|
| This work – in situ | 600 | 0.04 mbar | ∼2900 |
| This work – ex situ | 600 | 0.21 mbar | ∼1500 |
| Januschewsky et al.4 | 600 | 0.21 bar | ∼2000 |
| Wilson et al.16 | 725 | 0.01 bar | ∼2200 |
| Wilson et al.16 | 725 | 0.21 bar | ∼1100 |
| Kawada et al.17 | 600 | 0.1 mbar | ∼2000 |
| Rupp et al.19 | 600 | 0.04 mbar | ∼1300 |
| Rupp et al.15 | 600 | 0.4 mbar | ∼2500 |
| Sase et al.27 | 700 | 0.1 bar | ∼1050 |
| Sase et al.27 | 700 | 0.1 mbar | ∼2000 |
3.3 In situ analysis of oxygen surface exchange on pristine LSC thin films: temperature and grain size effects
To compare the properties of differently deposited pristine LSC thin films directly after deposition it is necessary to bring the samples prepared at different temperatures to the same measurement temperature. To minimize degradation, the samples were only measured at temperatures lower than the deposition temperature. Thereby the film deposited at 600 °C was measured at the four temperatures 600, 550, 500 and 450 °C while the sample prepared at 450 °C was only measured at 450 °C. The results are shown in Fig. 9. Interestingly, all data points lie close to the Arrhenius line given by the film deposited at 600 °C. Accordingly, we conclude that the preparation temperature has very little effect on the initial oxygen exchange properties, very fast oxygen exchange is found for all deposition temperatures. Hence, an overall analysis of all points was performed and lead to an activation energy EA = 1.05 eV for the oxygen exchange reaction of these freshly deposited films. Excluding the film prepared at 450 °C, which is slightly off, we get an activation energy of EA = 1.02 eV. This is unusually low compared to literature data obtained ex situ4,15,28–31 (see Table 2). In all cases the average measured surface exchange resistance is very low for all temperatures, ranging between RWE ∼ 1.3 Ω cm2 (at 600 °C and p(O2) = 0.04 mbar) and RWE ∼ 20.9 Ω cm2 (at 450 °C and p(O2) = 0.04 mbar) excluding the sample at 450 °C.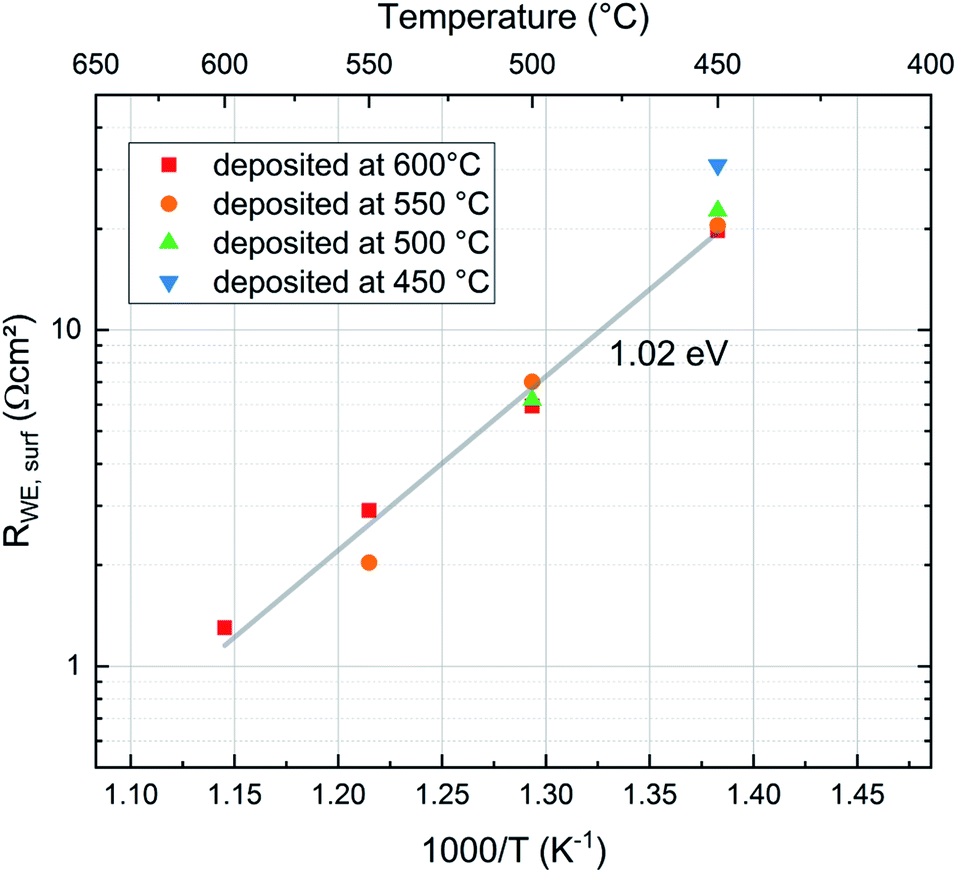 | ||
| Fig. 9 Comparison of the surface exchange resistance of pristine LSC films deposited at different temperatures (1.02 eV activation energy results without the film deposited at 450 °C, else EA = 1.05 eV). | ||
| Source | T [°C] | p(O2) | E A [eV] |
|---|---|---|---|
| This work – in situ | 450–600 | 0.04 mbar | 1.02–1.05 |
| This work – ex situ | 450–600 | 0.21 mbar | 1.31–1.59 |
| This work – ex situ | 450–600 | 0.04 mbar | 0.85–0.96 |
| Rupp et al.15 | 475–600 | 0.21 bar | 1.26 |
| Egger et al.28 | 525–725 | 0.001 bar | 1.44 |
| Egger et al.28 | 525–725 | 0.1 bar | 1.84 |
| Januschewsky et al.4 | <670 | 1 bar | 1.4 |
| Zhao et al.29 | 450–600 | 0.21 bar | 1.26–1.33 |
| Hayd et al.30 | 500–700 | 0.21 bar | 1.42 |
| Baumann et al.31 | 600–750 | 0.21 bar | 1.3 |
