
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese hexacyanoferrate reinforced by PEDOT coating towards high-rate and long-life sodium-ion battery cathode†
Xiao
Wang
a,
Baoqi
Wang
a,
Yuxin
Tang
b,
Ben Bin
Xu
 c,
Chu
Liang
d,
Mi
Yan
a and
Yinzhu
Jiang
*a
c,
Chu
Liang
d,
Mi
Yan
a and
Yinzhu
Jiang
*a
aSchool of Materials Science and Engineering, State Key Laboratory of Silicon Materials, Key Laboratory of Novel Materials for Information Technology of Zhejiang Province, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: yzjiang@zju.edu.cn
bInstitute of Applied Physics and Materials Engineering, University of Macau, Macau 999078, P. R. China
cSmart Materials and Surfaces Lab, Mechanical Engineering, Faculty of Engineering and Environment, Northumbria University, Newcastle upon Tyne NE1 8ST, UK
dCollege of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China
First published on 2nd January 2020
Abstract
Prussian blue analogues hold great promise as cathodes in sodium ion batteries. Among Prussian blue analogues, manganese hexacyanoferrate is desirable because of its high working voltage, as well as its high specific capacity and low cost. However, poor cycling stability and unsatisfactory rate capability of manganese hexacyanoferrate, which are mainly caused by poor intrinsic conductivity, phase transition, side reactions, and transition metal dissolution, extremely limit its practical application. In this work, we demonstrate a high-rate and long-life MnHCF@PEDOT sodium ion battery cathode through a facile in situ polymerization method. Benefitting from the synergistic effect of the inhibited Mn/Fe dissolution, suppressed phase transition, and improved capacitive storage, the composite electrode exhibits a high capacity of 147.9 mA h g−1 at 0.1C, 95.2 mA h g−1 at a high rate of 10C, and 78.2% capacity retention after 1000 cycles. Furthermore, even at a low temperature of −10 °C, MnHCF@PEDOT still delivers a high capacity of 87.0 mA h g−1 and maintains 71.5 mA h g−1 (82.2%) after 500 cycles.
Introduction
Advanced large-scale energy storage system (ESS) is much desirable to meet the rapid development of clean and sustainable energies such as solar and wind due to their instability and intermittence.1–4 Sodium-ion batteries (SIBs) are amongst the most attractive energy storage techniques due to the abundant and low-cost sodium resources, as well as their similar working principle to lithium-ion batteries (LIBs).5–8 Exploring a suitable cathode material is critical for the development of SIBs considering the larger ionic radius of sodium and the higher redox potential of Na/Na+ as compared to those of lithium.9–11 Prussian blue and its analogues (PBAs) have attracted extensive attention as cathode materials for SIBs because of their ease of synthesis, open framework structure, and high theoretical capacity (∼170 mA h g−1).12–20Among the PBAs, manganese hexacyanoferrate (MnHCF) exhibits a high redox potential in high spin MnHS configuration as compared to other transition metals, which makes MnHCF more competitive in delivering higher output voltage and energy density.20–25 However, MnHCF as cathode material suffers from poor cycling and high polarization on account of its poor conductivity and side reactions with the electrolyte, as well as the phase transition and dissolution of Mn during cycling.26–29 Many efforts have been devoted to improving the electrochemical performance of MnHCF, such as hetero transition metal doping,30,31 removal of interstitial H2O,32 and conductive polymer/carbon coating.33,34 However, realizing a high-rate and long life MnHCF-based cathode is still a tough challenge considering the cooperative Jahn–Teller distortion in the structure of Mn(III)-based electrodes.
In this work, we report a facile in situ polymerization method for the synthesis of MnHCF@PEDOT composite by employing MnHCF cubes as both template and oxidation initiator. The inhibited dissolution of Mn/Fe, suppressed phase transition, and improved capacitive storage that benefit from the PEDOT coating greatly promote the sodium storage performance. A highly reversible capacity of 147.9 mA h g−1 at 0.1C and an ultra-long cycling life (78.3% capacity retention after 1000 cycles at 10C) are demonstrated. Furthermore, the composite shows impressive low-temperature performance at −10 °C, delivering a high capacity of 87.7 mA h g−1 and a capacity retention of 82.2% after 500 cycles at 1C.
