
Infiltrating molecular gatekeepers with coexisting molecular solubility and 3D-intrinsic porosity into a microporous polymer scaffold for gas separation†
 *ab
*ab
Abstract
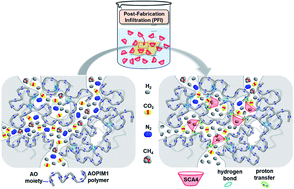
The inherently broad pore-size distribution in polymer membranes endows them with fewer size-selective microporous regions which could impair their performance and would require challenging size control at the angstrom level for enabling energy-efficient gas separation. Here, we successfully remodeled the non-selective microporous regions in polymer membranes with sub-angstrom size-sensitivity via an unconventional post-(membrane) fabrication infiltration (PFI) method based on a water-soluble member of the highly tunable and expandable organic macrocyclic family, namely, 4-sulfocalix[4]arene (SCA4). The small molecular size and attached multiple sulfonic groups of SCA4 molecules have enabled their complete solvation in common membrane-treating protic solvents, like methanol, such that they could molecularly infiltrate the entire microporous structure of already fabricated polymer membranes which plays the role of an interactive scaffold with extensive hydrogen or ionic bonding sites. Meanwhile, bearing an intrinsic size-sieving 3D open cavity, SCA4 molecules could act as molecular gatekeepers that effectively retard size-indiscriminative gas transport for realizing exceptional molecular-sieving properties towards efficient separation of multiple important gas pairs. This ultra-facile yet unconventional PFI design is free from the longstanding issues of interfacial nano-defects and pore blockage and is also potentially diversifiable by using a pool of other water-soluble and functionalizable macrocyclic counterparts for uncovering new composite-membrane design possibilities.
 Please wait while we load your content...
Please wait while we load your content...