These results indicate that the crystallite size of the film, which depends on the deposition temperature, has no significant effect on the surface exchange reaction properties of the films. This is expected since LSC is a mixed ionic electronic conducting material and the whole surface area of the electrode is available for the oxygen exchange reaction. On the other hand studies of other authors have shown that LSC thin films deposited at different temperatures show differences in degradation behaviour and thus also in their ex situ surface exchange resistance.6 Although the area specific grain boundary length in our experiments almost doubles when decreasing the deposition temperature from 600 °C to 450 °C, the surface exchange resistance shows no corresponding trend and seems to be not affected by the different grain sizes (see Fig. 10). Thus, we conclude that in pristine LSC films, grain boundaries do not contribute significantly to the oxygen exchange reaction. This is also in accordance with (ex situ) tracer exchange measurements done by Saranya et al.32
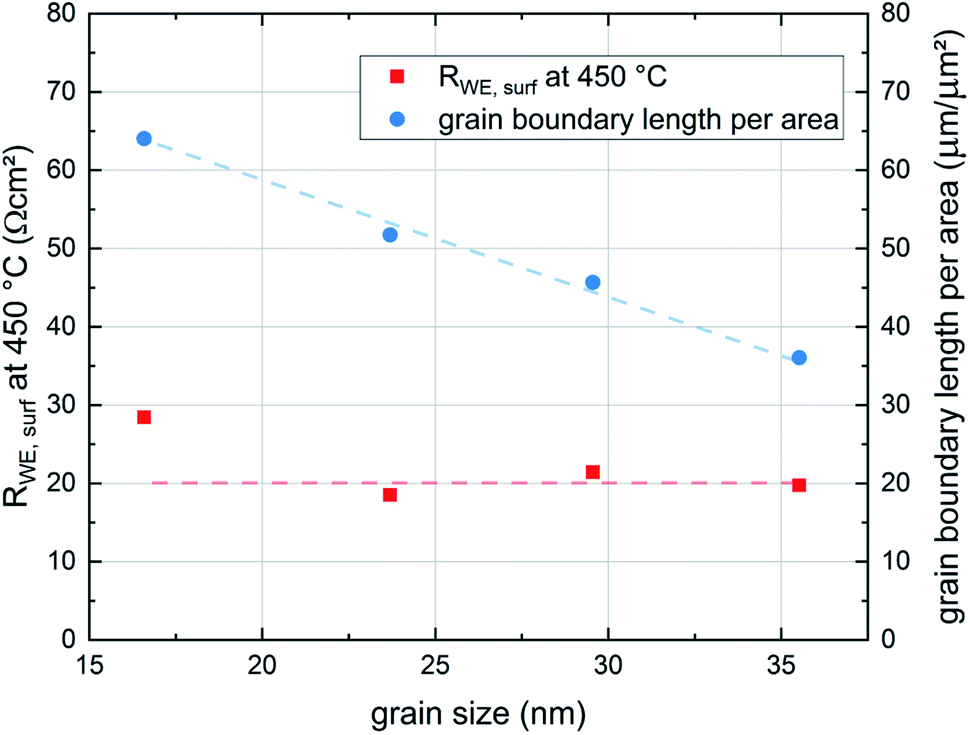 | ||
| Fig. 10 Dependence of the surface exchange resistance of the thin film grain size. To illustrate the impact of the grain size on the structure of the film the area specific grain boundary length is also plotted in the figure. Lines are a guide to the eye. | ||
3.4 Comparison of oxygen exchange properties measured in situ (pristine films) and ex situ
To gain information about the ex situ behavior of the thin films, another set of samples was deposited at the same conditions (all four deposition temperatures and 0.04 mbar p(O2)). Again the in situ surface exchange resistance data fit very well to the previously measured data. These samples were then measured during temperature cycling between 450 °C and 650 °C in a conventional setup after evacuation and filling with oxygen to reach the same operating pressure as in the PLD chamber. These measurements show a severe change in the surface exchange resistance between in situ and ex situ measurements (Fig. 11a). Already the very first ex situ measurements revealed more than an order of magnitude higher resistances than the in situ studies (“unknown degradation” in Fig. 11a). The heating/cooling cycle lead to only slight further degradation of the surface resistance and an average activation energy of 0.96 eV resulting without 650 °C data, which is close to the in situ measurements (1.02–1.05 eV). The small difference is at least partly attributed to the fact that the in situ measurements were only performed during cooling. This is a first indication that the severe difference between in situ and ex situ measurements due to “unknown” degradation possibly originates from the number of catalytically active sites but not a difference in the oxygen exchange mechanism itself.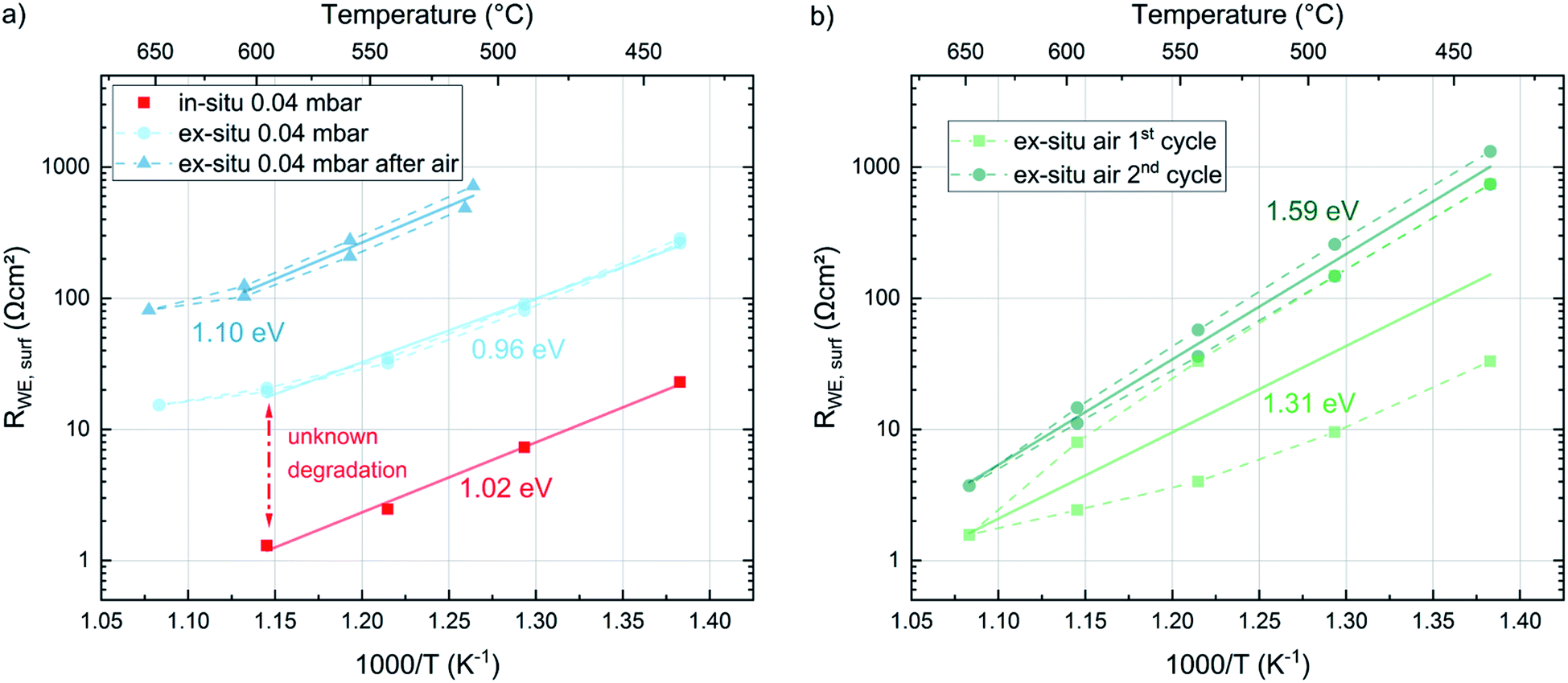 | ||
| Fig. 11 Arrhenius plots of the surface exchange resistance values averaged over the dense LSC thin films deposited at 450, 500, 550 and 600 °C, measured in different environments (all measurements at 0.04 mbar are shown in (a), measurements in air are shown in (b)). Arrhenius lines are labelled with the corresponding activation energies. | ||