Experimental
Synthesis of MnHCF and MnHCF@PEDOT
MnHCF was synthesized by a co-precipitation method as described in other reports with minor modifications.36,37 In a typical preparation, 3 mmol Na4Fe(CN)6·10H2O, 0.24 mol NaCl, and 12 mmol sodium citrate were dissolved in 100 mL deionized (DI) water with magnetic stirring to form solution A. 6 mmol MnSO4·4H2O was dissolved in 100 mL DI water with magnetic stirring to form solution B. Solution B was added into solution A (90 °C) by a peristaltic pump at a rate of 1 mL min−1 under magnetic stirring condition and N2 atmosphere. The white suspension was aged for 3 h under continuous stirring at 90 °C after solution B was used up. The obtained white precipitate was collected by centrifugation and washed by DI water and ethanol thoroughly before drying in vacuum at 110 °C for 24 h.A certain amount of 3,4-ethylenedioxythiophene (EDOT) was dispersed in 40 mL DI water and then stirred for 0.5 h. Meanwhile, 200 mg as-synthesized MnHCF and 100 mg (NH4)2S2O8 were dispersed in 10 mL DI water by magnetic stirring and ultrasonication. Then, MnHCF/(NH4)2S2O8 suspension was added into EDOT suspension immediately, followed by 10 h magnetic stirring at room temperature. The obtained greyish-green precipitate was collected by centrifugation and washed with DI water and ethanol thoroughly before drying in vacuum at 80 °C for 24 h. The as-synthesized powder was named MnHCF@PEDOT-10, MnHCF@PEDOT-20, and MnHCF@PEDOT-50 according to the amount of EDOT (10, 20, and 50 μL, respectively). As a comparison, bare PEDOT was prepared by the same method without the addition of MnHCF.
Materials' characterization
The X-ray diffraction (XRD) patterns were acquired on a Bruker D8 Advance X-ray diffractometer with Co Kα radiation (λ = 0.178897 nm) at a scan rate of 2° min−1. The samples for ex situ XRD were prepared in the coin cells during the 11th cycle at 1C. The molar ratios of Na, Mn, and Fe were measured by inductively coupled plasma-optical emission spectrometry (ICP-OES, Optima 8000) and the contents of C, H, and N were analyzed by elemental analysis (EA, Vario Micro). Raman spectra were collected by LabRAM HR Evolution Raman spectrometer using an Ar ion laser with the wavelength of 532 nm. The X-ray photoelectron spectra (XPS) were recorded on a Thermo Scientific K-Alpha + X-ray photoelectron spectrometer. Thermogravimetric (TG) analyses were carried out on TA-Q500 from 50 to 450 °C at a heating rate of 5 °C min−1 in air to determine the water content and the amount of PEDOT in the as-synthesized samples. Scanning electron microscopy (SEM, Hitachi S-4800) and transmission electron microscopy (TEM, JEM-2100) were used to characterize the morphologies of all the samples.Electrochemical measurements
The as-synthesized samples, Ketjen black, and polyvinylidene fluoride (PVDF) were mixed in the mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 and dissolved in N-methyl pyrrolidinone (NMP) to make a slurry. The slurry was cast on Al foil and then dried at 80 °C in vacuum for 24 h to prepare working electrodes. The average mass loading of the electrodes was about 2 mg cm−2. CR2025 coin cells using the as-prepared electrodes as cathodes were assembled in an Ar-filled glovebox with H2O and O2 contents less than 1 ppm. Metallic sodium, 1 M NaPF6 in ethylene carbonate/diethylene carbonate (EC/DEC, 1:1 in volume) with 5 vol% fluoroethylene carbonate (FEC) as the additive, and Whatman GF/D glass microfiber filters were used as anode, electrolyte, and separator, respectively. The galvanostatic charge–discharge test at different current densities was carried out on a Neware battery testing system at 30 °C with a voltage range of 2.0–4.0 V. The specific capacity was calculated based on the mass of the as-synthesized samples. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were carried out on a CHI604E electrochemical workstation. The voltage range of CV was 2.0–4.0 V and the scan rates ranged from 0.1 to 2 mV s−1. The frequency range of EIS was 10−2-105 Hz with an amplitude of 5 mV.