After the ex situ measurements at 0.04 mbar oxygen the setup was filled with air and the samples were measured again in ambient conditions. During these measurements an initial partial pressure induced decrease of the surface exchange resistance by a factor of 10 (at 600 °C) was observed (Fig. 11b). However, the two heating/cooling cycles shown in this figure indicate a very severe degradation in this atmosphere in contrast to the cycles in 0.04 mbar O2. This also complicates the determination of accurate activation energies. For averaged resistance values of two consecutive heating/cooling cycles, we get 1.31 eV and 1.59 eV respectively. Changing finally back to 0.04 mbar oxygen revealed a strongly increased resistance (due to the degradation in ambient air) but again a low activation energy (1.10 eV). Accordingly, we can conclude that the surface exchange kinetics has a substantially higher activation energy in air compared to 0.04 mbar O2, independent of the degradation state. However, it is still unknown, whether or not this activation energy change between 0.04 mbar O2 and air indicates a mechanism change. Also temperature and p(O2) dependent defect concentrations may cause a change for one and the same mechanism.33
The complete evolution of the surface exchange resistance over the course of the experiments is visualized in Fig. 12. Despite the continuous ex situ degradation we can estimate an ex situ partial pressure dependent change of RWE, surf in the range of a factor of ten between 0.21 bar and 0.04 mbar O2. When projecting the same dependence to the pristine films (in situ measured) we get a very low surface exchange resistance of about 0.1 Ω cm2 at 600 °C in air. Still, even after the first “unknown degradation” and about 36 h of measurements at 0.04 mbar O2 we find surface exchange resistance values between 1.7 and 2.9 Ω cm2ex situ in air and at 600 °C which is lower than most values reported in literature.19 This emphasizes that our LSC thin films are exceptionally active in situ and still kinetically very fast ex situ, despite the unknown degradation processes.
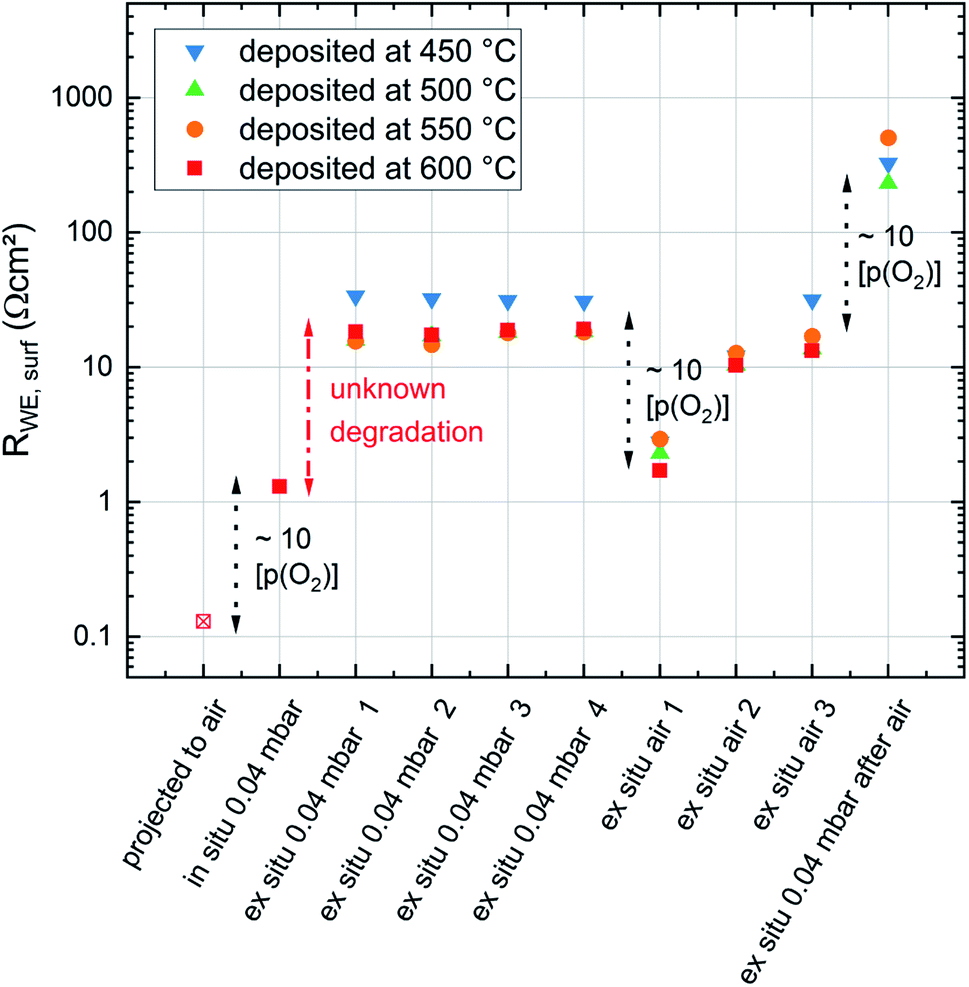 | ||
| Fig. 12 Evolution of the surface exchange resistance of dense LSC thin films deposited at different temperatures but all measured at 600 °C during different stages of the experiment. Significant changes of the surface exchange resistance were labelled according to their reason. | ||
For a further discussion of the degradation phenomena occuring in the experiments conducted in this study it is useful to consider three experimental stages: in situ measurements, the transfer to the ex situ setup and ex situ measurements (see Fig. 13). Already in situ, shortly after deposition, a first degradation of the LSC thin film surface activity can be observed. Since the oxygen partial pressure inside of the PLD chamber is very low and no other gases are present in considerable amounts, the degradation inside of the PLD chamber is likely due to internal driving forces leading to Sr segregation. For example the size mismatch of the doped cations and the thereby induced lattice strain, as well as electrostatic interactions may pull Sr ions out of the bulk towards the surface.8 This assumption is also supported by the results of XPS measurements which show an increased Sr content near the surface for some samples directly after deposition.6 In a representative measurement a film deposited at 450 °C was heated to 600 °C in the in situ setup and the degradation of the surface exchange resistance was measured (see Fig. 13). After 15 h, the initial resistance of 2 Ω cm2 increased by a factor of 4 and approached a stable value of 8 Ω cm2 in 0.04 mbar oxygen atmosphere.