:2:1 and dissolved in N-methyl pyrrolidinone (NMP) to make a slurry. The slurry was cast on Al foil and then dried at 80 °C in vacuum for 24 h to prepare working electrodes. The average mass loading of the electrodes was about 2 mg cm−2. CR2025 coin cells using the as-prepared electrodes as cathodes were assembled in an Ar-filled glovebox with H2O and O2 contents less than 1 ppm. Metallic sodium, 1 M NaPF6 in ethylene carbonate/diethylene carbonate (EC/DEC, 1:1 in volume) with 5 vol% fluoroethylene carbonate (FEC) as the additive, and Whatman GF/D glass microfiber filters were used as anode, electrolyte, and separator, respectively. The galvanostatic charge–discharge test at different current densities was carried out on a Neware battery testing system at 30 °C with a voltage range of 2.0–4.0 V. The specific capacity was calculated based on the mass of the as-synthesized samples. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were carried out on a CHI604E electrochemical workstation. The voltage range of CV was 2.0–4.0 V and the scan rates ranged from 0.1 to 2 mV s−1. The frequency range of EIS was 10−2-105 Hz with an amplitude of 5 mV.
Results and discussion
A facile co-precipitation method was carried out for the synthesis of bare MnHCF, which was used as the template and oxidation initiator for in situ polymerization of EDOT to obtain a series of MnHCF@PEDOT-10/20/50 composites with different amounts of EDOT. Fig. 1a shows the X-ray diffraction (XRD) patterns of the bare MnHCF and the MnHCF@PEDOT composites. Due to the lattice distortion caused by the high sodium content, bare MnHCF was crystallized into a monoclinic structure rather than a cubic one, which is consistent with the previous reports.20,23 The monoclinic phase of MnHCF was maintained after PEDOT coating, implying the reserved Na-rich nature. According to the TG profiles (Fig. S1†), ICP-OES measurement, and elemental analyses, the formula of MnHCF can be calculated as Na1.85Mn[Fe(CN)6]0.98□0.02·2.34H2O, while MnHCF@PEDOT-10, MnHCF@PEDOT-20, and MnHCF@PEDOT-50 correspond to the formulas of Na1.77Mn[Fe(CN)6]0.96□0.04·1.75H2O, Na1.71Mn[Fe(CN)6]0.94□0.06·1.66H2O, and Na1.56Mn[Fe(CN)6]0.90□0.10·1.37H2O with the mass fractions of the PEDOT coating layer as 5.4 wt%, 12.4 wt%, and 25.1 wt%, respectively. All the results clearly verify the Na-rich nature of these samples. | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of MnHCF, PEDOT, and MnHCF@PEDOT-10/20/50; (c and d) SEM and (e) TEM images of MnHCF; (f and g) SEM and (h) TEM images of MnHCF@PEDOT-20. | ||
Raman spectra was carried out to verify the PEDOT coating as shown in Fig. 1b. In the bare MnHCF, there are two sharp peaks at 2079 and 2123 cm−1, corresponding to the stretching vibrations of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–.37 In the bare PEDOT, the stretching of the symmetrical CαCβ band is observed at 1438 cm−1, and the peaks at 1502 and 1563 cm−1 are assigned to asymmetric CαCβ band stretching mode. Compared with the standard peaks, these main peaks of PEDOT in MnHCF@PEDOT composites shift upwards, indicating that PEDOT is well oxidized.35 The peak intensity of PEDOT increases while that of MnHCF decreases and the relative intensity of the peak at 2123 cm−1 weakens, indicating an excellent composite and the decrease in the average valence state of transition metals in MnHCF.37 In order to better understand the in situ polymerization process, XPS was carried out, as shown in Fig. S2.† During the in situ polymerization process, surface Fe3+ acts as an oxidant to induce EDOT to polymerize on the surface of MnHCF particles to PEDOT, while the valence of Mn remains unchanged. All these results clearly verify the tight binding between PEDOT and MnHCF, which is essential for improving the electronic conduction and enhancing the structure stability during cycling. Fig. 1c–h show the morphologies of bare MnHCF and MnHCF@PEDOT-20. The size of MnHCF particles varies from several hundred nanometers to several micrometers. Large particles have regular rectangular shape, while the shapes of most small particles are sphere-like, both of which exhibit smooth surface characteristics. Conversely, the surface of MnHCF@PEDOT-20 particles becomes rough due to the PEDOT coating, although the particle size does not change significantly. The above phenomenon is also verified through TEM, as shown in Fig. 1e and h. The edge of the MnHCF particles is smooth, while the edge of MnHCF@PEDOT-20 particles has a thin and rough PEDOT layer, manifesting the successful coating of PEDOT.