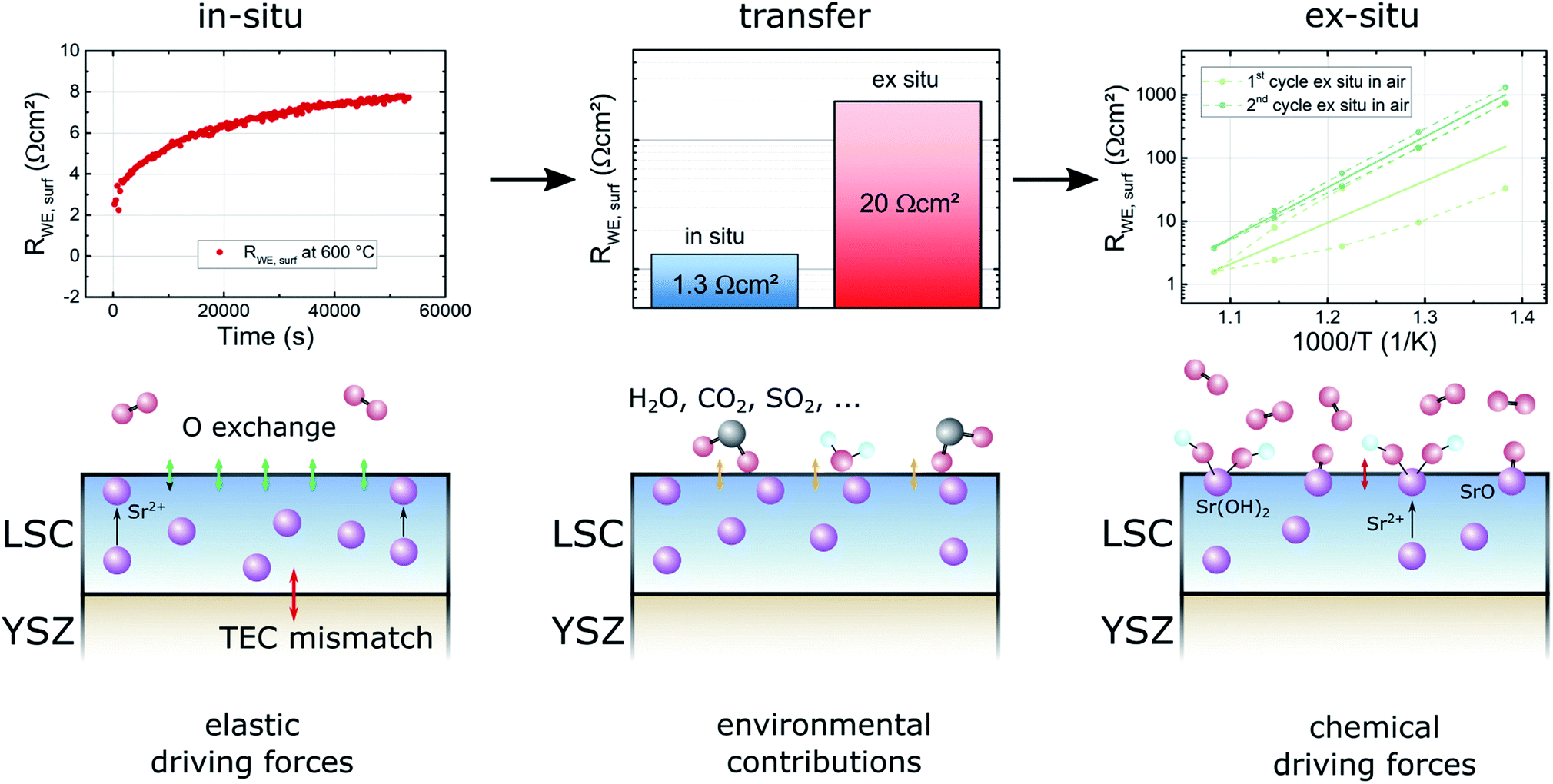 | ||
| Fig. 13 Overview of different degradation sources and driving forces in different experimental stages – in situ, during transfer and ex situ. Basic kinetic mechanisms are visualized next to exemplary measurements concerning the particular degradation processes. | ||
In the first ex situ measurement at low oxygen partial pressures the surface exchange resistance was usually between 20 and 30 Ω cm2. Since samples were usually removed from the PLD chamber within 30 min, there seem to be further adverse processes occurring during cooling in PLD atmosphere, during the transfer to the ex situ setup in air, or during heating up ex situ. These contributions are summarized as environmental contributions in Fig. 13.
During cooling and heating the samples are exposed to thermal stress which could lead to irreversible changes of the sample surface structure. Several authors report that it is possible that the sample surface cracks due to different thermic expansion coefficients of the film and the electrolyte materials.15,34 However, such cracks could not be found in AFM imaging. Furthermore, cracking of dense model electrodes would also lead to an increase of the free surface area, thus rather promoting the oxygen exchange reaction than impeding it. For these reasons thermal stress does not seem to be the reason for the observed degradation phenomenon between in situ and ex situ measurements.
During the transfer to the ex situ setup and in the ex situ setup itself the sample surface is exposed to lab air and a new measurement atmosphere for several hours. The most likely reason for the much higher surface exchange resistance during the first ex situ measurements is the adsorption of gas molecules on the surface, related to classic poisoning effects during operation.7,11 Such adsorbed molecules may facilitate or accelerate the formation of a number of species already found in ex situ annealing experiments like SrO,6 SrOH2,6 SrCO3,35 SrSO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,36,37 which could also form during the first heating and impede the initially very fast oxygen exchange reaction.
11,36,37 which could also form during the first heating and impede the initially very fast oxygen exchange reaction.
In the final stage of the experiments, during ex situ measurements in air, a further strong degradation of the surface activity was observed. We strongly expect that the degradation effects in this phase are dominated by oxide or hydroxide formation processes which were also observed in the XPS results from Cai et al.,6 indicating that SrO or Sr(OH)2 phases form on LSC surfaces exposed to high temperatures (600 °C) and air.
We thus conclude that not only our ex situ properties but also most other ones reported so far on thin film LSC do not fully exploit the kinetic capability of LSC surfaces. This again highlights the necessity to futher explore the in situ/ex situ difference and ways how to avoid it. While Sr segregation due to elastic driving forces can hardly be avoided in pure LSC, yet unknown processes seem to happen during the first exposure to air and the exposure to the gas in the measurement setup. A parallel study on La0.6Sr0.4FeO3 (with similar effects) indicates that sulfur, which is also known to have a severe effect on the surface exchange kinetics in the form of SO2 poisoning during operation11 may play a key role in the processes during the transfer from in situ to ex situ setups.
3.5 Chemical capacitances measured in situ and ex situ
With regard to the chemical capacitance of the films, the ex situ measurements at PLD conditions show a significant increase of Cchem compared to the in situ measurements, especially at higher temperatures (Fig. 14). Since the chemical capacitance is a bulk property, these changes are not necessarily connected to the unknown degradation of RWE, surf. A severe contribution due to asymmetrical heating of the sample in the in situ measurement setup is excluded, as the highest thermovoltage induced in the single crystal at 600 °C was ∼35 mV which does not shift the chemical potential of oxygen in LSC substantially. When the ex situ setup is filled with air, the chemical capacitance decreases to ∼1500 F cm−3 at 600 °C, a value which is in the range of literature values at comparable conditions (refer to Table 1). As there are very few other data available on the chemical capacitance of LSC thin films at such conditions, it is still difficult to make a conclusive statement about the reasons behind the difference between in situ and ex situ measurements. Similar measurements on the chemical capacitance of 1.5 μm thin films at such oxygen partial pressures were only done by Kawada et al. who found lower capacitances (∼2200 F cm−3 at 600 °C and 0.04 mbar p(O2)).17 One possible reason for the higher ex situ capacitance is that, due to compressive strain introduced during the PLD process,38 the formation of oxygen vacancies in our thin films is significantly inhibited which would lead to a decreased concentration of oxygen vacancies and thus of Cchem for the unrelaxed in situ films.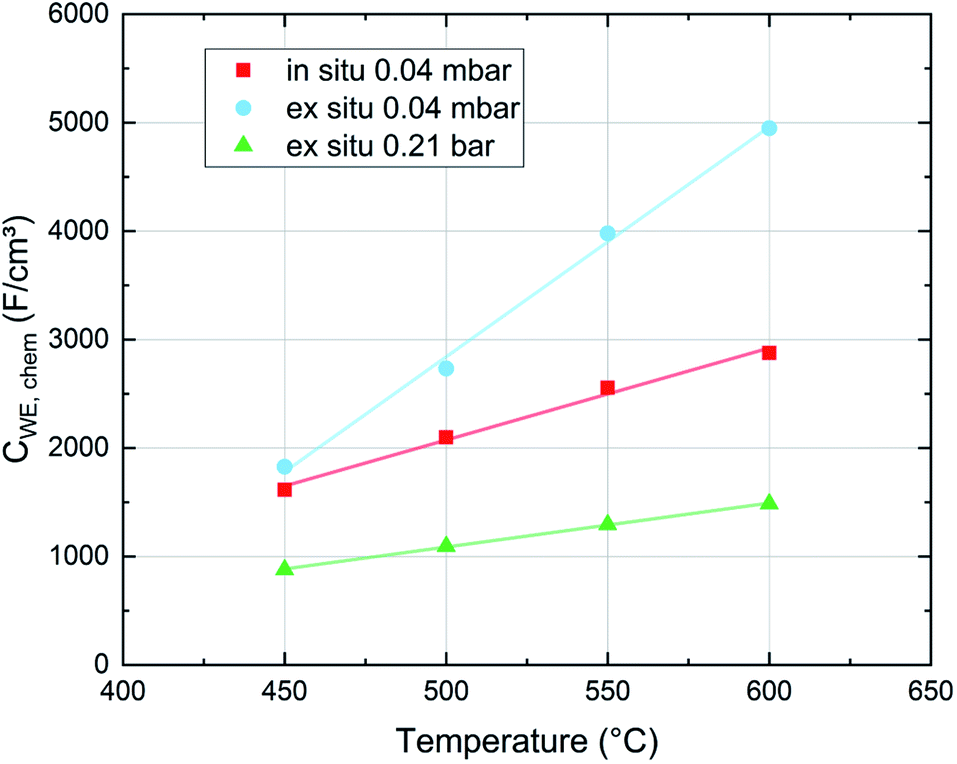 | ||
| Fig. 14 Chemical capacitance values of dense LSC thin films averaged for films measured in situ and ex situ at different oxygen partial pressures and different temperatures. Each value is an average of films deposited at 450, 500, 550 and 600 °C. | ||
For further discussion we may relate the chemical capacitance to the defect chemistry of the material. The capacitance of a film with diluted defects can be interpreted with regard to the chemical composition according to:33,39
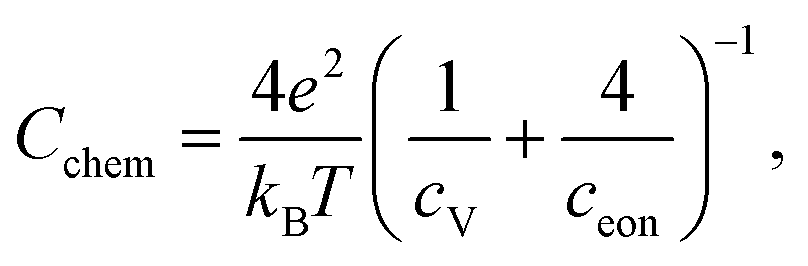 | (4) |
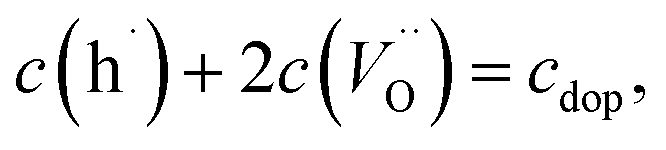 | (5) |
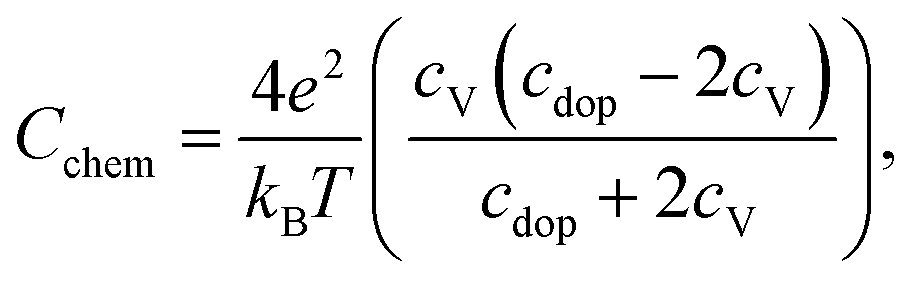 | (6) |
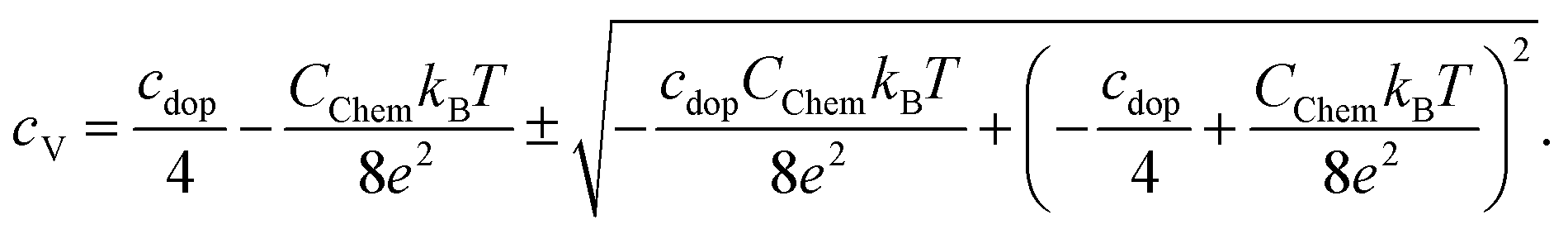 | (7) |