N–.37 In the bare PEDOT, the stretching of the symmetrical CαCβ band is observed at 1438 cm−1, and the peaks at 1502 and 1563 cm−1 are assigned to asymmetric CαCβ band stretching mode. Compared with the standard peaks, these main peaks of PEDOT in MnHCF@PEDOT composites shift upwards, indicating that PEDOT is well oxidized.35 The peak intensity of PEDOT increases while that of MnHCF decreases and the relative intensity of the peak at 2123 cm−1 weakens, indicating an excellent composite and the decrease in the average valence state of transition metals in MnHCF.37 In order to better understand the in situ polymerization process, XPS was carried out, as shown in Fig. S2.† During the in situ polymerization process, surface Fe3+ acts as an oxidant to induce EDOT to polymerize on the surface of MnHCF particles to PEDOT, while the valence of Mn remains unchanged. All these results clearly verify the tight binding between PEDOT and MnHCF, which is essential for improving the electronic conduction and enhancing the structure stability during cycling. Fig. 1c–h show the morphologies of bare MnHCF and MnHCF@PEDOT-20. The size of MnHCF particles varies from several hundred nanometers to several micrometers. Large particles have regular rectangular shape, while the shapes of most small particles are sphere-like, both of which exhibit smooth surface characteristics. Conversely, the surface of MnHCF@PEDOT-20 particles becomes rough due to the PEDOT coating, although the particle size does not change significantly. The above phenomenon is also verified through TEM, as shown in Fig. 1e and h. The edge of the MnHCF particles is smooth, while the edge of MnHCF@PEDOT-20 particles has a thin and rough PEDOT layer, manifesting the successful coating of PEDOT.
The galvanostatic charge–discharge profiles at 0.1C (1C = 150 mA g−1) are shown in Fig. 2a. MnHCF displays two long voltage plateaus at ∼3.4 V and ∼3.7 V during charge but only one voltage plateau at ∼3.4 V during discharge. The charge and discharge capacities of MnHCF are 162.5 and 130.2 mA h g−1, respectively, manifesting severe irreversibility during the initial charge/discharge process. For MnHCF@PEDOT, another voltage plateau at ∼3.3 V is observed during discharge possibly owing to the improved conductivity after PEDOT coating. With the increasing PEDOT mass fraction, charge/discharge capacities increase initially and then decrease, and MnHCF@PEDOT-20 shows the highest charge capacity of 167.7 mA h g−1 and the highest discharge capacity of 147.9 mA h g−1, with an improved coulombic efficiency (CE) of 88.2%. However, MnHCF@PEDOT-50 exhibits a lower capacity than MnHCF since PEDOT has little Na storage activity (Fig. S4†). As the current rate increases to 1C (Fig. 2b), MnHCF suffers more severe polarization during cycling, accompanied by voltage plateaus changing into slopes. However, MnHCF@PEDOT still displays two distinct voltage plateaus during charge and discharge, and the polarization is much lower. Furthermore, MnHCF@PEDOT-20 delivers the highest charge capacity of 133.9 mA h g−1 and discharge capacity of 125.7 mA h g−1, corresponding to a CE of 93.9%. When the current density reaches 10C, specific capacities of all the MnHCF@PEDOT samples are much higher than those of bare MnHCF, as shown in Fig. 2c.
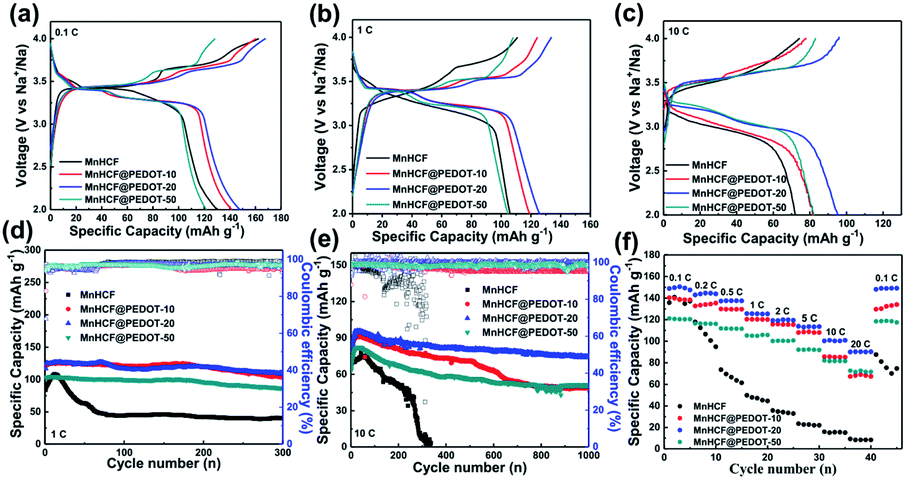 | ||
| Fig. 2 Galvanostatic charge–discharge profiles at (a) 0.1C, (b) 1C, and (c) 10C cycled between 2.0 and 4.0 V; cycling performance and coulombic efficiency at (d) 1C and (e) 10C; (f) rate capability at various rates from 0.1C to 20C. | ||
Fig. 2d compares the cycling performance of MnHCF and MnHCF@PEDOT at 1C. The capacity of MnHCF increases continuously in the first ten cycles, indicating the existence of an activation process but the activation in MnHCF@PEDOT is inapparent. The highest discharge capacity of 106.0 mA h g−1 is observed at the 10th cycle for MnHCF and only a capacity of 44.3 mA h g−1 remains after 100 cycles. On the contrary, MnHCF@PEDOT-20 delivers the highest discharge capacity of 125.7 mA h g−1 at the 10th cycle, and retains 117.0 mA h g−1 (93.1%) after 100 cycles and 109.2 mA h g−1 (86.9%) even after 300 cycles, manifesting that PEDOT significantly improves the cycling stability of MnHCF. The cycling performances of all the MnHCF@PEDOT samples are also more outstanding than those of bare MnHCF at 10C, as shown in Fig. 2e. MnHCF@PEDOT-20 presents the highest discharge capacity of 95.2 mA h g−1 and retains 74.4 mA h g−1 (78.2%) after 1000 cycles. In contrast, the capacity of bare MnHCF decays rapidly to 0 after 330 cycles. Although MnHCF@PEDOT-10 has a capacity close to that of MnHCF@PEDOT-20, its capacity fading is worse and only retains 48.5 mA h g−1 (53.4%) after 1000 cycles probably due to imperfection of the PEDOT coating layer.
Ex situ XRD was applied to track the structural evolution upon Na+ extraction and insertion of MnHCF and MnHCF@PEDOT-20. As shown in Fig. 3a and b, due to the cooperative Jahn–Teller distortion of Mn3+ in the MnN6 octahedra, the MnHCF electrode experiences a two-phase transition between the cubic and tetragonal phases during the cycle, while the MnHCF@PEDOT-20 electrode maintains the monoclinic phase with a peak shift of only 0.27°, demonstrating stability in the structure during cycling. The PEDOT coating suppresses the phase transition during the cycle and reduces the volume change, thereby retarding the dissolution of the electrode and improving the cycle stability of the electrode. On the other hand, it is known that in the presence of water, NaPF6 salt is likely to decompose and generated hazardous substances such as HF, which accelerates the deterioration of the surface structure of MnHCF and leads to the dissolution of Mn/Fe, and ultimately leads to performance degradation during cycling. In order to determine the inhibition of the coating on the dissolution of Mn/Fe, ICP-OES was used to test the contents of Mn and Fe in the electrolyte after 100 cycles. An amplified half-cell was made, as shown in Fig. S5,† to check the concentrations of transition metals in the electrolyte. After 100 cycles, the contents of Mn and Fe in the electrolyte of bare MnHCF are 2.26 and 0.267 mg L−1, respectively, as shown in Fig. 3c. For comparison, the contents of Mn and Fe in the electrolyte of MnHCF@PEDOT-20 are only 0.586 and 0.143 mg L−1, respectively. It shows that the excellent cycling stability of MnHCF@PEDOT-20 can be attributed to the inhibition of side reactions and dissolution of transition metals, especially Mn, by PEDOT coating. In addition, as shown in Fig. S6,† after 1000 cycles at 10C or etching in 0.1 M HF solution at 100 °C for 4 h, the morphology of MnHCF@PEDOT-20 is well maintained, while that of MnHCF is destroyed, further indicating that the coating can well protect MnHCF from HF etching and thus, can inhibit the dissolution of Mn/Fe to maintain structural stability.