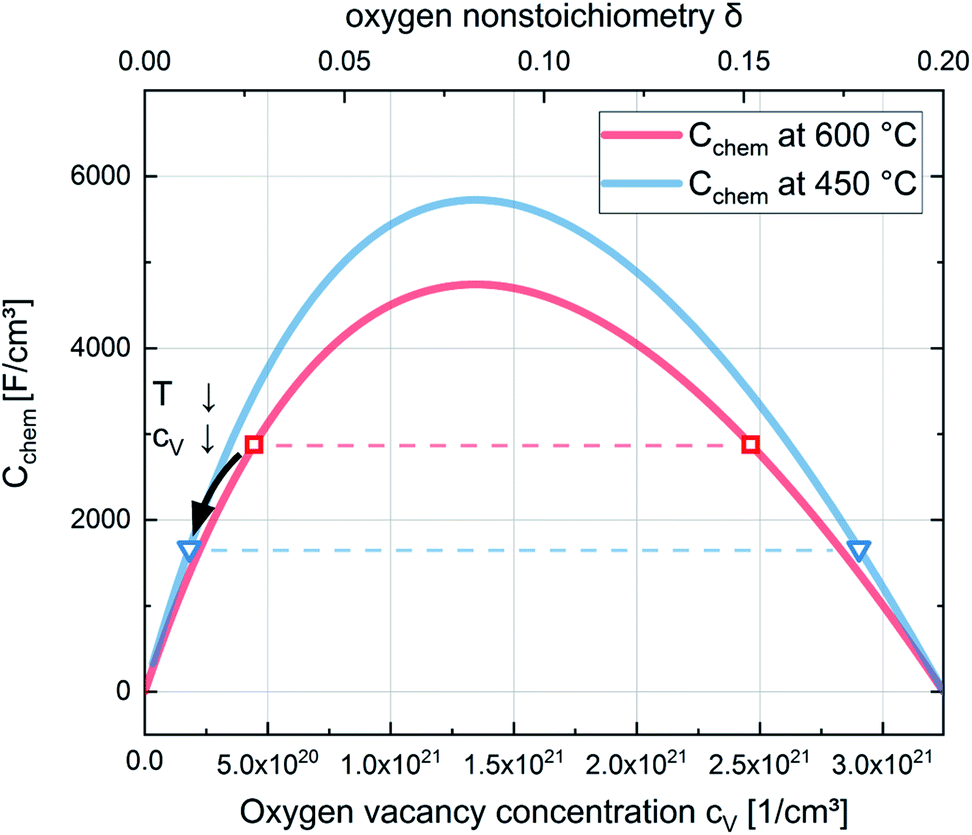 | ||
| Fig. 15 Chemical capacitances according to eqn (6) for 600 °C and 450 °C and defect concentrations of La0.6Sr0.4CoO3−δ calculated from the chemical capacitance measured in situ in 0.04 mbar O2. The symbols (squares for 600 °C and triangles for 450 °C) indicate the two possible cV values leading to the measured Cchem (according to eqn (7)). The temperature dependence indicates the lower cV values as the relevant ones. | ||
Regarding the correlation of chemical capacitance and oxygen vacancy concentration, every chemical capacitance can thus be caused by two different values of cV. The measured temperature dependence of Cchem, together with the known positive vacancy formation enthalpy, identifies the lower values as the correct ones (see arrow in Fig. 15), i.e. 2 × 1020 cm−3 at 450 °C and 5 × 1020 cm−3 at 600 °C. These values correspond to oxygen nonstoichiometries of 0.01 at 450 °C and 0.027 at 600 °C. In Fig. 16 oxygen nonstoichiometries resulting for in situ measurements, as well as ex situ measurements at 0.04 mbar O2 and ambient conditions are shown in more detail. Furthermore oxygen nonstoichiometry measurements of several other authors on bulk LSC are displayed for comparison. The ex situ measurement at 0.04 mbar p(O2) and 600 °C could not be analyzed in the same manner as we seemingly approach a region where the dilute limit is not valid anymore and the interaction between charge carriers cannot be neglected; the measured 5000 F cm−3 is already slightly higher than the nominal maximum possible for a dilute model at 600 °C (4743 F cm−3).
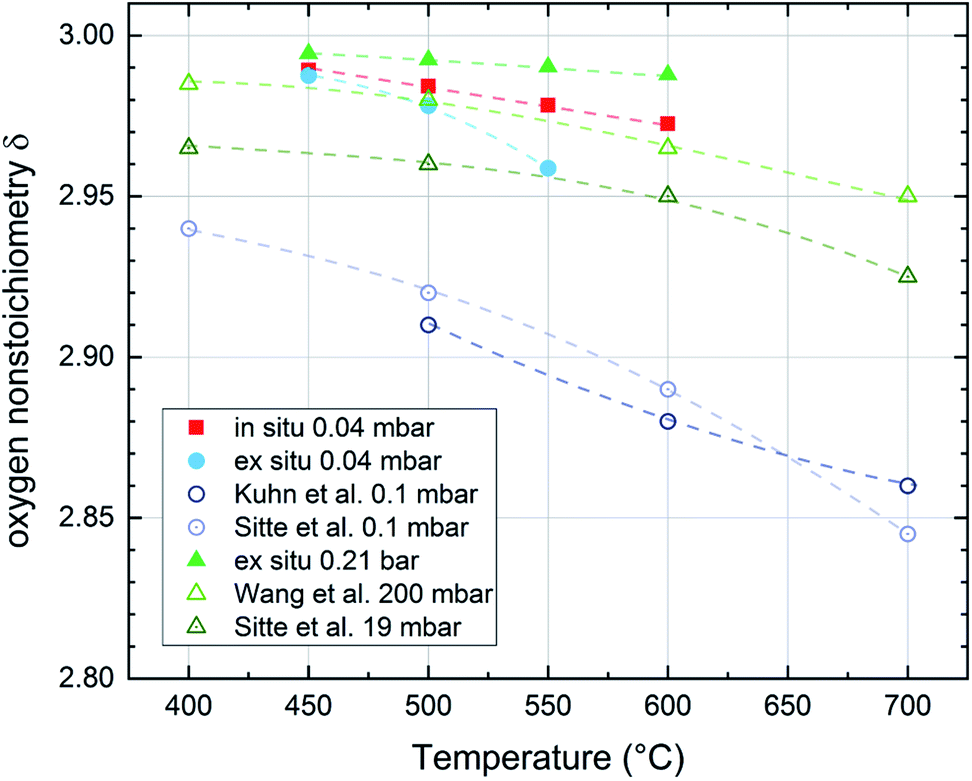 | ||
| Fig. 16 Oxygen nonstoichiometry of La0.6Sr0.4CoO3−δ films during in situ and ex situ measurements compared to bulk measurements of several authors in different pressure and temperature regimes (Kuhn et al.,41 Sitte et al.,42 Wang et al.43). Lines are a guide to the eye. | ||
For both the low pressure and the ambient regime the comparison of the oxygen nonstoichiometries of our thin films to bulk measurements of other authors shows that the thin films tend to exhibit a lower oxygen vacancy concentration than bulk samples at comparable conditions.41–43 This also supports the conclusion of Kawada et al.17 that the oxygen vacancy formation enthalpy in thin films is higher than for bulk samples of the same composition.
4 Conclusions
With IPLD a novel measurement technique is employed to characterize growing dense LSC thin films directly during pulsed laser deposition. This technique allows the investigation of LSC thin films at very early stages after deposition. These pristine LSC films, unaltered by degradation processes show very low surface exchange resistance (RWE ∼ 1.3 Ω cm2 for 600 °C and p(O2) = 0.04 mbar) and an unusually low activation energy (1.02–1.05 eV). The same films measured ex situ in comparable conditions show more than 10 times higher surface exchange resistance values, but a comparable activation energy. This points toward a yet unknown degradation mechanism affecting pristine LSC surfaces during the first exposure to air or measurement environment. When measured ex situ in air the resistance drops again but degradation happens much faster. The activation energy increases to higher values (in the 1.3–1.6 eV range), indicating a p(O2) dependent activation energy of the surface exchange resistance of LSC thin films. It is worth emphasizing that even ex situ the films exhibit high electrochemical activity compared to most literature data, at least before further degradation. Grain size does not seem to affect the oxygen exchange reaction of LSC thin films at all since films deposited at different temperatures (resulting in different grain sizes) show very similar in situ properties. Furthermore, with the method described in this study, it is possible to exactly measure the chemical capacitance of freshly deposited thin films and to assess their otherwise hardly accessible defect chemistry. The results of these measurements suggest that thin films tend to have a lower oxygen vacancy concentration compared to bulk measurements at comparable conditions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support by Austrian Science Fund (FWF) projects P4509-N16 and P31654-N37 is gratefully acknowledged.References
- J. W. Fergus, Materials challenges for solid-oxide fuel cells, Journal of Metals, 2007, 59(12), 56–62 CAS.
- K. Huang and J. B. Goodenough, Solid oxide fuel cell technology: principles, performance and operations, Elsevier, 2009 Search PubMed.
- S. B. Adler, Factors governing oxygen reduction in solid oxide fuel cell cathodes, Chem. Rev., 2004, 104(10), 4791–4844 CrossRef CAS PubMed.
- J. Januschewsky, M. Ahrens, O. Alexander, K. Frank and J. Fleig, Optimized La0.6Sr0.4CoO3−δ Thin-Film Electrodes with Extremely Fast Oxygen-Reduction Kinetics, Adv. Funct. Mater., 2009, 19(19), 3151–3156 CrossRef CAS.
- M. Kubicek, A. Limbeck, T. Froemling, H. Hutter and J. Fleig, Relationship between cation segregation and the electrochemical oxygen reduction kinetics of La0.6Sr0.4CoO3−δ thin film electrodes, J. Electrochem. Soc., 2011, 158(6), B727–B734 CrossRef CAS.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chemical Heterogeneities on La0.6Sr0.4CoO3−δ Thin Films Correlations to Cathode Surface Activity and Stability, Chem. Mater., 2012, 24(6), 1116–1127 CrossRef CAS.
- E. Bucher, S. Werner, F. Klauser and E. Bertel, Impact of humid atmospheres on oxygen exchange properties, surface-near elemental composition, and surface morphology of La0.6Sr0.4CoO3−δ, Solid State Ionics, 2012, 208, 43–51 CrossRef CAS.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. C. Jung, Sr segregation in perovskite oxides: why it happens and how it exists, Joule, 2018, 2(8), 1476–1499 CrossRef CAS.
- G. M. Rupp, H. Tellez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, Surface chemistry of La0.6Sr0.4CoO3−δ thin films and its impact on the oxygen surface exchange resistance, J. Mater. Chem. A, 2015, 3(45), 22759–22769 RSC.
- E. Bucher, M. Yang and W. Sitte, In Situ Investigations of the Chromium-Induced Degradation of the Oxygen Surface Exchange Kinetics of IT-SOFC Cathode Materials La0.6Sr0.4CoO3−δ and La0.58Sr0.4Co0.2Fe0.8O3−δ, J. Electrochem. Soc., 2012, 159(5), B592–B596 CrossRef CAS.
- E. Bucher, C. Gspan and S. Werner, Degradation and regeneration of the SOFC cathode material La0.6Sr0.4CoO3−δ in SO2-containing atmospheres, Solid State Ionics, 2015, 272, 112–120 CrossRef CAS.
- F. S. Baumann, J. Fleig, H.-U. Habermeier and J. Maier, Impedance spectroscopic study on well-defined (La, Sr)(Co, Fe)O3−δ model electrodes, Solid State Ionics, 2006, 177(11–12), 1071–1081 CrossRef CAS.
- A. K. Opitz and J. Fleig, Investigation of O2 reduction on Pt/YSZ by means of thin film microelectrodes: The geometry dependence of the electrode impedance, Solid State Ionics, 2010, 181(15–16), 684–693 CrossRef CAS.
- F. Zhao, R. Peng and C. Xia, A La0.6Sr0.4CoO3−δ-based electrode with high durability for intermediate temperature solid oxide fuel cells, Mater. Res. Bull., 2008, 43(2), 370–376 CrossRef CAS.
- G. M. Rupp, A. Schmid, A. Nenning and J. Fleig, The superior properties of La0.6Ba0.4CoO3−δ thin film electrodes for oxygen exchange in comparison to La0.6Sr0.4CoO3−δ, J. Electrochem. Soc., 2016, 163(6), F564–F573 CrossRef CAS.
- J. R. Wilson, M. Sase, T. Kawada and S. B. Adler, Measurement of Oxygen Exchange Kinetics on Thin-Film La0.6Sr0.4CoO3−δ Using Nonlinear Electrochemical Impedance Spectroscopy, Electrochem. Solid-State Lett., 2007, 10(5), B81–B86 CrossRef CAS.
- T. Kawada, J. Suzuki, M. Sase, A. Kaimai, K. Yashiro, Y. Nigara, J. Mizusaki, K. Kawamura and H. Yugami, Determination of oxygen vacancy concentration in a thin film of La0.6Sr0.4CoO3−δ by an electrochemical method, J. Electrochem. Soc., 2002, 149(7), E252–E259 CrossRef CAS.