 | ||
| Fig. 3 Ex-situ XRD patterns of (a) MnHCF and (b) MnHCF@PEDOT-20; (c) contents of Mn/Fe in the electrolyte after 100 cycles. | ||
The rate capability of MnHCF and MnHCF@PEDOT is shown in Fig. 2f. As the current rate increases from 0.1 to 0.2, 0.5, 1, 2, 5, 10, and 20C, the reversible specific capacity of MnHCF@PEDOT-20 slightly decreases from 148.8 to 142.9, 137.3, 125.6, 119.3, 113.8, 100.3, and 90.1 mA h g−1, respectively. Moreover, the capacity recovers to 148.2 mA h g−1 when the current rate returns to 0.1C. MnHCF@PEDOT-10 and MnHCF@PEDOT-50 also show good rate capability. On the contrary, the specific capacity of bare MnHCF decays rapidly with increasing current rate. Only a capacity of 8.4 mA h g−1 can be obtained at 20C and the capacity is only 87.5 mA h g−1 when the current rate returns to 0.1C. It can be observed that there is a significant improvement in the rate performance of MnHCF, which might be ascribed to the promotion in electronic conduction of the electrode materials caused by tightly integrated PEDOT coating. Such electrochemical performance of MnHCF@PEDOT-20 exceeds most state-of-the-art MnHCF electrodes for SIBs, as summarized in Table S1.† EIS tests of MnHCF and MnHCF@PEDOT electrodes were carried out to further elucidate the effect of PEDOT on the electrochemical properties of the electrodes. As shown in Fig. 4, an equivalent circuit is used to fit the Nyquist plot; the Rct of MnHCF, MnHCF@PEDOT-10, MnHCF@PEDOT-20, MnHCF@PEDOT-50, and PEDOT is 602.9, 427.5, 373.8, 343.8, and 285.8 Ω, respectively. With the increase in the PEDOT content, the Rct of MnHCF@PEDOT decreases due to promotion of the electronic conduction of the electrode, resulting in a boosted rate performance.
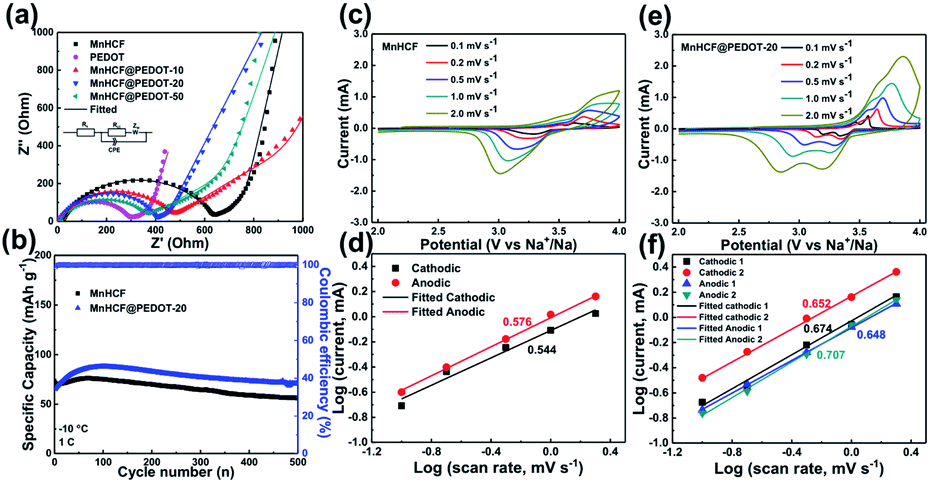 | ||
| Fig. 4 (a) EIS spectra at the 10th cycle in a fully discharged state in the frequency range of 10−2 to 105 Hz; (b) Cycling performance and CE at −10 °C, 1C; CV curves at scan rates ranging from 0.1 to 2.0 mV s−1 of (c) MnHCF and (e) MnHCF@PEDOT-20; the b-value calculation with the peak currents and the scan rates for (d) MnHCF and (f) MnHCF@PEDOT-20. | ||
The CV tests of bare MnHCF and MnHCF@PEDOT-20 at different sweep rates were conducted to further elucidate the electrochemical process of the electrodes. As shown in Fig. S7,† MnHCF shows three cathodic peaks and two anodic peaks at a sweep rate of 0.1 mV s−1, and the anodic peak at 3.55 V is very weak, while MnHCF@PEDOT-20 shows two pairs of sharp redox peaks, which correspond to the GCD results. The cathodic peak at 3.71 V and the small anodic peak at 3.55 V of MnHCF is supposed to result from the polarization caused by the poor conductivity of MnCHF after partial Na extraction.34,38 As shown in Fig. 4c and e, with the increase in the sweep rate, MnHCF exhibits only one pair of redox peaks, while MnHCF@PEDOT-20 still exhibits two pairs of redox peaks. The linear fitting results of log(peak current densities, i) and log(scanning rates, v) are shown in Fig. 4d and f, according to the above CV curves. According to eqn (1), b value can be obtained from the slope rate of the fitting line of log(i) vs. log(ν).39
| i = aνb | (1) |
Conclusions
In summary, a series of MnHCF@PEDOT composites were successfully prepared through co-precipitation followed by a facile in situ polymerization method. Such a tightly bound PEDOT surface layer on MnHCF boosts the electronic conduction and capacitive storage, and on the other hand, effectively suppresses phase transition, and inhibits the side reactions and the transition metal dissolution when used as a cathode for SIBs. The resultant MnHCF@PEDOT-20 composite exhibits excellent rate capability with a capacity as high as 147.9 mA h g−1 at 0.1C, 95.2 mA h g−1 at 10C, and retains 90.2 mA h g−1 at 20C, and a long cycle life with a retained capacity of 74.4 mA h g−1 (78.2%) at 10C after 1000 cycles. Even at a low temperature of −10 °C, MnHCF@PEDOT-20 delivers a high capacity of 87.0 mA h g−1 and retains 71.5 mA h g−1 (82.2%) after 500 cycles. The present work offers a rational strategy for improving the electrochemical performance of PBAs for rechargeable batteries.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was supported by National Natural Science Foundation of China (Grant No. 51722105), Zhejiang Provincial Natural Science Foundation of China (LR18B030001), National Key Research and Development Program (Grant No. 2016YFB0901600) and the Fundamental Research Funds for the Central Universities (2018XZZX002-08). Ben Bin Xu would gratefully acknowledge the supports from the Engineering and Physical Sciences Research Council (EPSRC) through grants-EP/N007921 and EP/N032861.Notes and references
- Z. Xing, S. Wang, A. Yu and Z. Chen, Nano Energy, 2018, 50, 229–244 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 RSC.
- N. Ortiz-Vitoriano, N. E. Drewett, E. Gonzalo and T. Rojo, Energy Environ. Sci., 2017, 10, 1051–1074 RSC.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- F. Xie, Z. Xu, A. C. S. Jensen, H. Au, Y. Lu, V. Araullo-Peters, A. J. Drew, Y. S. Hu and M. M. Titirici, Adv. Funct. Mater., 2019, 29, 1901072 CrossRef.
- M. J. Piernas-Muñoz, E. Castillo-Martínez, J. L. Gómez-Cámer and T. Rojo, Electrochim. Acta, 2016, 200, 123–130 CrossRef.
- B. Wang, Y. Han, Y. Chen, Y. Xu, H. Pan, W. Sun, S. Liu, M. Yan and Y. Jiang, J. Mater. Chem. A, 2018, 6, 8947–8954 RSC.
- J. Song, B. Xiao, Y. Lin, K. Xu and X. Li, Adv. Energy Mater., 2018, 8, 1703082 CrossRef.
- W. Luo, Y. Zhang, S. Xu, J. Dai, E. Hitz, Y. Li, C. Yang, C. Chen, B. Liu and L. Hu, Nano Lett., 2017, 17, 3792–3797 CrossRef CAS PubMed.
- J. Conder and C. Villevieille, Chem. Commun., 2019, 55, 1275–1278 RSC.
- L. Fang, Z. Lan, W. Guan, P. Zhou, N. Bahalawane, W. Sun, Y. Lu, C. Liang, M. Yan and Y. Jiang, Energy Storage Materials, 2019, 18, 107–113 CrossRef.
- Y. Cao, L. Xiao, W. Wang, D. Choi, Z. Nie, J. Yu, L. V. Saraf, Z. Yang and J. Liu, Adv. Mater., 2011, 23, 3155–3160 CrossRef CAS PubMed.
- H. Kim, R. A. Shakoor, C. Park, S. Y. Lim, J.-S. Kim, Y. N. Jo, W. Cho, K. Miyasaka, R. Kahraman, Y. Jung and J. W. Choi, Adv. Funct. Mater., 2013, 23, 1147–1155 CrossRef CAS.
- Y. Fang, L. Xiao, X. Ai, Y. Cao and H. Yang, Adv. Mater., 2015, 27, 5895–5900 CrossRef CAS PubMed.
- H. Kim, I. Park, S. Lee, H. Kim, K.-Y. Park, Y.-U. Park, H. Kim, J. Kim, H.-D. Lim, W.-S. Yoon and K. Kang, Chem. Mater., 2013, 25, 3614–3622 CrossRef CAS.
- Y. Jiang, S. Yu, B. Wang, Y. Li, W. Sun, Y. Lu, M. Yan, B. Song and S. Dou, Adv. Funct. Mater., 2016, 26, 5315–5321 CrossRef CAS.
- Y. You, H. R. Yao, S. Xin, Y. X. Yin, T. T. Zuo, C. P. Yang, Y. G. Guo, Y. Cui, L. J. Wan and J. B. Goodenough, Adv. Mater., 2016, 28, 7243–7248 CrossRef CAS PubMed.
- Y. Qiao, G. Wei, J. Cui, M. Zhang, X. Cheng, D. He, S. Li and Y. Liu, Chem. Commun., 2019, 55, 549–552 RSC.
- B. Wang, Y. Han, X. Wang, N. Bahlawane, H. Pan, M. Yan and Y. Jiang, iScience, 2018, 3, 110–133 CrossRef CAS PubMed.
- B. Wang, S. Liu, B. Pan, W. Sun, Y. Tang, H. Pan, M. Yan and Y. Jiang, ChemSusChem, 2019, 7, 2415–2420 Search PubMed.
- L. Wang, J. Song, R. Qiao, L. A. Wray, M. A. Hossain, Y. D. Chuang, W. Yang, Y. Lu, D. Evans, J. J. Lee, S. Vail, X. Zhao, M. Nishijima, S. Kakimoto and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2548–2554 CrossRef CAS PubMed.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, Angew. Chem., 2013, 52, 1964–1967 CrossRef CAS PubMed.
- P. Ge, S. Li, H. Shuai, W. Xu, Y. Tian, L. Yang, G. Zou, H. Hou and X. Ji, Adv. Mater., 2019, 31, e1806092 CrossRef PubMed.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, J. Power Sources, 2018, 378, 322–330 CrossRef CAS.
- X. Jiang, H. Liu, J. Song, C. Yin and H. Xu, J. Mater. Chem. A, 2016, 4, 16205–16212 RSC.
- H. Kim, G. Yoon, I. Park, K.-Y. Park, B. Lee, J. Kim, Y.-U. Park, S.-K. Jung, H.-D. Lim, D. Ahn, S. Lee and K. Kang, Energy Environ. Sci., 2015, 8, 3325–3335 RSC.
- C. Wang, L. Xing, J. Vatamanu, Z. Chen, G. Lan, W. Li and K. Xu, Nat. Commun., 2019, 10, 3423 CrossRef PubMed.
- S. Zhang, H. Gu, H. Pan, S. Yang, W. Du, X. Li, M. Gao, Y. Liu, M. Zhu and L. Ouyang, Adv. Energy Mater., 2017, 7, 1601066 CrossRef.
- Y. Moritomo, S. Urase and T. Shibata, Electrochim. Acta, 2016, 210, 963–969 CrossRef CAS.
- D. Yang, J. Xu, X. Z. Liao, Y. S. He, H. Liu and Z. F. Ma, Chem. Commun., 2014, 50, 13377–13380 RSC.
- J. Song, L. Wang, Y. Lu, J. Liu, B. Guo, P. Xiao, J. J. Lee, X. Q. Yang, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2658–2664 CrossRef CAS PubMed.
- W. Li, S. Chou, J. Wang, J. Wang, Q. Gu, H. Liu and S. Dou, Nano Energy, 2015, 13, 200–207 CrossRef CAS.
- Y. Mao, Y. Chen, J. Qin, C. Shi, E. Liu and N. Zhao, Nano Energy, 2019, 58, 192–201 CrossRef CAS.
- S. Kulandaivalu, Z. Zainal and Y. Sulaiman, Int. J. Polym. Sci., 2016, 2016, 1–12 CrossRef.
- S. Yu, Y. Li, Y. Lu, B. Xu, Q. Wang, M. Yan and Y. Jiang, J. Power Sources, 2015, 275, 45–49 CrossRef CAS.
- Y. Tang, W. Zhang, L. Xue, X. Ding, T. Wang, X. Liu, J. Liu, X. Li and Y. Huang, J. Mater. Chem. A, 2016, 4, 6036–6041 RSC.
- Y. Liu, D. He, R. Han, G. Wei and Y. Qiao, Chem. Commun., 2017, 53, 5569–5572 RSC.
- Y. Jiang, Y. Li, P. Zhou, Z. Lan, Y. Lu, C. Wu and M. Yan, Adv. Mater., 2017, 29, 1606499 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TG, XPS, SEM, galvanostatic charge–discharge profile, XRD, and CV test. See DOI: 10.1039/c9ta12376h |
| This journal is © The Royal Society of Chemistry 2020 |