- G. M. Rupp, A. K. Opitz, A. Nenning, A. Limbeck and J. Fleig, Real-time impedance monitoring of oxygen reduction during surface modification of thin film cathodes, Nat. Mater., 2017, 16(6), 640 CrossRef CAS PubMed.
- G. M. Rupp, M. Kubicek, A. K. Opitz and J. Fleig, In Situ Impedance Analysis of Oxygen Exchange on Growing La0.6Sr0.4CoO3−δ Thin Films, ACS Appl. Energy Mater., 2018, 1(9), 4522–4535 CrossRef CAS PubMed.
- M. P. Pechini, Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor, US Pat. 3,330,697. 1967.
- G. M. Rupp, A. Limbeck, M. Kubicek, A. Penn, M. Stöger-Pollach, G. Friedbacher and J. Fleig, Correlating surface cation composition and thin film microstructure with the electrochemical performance of lanthanum strontium cobaltite (LSC) electrodes, J. Mater. Chem. A, 2014, 2(19), 7099–7108 RSC.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, The HighScore suite, Powder Diffr., 2014, 29(S2), S13–S18 CrossRef CAS.
- K. Byrappa and T. Ohachi. Crystal growth technology. Elsevier, 2003 Search PubMed.
- J. Jamnik and J. Maier, Generalised equivalent circuits for mass and charge transport: chemical capacitance and its implications, Phys. Chem. Chem. Phys., 2001, 3(9), 1668–1678 RSC.
- M. E. Orazem and T. Bernard, Electrochemical impedance spectroscopy, John Wiley & Sons, 2017 Search PubMed.
- G. M. Rupp, M. Glowacki and J. Fleig, Electronic and ionic conductivity of La0.95Sr0.05Ga0.95Mg0.05O3−δ (LSGM) single crystals, J. Electrochem. Soc., 2016, 163(10), F1189–F1197 CrossRef CAS.
- M. Sase, J. Suzuki, K. Yashiro, T. Otake, A. Kaimai, T. Kawada, J. Mizusaki and H. Yugami, Electrode reaction and microstructure of La0.6Sr0.4CoO3−δ thin films, Solid State Ionics, 2006, 177(19–25), 1961–1964 CrossRef CAS.
- A. Egger, E. Bucher, M. Yang and S. Werner, Comparison of oxygen exchange kinetics of the IT-SOFC cathode materials La0.5Sr0.5CoO3−δ and La0.6Sr0.4CoO3−δ, Solid State Ionics, 2012, 225, 55–60 CrossRef CAS.
- Z. Zhao, L. Liu, X. Zhang, W. Wu, B. Tu, D. Ou and M. Cheng, A comparison on effects of CO2 on La0.8Sr0.2MnO3+δ and La0.6Sr0.4CoO3−δ cathodes, J. Power Sources, 2013, 222, 542–553 CrossRef CAS.
- H. Jan, D. Levin, U. Guntow, D. Gerthsen and E. Ivers-Tiffée, Nanoscaled La0.6Sr0.4CoO3−δ as intermediate temperature solid oxide fuel cell cathode: Microstructure and electrochemical performance, J. Power Sources, 2011, 196(17), 7263–7270 CrossRef.
- F. S. Baumann, J. Fleig, G. Cristiani, B. Stuhlhofer, H.-U. Habermeier and J. Maier, Quantitative comparison of mixed conducting SOFC cathode materials by means of thin film model electrodes, J. Electrochem. Soc., 2007, 154(9), B931–B941 CrossRef CAS.
- A. M. Saranya, A. Morata, D. Pla, M. Burriel, F. Chiabrera, I. Garbayo, A. Hornés, J. A. Kilner and T. Albert, Unveiling the Outstanding Oxygen Mass Transport Properties of Mn-Rich Perovskites in Grain Boundary-Dominated La0.8Sr0.2(Mn1−xCox)0.85O3±δ Nanostructures, Chem. Mater., 2018, 30(16), 5621–5629 CrossRef CAS PubMed.
- A. Schmid, G. M. Rupp and J. Fleig, Voltage and partial pressure dependent defect chemistry in (La, Sr)FeO3−δ thin films investigated by chemical capacitance measurements, Phys. Chem. Chem. Phys., 2018, 20(17), 12016–12026 RSC.
- C. Peters, Grain-size effects in nanoscaled electrolyte and cathode thin films for solid oxide fuel cells (SOFC), KIT Scientific Publishing, 2009, vol. 15 Search PubMed.
- D. Kim, J. W. Park, B.-H. Yun, J. H. Park and K. T. Lee, Correlation of Time-Dependent Oxygen Surface Exchange Kinetics with Surface Chemistry of La0.6Sr0.4Co0.2Fe0.8O3-δ Catalysts, ACS Appl. Mater. Interfaces, 2019, 11(35), 31786–31792 CrossRef CAS PubMed.
- C. Jeffrey, K. Develos-Bagarinao, S.-S. Liu, H. Kishimoto, T. Ishiyama, K. Yamaji, T. Horita and H. Yokokawa, Sulfur poisoning behavior of La1-xSrxCo1-yFeyO3-δ thin films with different compositions, J. Alloys Compd., 2018, 748, 608–619 CrossRef.
- E. Bucher, C. Gspan, F. Hofer and S. Werner, Sulphur poisoning of the SOFC cathode material La0.6Sr0.4CoO3-δ, Solid State Ionics, 2013, 238, 15–23 CrossRef CAS.
- E. J. Crumlin, S.-J. Ahn, D. Lee, E. Mutoro, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Oxygen electrocatalysis on epitaxial La0.6Sr0.4CoO3-δ perovskite thin films for solid oxide fuel cells, J. Electrochem. Soc., 2012, 159(7), F219–F225 CrossRef CAS.
- J. Fleig, G. M. Rupp, A. Nenning and A. Schmid, Towards an Improved Understanding of Electrochemical Oxygen Exchange Reactions on Mixed Conducting Oxides, ECS Trans., 2017, 93–108 CrossRef CAS.
- T. Ishihara, Perovskite oxide for solid oxide fuel cells, Springer Science & Business Media, 2009 Search PubMed.
- M. Kuhn, S. Hashimoto, K. Sato, K. Yashiro and J. Mizusaki, Oxygen nonstoichiometry and thermo-chemical stability of La0.6Sr0.4CoO3−δ, J. Solid State Chem., 2013, 197, 38–45 CrossRef CAS.
- W. Sitte, E. Bucher and W. Preis, Nonstoichiometry and transport properties of strontium-substituted lanthanum cobaltites, Solid State Ionics, 2002, 154, 517–522 CrossRef.
- S. Wang, R. Zheng, A. Suzuki and T. Hashimoto, Preparation, thermal expansion and electrical conductivity of La0.6Sr0.4Co1−xGaxO3−δ (x=0.0–0.4) as a new cathode material of SOFC, Solid State Ionics, 2004, 157–162 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |