
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using nanoconfinement to inhibit the degradation pathways of conversion-metal oxide anodes for highly stable fast-charging Li-ion batteries†
Benjamin
Ng
,
Xiong
Peng
,
Ehsan
Faegh
and
William E.
Mustain
 *
*
Department of Chemical Engineering, Swearingen Engineering Center, University of South Carolina, Columbia, SC 29208, USA. E-mail: mustainw@mailbox.sc.edu; Tel: +1-803-576-6393
First published on 17th January 2020
Abstract
Nanostructured hybrids that physically encapsulate highly morphable, high capacity Li-ion battery anodes can potentially enable much longer cycle life than straightforward deployment of the same chemistry. This study demonstrates improved reaction reversibility (longer cycle life) and increased reaction rate (faster charge/discharge) for conversion metal oxides (MOs) reacting with Li-ions in a new architecture where they are physically confined inside of nanostructured carbon. The specific system of focus is nickel oxide (NiO) nanoparticles that are selectively deposited inside of, and fully encapsulated by, carbon nanotubes (CNTs). The physical and electrochemical behavior of the resulting nano-confined NC-NiO@CNT10 anode shows that confinement, and more specifically isolation from the electrolyte, eliminates the primary mechanisms for degradation and coulombic efficiency loss in MO anodes. Importantly, the elimination of these parasitic degradation mechanisms allows a nano-confined NC-NiO@CNT10 anode to achieve a stable reversible capacity of ca. 700 mA h g−1 and >99.9% coulombic efficiency, even after 2000 deep charge/discharge cycles (0–100% state-of-charge) at 1C. These findings provide a blueprint for future understanding of the role of material confinement in controlling reactions that are not only applicable to achieving long-term cycle performance for future high rate MO@CNT anodes in Li-ion batteries, but other chemical and electrochemical systems as well.
1. Introduction
The demands of emerging portable electronics, electric vehicles (EVs), and space technologies have ushered in a new paradigm for engineers and scientists to develop safe, durable, and lightweight rechargeable batteries.1–4 Li-ion batteries (LIBs) have become the backbone of our modern, electrified, interconnected society and have allowed device manufacturers to push the performance boundaries of cell phones, laptops, and other devices. The emergence of enhanced capabilities (e.g. GPS, music players, web browsing, video chats) and processing power (e.g. emerging 5G networks, cloud computing) is expected to drain existing onboard batteries at an alarming rate – thereby creating an urgency for the development of advanced high energy density and safe materials.5,6 In addition, high rate charging is highly desirable – especially in the electric vehicle sector that is looking to compete with the timescale for refueling both gasoline and H2-fueled cars (∼5 min, a battery charging rate ∼10C). During such rapid charging of existing Li-ion batteries (LIBs), over-polarization at the anode drives Li deposition and dendrite formation, which can cause internal short circuiting leading to thermal runaway and catastrophic failure of the battery pack.7,8 Therefore, new LIB anode materials are needed that are safer (no Li plating during fast charging), have higher capacities (>600 mA h g−1), and are durable (>1000 deep cycles).9–11One pathway for materials development, to achieve high rate charging at lower overpotentials (reducing the driving force for Li-plating), is to create nano-sized electroactive materials. Small particle sizes allow for enhanced reaction kinetics and rapid diffusion.12,13 Small size can simultaneously enhance the ability of active particles to accommodate strain, as well as significantly increase the number of charge carriers near the electrode–electrolyte interface. However, batteries employing nano-sized active materials are typically penalized with increased irreversible capacity loss, as well as decreased stability and reversibility. The result is typically electrodes with low coulombic efficiency (CE), mediocre capacity, and limited cycle life.
A second material pathway, to avoid Li plating, is to search for materials with a reversible potential substantively higher than the Li/Li+ redox couple to provide a buffer in its operating potential. Though materials with higher anode potentials might have lower operating voltages, materials with much higher theoretical capacity than commercial graphite will more than compensate for any energy density concerns, and the ability to safely operate at higher discharge rates can also result in comparable or superior power density. The desire to have nanostructured active materials with a higher anode potential during charge/discharge leads to the consideration of a whole new set of chemistries that have never before been deployed in commercial LIBs such as metal hydrides, nitrides, oxides, fluorides, phosphides, and sulfides14–20 – each of these conversion materials has its own advantages and challenges, but the most widely studied family of conversion anodes is metal oxides (MOs). The primary advantages that MO materials have over the standard graphite intercalation compounds (GICs) are: (1) multiple electrons per repeat unit (e.g. 2e−/NiO, resulting in a theoretical capacity of 718 mA h g−1) compared to one electron per six carbons (372 mA h g−1 theoretical capacity) – giving MO-anode LIBs a much higher theoretical energy density than graphite-anode LIBs; and (2) a redox potential approximately 1 V greater than Li/Li+ redox couple, which makes Li plating thermodynamically unfavorable.
MOs also have advantages over the most widely touted graphite alternative, Si: (1) MOs can be operated between 0–100% state of charge at high rates with as little as 5–10 wt% carbon added to the electrode,21 where Si electrodes typically have nearly 50% added carbon22–25 to achieve high performance and employ voltage cutoff strategies. In combination, these significantly limit the achievable capacity of Si-based anodes; (2) the volumetric expansion of MOs is much less than Si, typically ∼60%, versus ∼300% for Si;26–28 and (3) MOs are air stable and easy to process, unlike Si (and other more widely discussed anode replacements, such as Li metal).
However, MOs do suffer from intrinsically low electronic conductivities (10−8 to 10−3 Ω−1 cm−1)29 and there exists several degradation mechanisms that are inherent to their chemistry (eqn (1)).
| MxOy + 2yLi+ + 2e− ⇌ xM + yLi2O | (1) |
There have been several approaches in the literature aimed at minimizing the effects of these MO degradation mechanisms. Many authors have created highly tailored architectures including anchored-materials,44 shelled-materials (e.g. single-shell,45–48 yolk–shell,49,50 multi-shell51–53), and hierarchical frameworks.54–56 For instance, Deng et al.44 have demonstrated that ZnCo2O4 with cobalt–boride (Co–B) anchors can reduce degradation, volume change and phase segregation. This is accomplished by utilizing Co–B as an intra-particle “glue” that promotes enhanced intra-particle electron conduction and faster Li+ diffusion to/from the ZnCo2O4 structure. The result is a mixed MO that is capable of delivering stable gravimetric capacities over 1000 cycles. However, this design is likely to encounter issues at high loadings where long-range electronic and thermal conductivity play a significant role in performance as active materials are integrated into relatively thick electrodes for practical cells. Therefore, the integration of MO materials into highly conductive, well integrated networks is important for practical deployment in the future.
In another study, Lu et al.50 synthesized an elaborate tri-metallic NiCo2V2O8 yolk-double shelled MO anode that was able to achieve an outstanding 1228 mA h g−1 for 500 cycles. However, the material does suffer nearly 40% irreversible capacity loss – which in a finite Li system, leads to deadweight in the cathode or requires additional processing steps to allow for the prelithiation of the anode. In other words, creating Li-ion anodes with low initial irreversible capacity loss is crucial to practical application of MOs in full cells. Additionally, bi-metallic or multi-metallic MOs may have unique limitations during long-term cycling, where it appears that repetitive displacement and replacement of oxygen leads to phase segregation of the anode into two distinct MOs (i.e. M1M2Ox → M1Oy + M2Oz where M1 or M2 = Ni, Co, V, Zn, Mn, etc.). Another important consideration is that the electronic conductivities of these shelled-materials are typically low, which decreases electrode-level conductance.
There have also been reports of graphene-encapsulated MOs. Xie et al.52 was able to encapsulate ZnO–Mn–C with graphene and achieved 843 mA h g−1 capacity over 1000 cycles, which was attributed to a boost in the electronic conductivity and reduced phase segregation between the charge/discharge products. However, the graphene coating does not fully protect the active material. The result is an exposed MO with a high surface area supporting matrix that leads to high irreversible capacity loss of 32.7% in the first cycle as well as significant overgrowth of the SEI layer (and depletion of the electrolyte). Unfortunately, high initial irreversible capacity loss has been generalizable to other carbon-encapsulation approaches as well, whether the carbon-encapsulant surface is SEI-reactive (low-crystallized carbons >25% irreversible capacity loss57) or practically SEI-inactive (crystalline graphite ∼10–15% irreversible capacity loss58).
Therefore, Li-ion cells employing MO anodes must overcome the low (particle-level) and long-range (electrode-level) electronic conductivity problems, long-term phase segregation, and irreversible capacity loss due to uncontrolled overgrowth of the SEI. One possible approach is physically confining the MO from the electrolyte in a highly entangled, high-aspect ratio conductive matrix (e.g. high crystallinity carbon nanotubes), which has the potential to significantly reduce the effects of all the “inherent” degradation mechanisms listed above – increasing their cyclability and coulombic efficiency – and limit irreversible capacity loss in the first cycle. Doing so with a low loading of the conductive material (∼10%) can provide a practical pathway forward for these materials – resulting in a low cost, long-life, high rate active material with no Li plating during fast charge, which would have a significant impact on the LIB technology. This study will provide the first steps in this direction – using NiO confined inside of carbon nanotubes (NiO@CNT) as a proof-of-concept. The physical and electrochemical properties of the NiO@CNT material will be investigated. It will be shown that nano-confinement is a convincing panacea to eliminate the existing degradation pathways, allowing for these materials to be extensively cycled with full material utilization and fast-charged.
2. Materials and methods
2.1 Reagents
Multi-walled carbon nanotubes (CNTs) (average D = 10 nm, P/N 412![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 988 and average D = 50 nm, P/N 901002), pluronic P123 triblock copolymer (PEG-PPG-PEG, Mn ∼ 5800), tetraethyl orthosilicate (TEOS, >99.0%), hydrochloric acid (HCl, ACS reagent grade, 37%) and nitric acid (HNO3, ACS reagent grade, 70%) were purchased from Sigma Aldrich. Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 99%) and potassium hydroxide (NaOH) were purchased from Acros Organics through Fisher Scientific. Vulcan XC-72R was purchased from Cabot. The CNTs were purified with acid treatment and high temperature defunctionalization (detailed in Section 2.2). Li(Ni0.5Mn0.3Co0.2)O2 powder was purchased from MTI corporation. Ultrapure deionized water (18.2 MΩ cm) was supplied by a lab-scale Millipore Milli-Q Integral system with E-POD. All other chemicals were used as received.
988 and average D = 50 nm, P/N 901002), pluronic P123 triblock copolymer (PEG-PPG-PEG, Mn ∼ 5800), tetraethyl orthosilicate (TEOS, >99.0%), hydrochloric acid (HCl, ACS reagent grade, 37%) and nitric acid (HNO3, ACS reagent grade, 70%) were purchased from Sigma Aldrich. Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 99%) and potassium hydroxide (NaOH) were purchased from Acros Organics through Fisher Scientific. Vulcan XC-72R was purchased from Cabot. The CNTs were purified with acid treatment and high temperature defunctionalization (detailed in Section 2.2). Li(Ni0.5Mn0.3Co0.2)O2 powder was purchased from MTI corporation. Ultrapure deionized water (18.2 MΩ cm) was supplied by a lab-scale Millipore Milli-Q Integral system with E-POD. All other chemicals were used as received.
2.2. Synthesis of NiO/C, ID-NiO/CNT, and NC-NiO@CNT
Three unique NiO-carbon composite materials were synthesized: (1) unconstrained deposition of NiO on the surface of carbon black (Vulcan XC-72R), which will be denoted as NiO/C; (2) NiO indiscriminately deposited on both the inside and outside of CNTs, denoted as ID-NiO/CNT; and (3) NiO nano-confined exclusively on the inside of CNTs, denoted as NC-NiO@CNTx (where x = the CNT diameter in nm).NiO/C was synthesized through a mesoporous silica (SBA-15) template-assisted synthesis. First, the SBA-15 template was produced using the same procedure as our previous publications.14,59,60 In a typical synthesis, 6 g of pluronic P123 and 13.6 mL TEOS was first dissolved in 180 mL of 2 M HCl solution and successively heated at 45 °C (20 hours) and 100 °C (24 hours). The resulting SBA-15 template was washed with a DI water/ethanol mixture and dried under vacuum. The dried SBA-15 was then calcined in air at 500 °C for 3 hours. Next, 500 mg of nickel nitrate hexahydrate was dissolved to create an aqueous 0.5 M Ni(NO3)2 solution. The solution was added dropwise to the SBA-15 until a ratio of 6.3 mL gSBA-15−1 was obtained. The Ni(NO3)2/SBA-15 gel was magnetically stirred and then calcined in air for 3.5 hours at 400 °C. The resulting powder was immersed into an aqueous 5 M KOH solution for 12 hours at 100 °C to etch away the SBA-15 template. The supernatant was disposed, and the remaining solids were rinsed three times with excess DI water, centrifuged at 4500 rpm using a Sorvall ST-8 centrifuge, and then dried for 8 hours at 70 °C under vacuum. Finally, the templated NiO was added to a mortar along with Vulcan XC-72R in a 7:1 mass ratio and ground with a pestle for 30 minutes.
ID-NiO/CNT was synthesized by dispersing 200 mg of CNT50 into 20 mL nitric acid with a magnetic stirring rod. The CNT50 dispersion was refluxed at 120 °C for 30 minutes to purify the CNTs, which also opened their closed-ends. Next, the CNT50 dispersion was diluted slowly under magnetic stirring with a total of 80 mL of DI water. The diluted dispersion was then transferred to a 50 mL centrifuge tube and the solids were separated by centrifugation at 4500 rpm for 10 min. After decanting the liquid, an additional 40 mL of DI water was added to the centrifuge tube, which was then sonicated and centrifuged (repeated three times with the liquid being decanted off each time). The solids were vacuum dried at 70 °C for 24 hours and defunctionalized for 4 hours at 1500 °C in a tube furnace under an argon atmosphere to ensure high CNT crystallinity. Next, 30 mL of a 10 M nickel nitrate hexahydrate (Ni(NO3)2·6H2O) in 70% nitric acid solution was prepared by stirring for 1 h. 100 mg of the defunctionalized CNT50 was added to the solution and refluxed at 120 °C for 12 hours. The solid precipitate was vacuum filtered with a G2-grade glass fiber membrane (Fisherbrand) in a Büchner funnel, vacuum dried at 70 °C for 24 hours, and then placed in a tube furnace and heated to 450 °C for 4 hours under argon atmosphere. The final ID-NiO/CNT was obtained after grinding with a mortar and pestle for 30 minutes to break up any agglomerates.
NC-NiO@CNTx anodes were synthesized by dispersing 200 mg of CNTx into 20 mL of 70% nitric acid under continuous stirring. The CNT dispersion was refluxed at 120 °C for 30 minutes. The temperature was reduced to room temperature and then slowly diluted with 80 mL of DI water. The diluted dispersion was centrifuged at 4500 rpm for 10 minutes, then decanted and refilled with 40 mL of DI water, and finally sonicated. The process was repeated three times. The supernatant was removed, and the remaining CNTx solids were dried at 70 °C for 24 hours under vacuum. The dried solids were thermally treated and defunctionalized for high CNT crystallinity in a tube furnace under continuous argon flow for 4 hours at 1500 °C. The samples were re-dispersed in a 30 mL 70% nitric acid and 10 M Ni(NO3)2 solution. Then, the 10 M Ni(NO3)2–CNTx solution was refluxed for 12 hours at 120 °C to allow for complete permeation of the nickel precursor into the CNTx. After filtration with a G2-grade glass fiber filter, the samples were dried under vacuum at 70 °C for 24 hours and pyrolyzed at 450 °C for 4 hours under continuous argon flow. 300 mg of the pyrolyzed solids was dispersed in 50 mL of 0.25 M nitric acid and stirred for 1 hour. The remaining solids were collected by vacuum filtration with G2-grade glass fiber circles in a Büchner funnel (the liquid filtrate was bluish-green), dried under vacuum at 70 °C for 24 hours, and then heated in a tube furnace at 450 °C for 2 hours in an argon atmosphere. The final NC-NiO@CNT50 (i.e. CNTs with 50 nm diameter) and NC-NiO@CNT10 (i.e. CNTs with 10 nm diameters) active materials were obtained after grinding with a mortar and pestle for 30 minutes.
2.3. Electrode fabrication and coin cell assembly
NiO/C, ID-NiO/CNT, and NC-NiO@CNTx anodes were prepared by mixing 95% active material with 5% polyvinylidene fluoride binder (PVDF, Kynar Blend). A transfer micropipette was used to measure 400 μL of N-methyl-pyrrolidone (NMP, Acros, 99.5% Extra Dry) solvent, which was used as a dispersing agent for 100 mg of total solids to obtain an anode ink with moderate viscosity. The final ink was obtained after homogenization by 3 rounds of sonication (20 minutes) and continuous stirring (8 hours). The homogenized ink was uniformly sprayed onto a 50 μm thickness Cu current collector (Alfa Aesar, Catalog No. AA42972FI) with an Iwata-Medea Eclipse HP-CS sprayer. The resulting electrodes were dried in a vacuum oven at 75 °C for 24 hours, pressed at 1500 lbs with a hydraulic press (MTI 5T Max. Manual Mechanical Press), and calendared (MTI Electric Roller-MSK-MR100DC). Electrodes used in half-cell studies had an active loading between 0.5 and 1.5 mg cm−2. Full cells had a higher target anode mass loading of 5 mg cm−2 and the cathode target mass loading was 18 mg cm−2 (target N/P ratio ∼1.1). The full cell cathode active material:carbon:binder ratio was 8:1:1.
Coin cells were assembled in an argon (Ar, UHP Praxair) filled glove box (O2 and H2O <0.1 ppm, MBraun Labmaster SP) in a half cell configuration with a Li metal counter/reference electrode. All electrochemical tests were conducted with CR2032 coin cells (Hohsen Corp.), 1.5 cm diameter lithium metal (99.9%, Alfa Aesar) counter electrodes, Celgard 2320 tri-layer PP/PE/PP membrane separator, and 1 M lithium hexafluorophosphate (LiPF6, Acros 98%) salt in a 1:1:1 volumetric mixture of ethylene carbonate (EC, Acros 99+%): dimethyl carbonate (DMC, Acros 98+%): diethyl carbonate (DEC, Acros 99+%) electrolyte. A typical coin cell assembly began with a lithium counter electrode centered and flattened onto the coin cell base. A micropipette was used to transfer 15 μL of electrolyte to both sides of the Celgard separator, which was then placed, centered, onto the lithium electrode. The gasket was placed to position the working electrode (NiO/C, ID-NiO/CNT, NC-NiO@CNTx) directly in the center of the assembly. The spacer disk, spring, and the anodic cap of the coin cell were placed on top and the cell was finished by crimping with an MTI hydraulic press (MSK-110) to a pressure of 750 PSI. All sealed coin cells were inspected and safely transferred out of the glove box for electrochemical testing.
To assemble full cells, the anodes were first prelithiated to control the state of charge of the anode upon assembly and to avoid any irreversible capacity loss in the initial cycles. The anode electrode was prelithiated by first immersing the electrode in a 1:1:1 EC: DMC, DEC, 1 M LiPF6 electrolyte for 72 hours to allow for full electrolyte penetration. Then, the anode was cycled in the same electrolyte 10 times between 0.001 and 3.0 V versus a Li foil. The first cycle rate was C/20 and subsequent cycles was 1C. Then, the prelithiated anode (5.16 mg cm−2) was paired with a Li(Ni0.5Mn0.3Co0.2)O2 (18.23 mg cm−2) cathode using the same procedure as the half-cell experiments. Before cycling, full cells were aged for 48 hours at 40 °C inside a Tenney temperature control chamber to allow the electrolyte to fully permeate into the electrodes. After removal from the Tenney chamber, the cell was given an additional 8 hour relaxation to equilibrate to ambient temperature before testing.
2.4. Chemical and structural characterization
An extensive suite of characterization tools was used to determine the structural and chemical state of the active materials before and after cycling. For materials characterized at the end of cycling, the Li-ion cells were disassembled in the same MBraun Ar-filled glovebox as above. The electrodes were washed with dimethyl carbonate (DMC) to remove residual Li salts and dried for 1 hour under vacuum at room temperature. X-Ray Diffraction (XRD) patterns were collected between 10 and 90 2θ degrees at a scan rate of 0.0285 degrees per second using a Rigaku Miniflex II at room temperature equipped with a high sensitivity D/tex Ultra Si slit detector. The Miniflex II had a Cu K(α) radiation source (λ = 0.15405 nm) that was operated at 30 mA and 15 kV. Thermo-gravimetric analysis (TGA) was done using a NETZSCH STA 449. The TGA traces were collected from room temperature to 1000 °C. Each sample was purged of moisture for one hour at 120 °C under UHP N2. Scanning transmission electron microscopy (S/TEM) images were collected using a FEI Talos S/TEM and Hitachi H8000 TEM with integrated energy dispersive X-ray spectroscopy (EDS). X-ray photoelectron spectroscopy (XPS) measurements were performed with a Kratos AXIS Ultra DLD XPS system.Pre-/post-cycled TEM images were collected using the identical-location TEM technique from our previous publication.61 Generally, a Cu TEM finder grid (3 mm diameter, 100 mesh, Ted Pella, Inc) was dropped with 1 μL of diluted active material ink (1:10 ratio), drained, repeated three times, and dried for 24 hours under ambient conditions. After TEM imaging, the TEM grid/active material was cycled with a custom-designed Teflon-shrouded Cu electrode with a Teflon cap that applies pressure to the outer ring of the Cu grid. A 3-electrode system was employed to ensure high-fidelity voltage measurements (i.e. mitigate external factors that contribute to high overpotentials) inside the Ar-filled MBraun glovebox. The 3-electrode system (working electrode: NiO/C, ID-NiO/CNT, NC-NiO@CNT; counter electrode: Li foil; reference electrode: Li foil) was separated with a Celgard separator inside a beaker and filled with 1 M LiPF6 in 1:1:1 volumetric mixture of EC/DMC/DEC. After electrochemical treatment, described in the next section, the working electrodes were rinsed with DMC by submersion for 30 minutes, and dried under ambient glovebox conditions for 24 hours. Afterwards, the post-cycled electrodes were transferred to an Ar-filled Kratos AXIS Ultra multipurpose transfer vessel and removed from the glovebox for further characterization (e.g. XPS, TEM, SEM).
2.5. Electrochemical testing
Electrochemical measurements were done on both coin-cells and the identical location TEM working electrodes. For the coin cell experiments, chronopotentiometric charge/discharge experiments were performed using an Arbin MSTAT battery test station. The voltage window for half-cells charge/discharge measurements were 0.001–3.0 V. Half cells were tested at multiple rates between C/10 and 5C, though 1C was the most common. The C-rates for the charge/discharge were determined based on the NiO mass, and all reported capacities are normalized to the total material mass (NiO + C + PVDF). The voltage window for full-cells were set to 0.001–4.0 V and held at the upper cutoff voltage until a C/20 current taper was realized. Full cells were tested at a C/2 rate. The coin cells were also exposed to cyclic voltammetry and Electrochemical Impedance Spectroscopy (EIS) using an Autolab PGSTAT302N Potentiostat (Metrohm USA). Cyclic voltammograms (CVs) were collected in the same voltage window as the charge/discharge experiments with a 0.1 mV s−1 scan rate. EIS experiments were done at open circuit with a 5 mV amplitude after full discharge (3 V) at multiple frequencies between 1 MHz and 0.01 Hz. The open circuit potential was assumed to be reached was reached when dV/dt < 1 μV s−1. All of the electrochemical measurements were made at room temperature. The electrodes used for identical location TEM electrodes were exposed to either 10 or 100 CV cycles (more information is available in Fig. S1 and subsequent discussion in the ESI† file) between 0.001–3.0 V at a scan rate of 0.8 mV s−1.3. Results and discussion
3.1 Behavior of non-confined MO nanoparticles
Fig. 1a is an SEM image of the NiO/C macrostructure, showing that the material synthesized consisted of large agglomerations of NiO nanoparticles with good porosity. Fig. 1b presents transmission electron microscopy (TEM) images and energy dispersive X-ray (EDS) spectroscopy of pre-cycled NiO/C and shows the successful synthesis of ordered mesoporous NiO via the SBA-15 template-assisted synthesis. The TEM images of NiO/C indicate highly distinctive boundaries between each NiO nanoparticle. In addition, high angle annular dark-field (HAADF) imaging and elemental mapping of NiO/C shows that the majority of the NiO is distributed on Vulcan carbon, though some NiO particles extend beyond the main carbon cluster. The images and EDS mapping shows that the MO particles were not always directly bound to the surface, but existed as a physical mixture with the carbon. The X-ray diffraction (XRD) pattern for the NiO/C (blue line) is presented in Fig. 1c. Characteristic peaks for NiO were observed at 37°, 44°, 63°, 75°, and 80° (2θ), corresponding to its (111), (200), (220), (311), and (222) Miller indices, respectively.62 Additionally, two carbon-related peaks were observed at 26° and 44°, which are associated with the graphitic (002) and C (100) Miller planes, respectively.63 No secondary phases or peaks ascribed to contaminants were observed. The Scherrer equation (Tables S1 and S2 in the ESI† file) was applied to calculate the average crystallite domain size from all planes for NiO/C, which was 23.4 nm.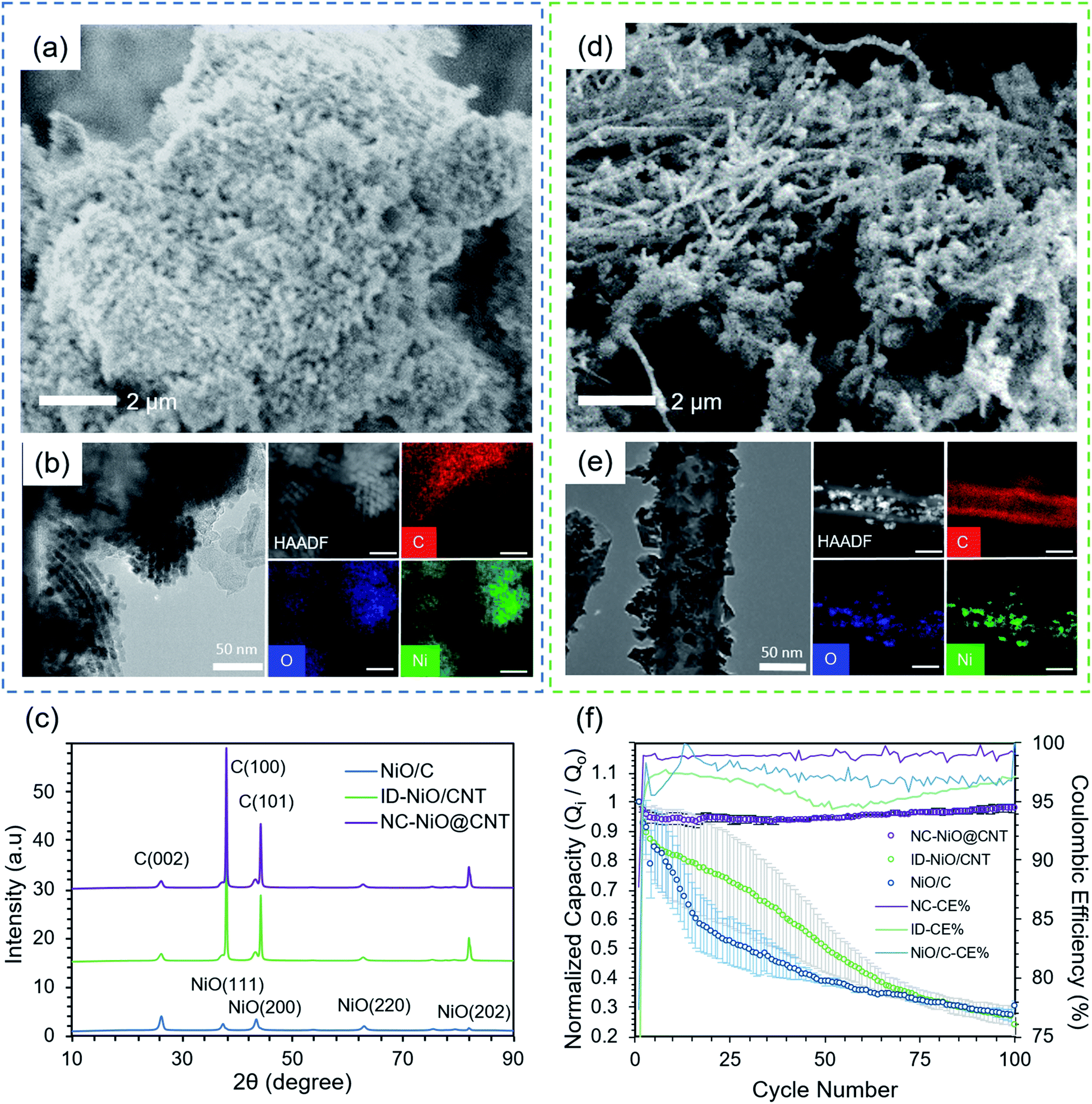 | ||
| Fig. 1 Physical and electrochemical characterization. (a) SEM, (b) TEM/EDS of NiO/C. (c) X-Ray Diffraction (XRD) patterns comparing the crystallography (e.g. crystallite domain size, structure) of NiO/C (blue line), ID-NiO/CNT (green line), and NC-NiO@CNT50 (purple line). (d) SEM and (e) TEM/ESC of ID-NiO/CNT. (f) Comparing the capacity retention of NiO/C (blue line), ID-NiO/CNT (green line), and NC-NiO@CNT50 (purple line) at 1C charge/discharge rate to observe reaction and cell-level reversibility. | ||
Fig. 1d is an SEM image of NiO indiscriminately deposited and anchored onto the surface of the 50 nm diameter CNTs, ID-NiO/CNT. The image clearly shows a semi-entangled carbon nanotube network with a high degree of surface NiO deposition. As shown in the SEM image, the MO particles are tethered to the CNT surface as both dispersed particles as well as larger agglomerates. In addition, TEM and EDS in Fig. 1e, were used to analyze the nanostructure and spatial arrangement of NiO and CNT in the ID-NiO/CNT material before any electrochemical testing. Since HAADF is able to contrast by atomic weight (high Z-contrasted material is ascribed to Ni, whereas the lighter Z-contrasted material is C), it was confirmed that surface of the CNTs was covered extensively by NiO, though it should be noted that some NiO was also deposited inside of the CNTs. The XRD pattern for ID-NiO/CNT (green line) has similar characteristic peaks for NiO and C, NiO: 37° (111), 44° (200), 63° (220), and 75° (222); C: 26° (002) and 44° (100). The average crystallite domain size from all planes for ID-NiO/CNT was found to be 18.6 nm and smaller than that of NiO/C.
Although their physical structures are different, one important similarity between the NiO/C and ID-NiO/CNT materials are that they are both comprised of NiO particles that are completely exposed to the electrolyte in operating cells. Therefore, it can be expected that all four of the degradation mechanisms discussed in the Introduction will be active for these materials and it is likely that their behavior in operating LIBs would be similar. Fig. 1f presents the normalized capacity retention (Q/Qo) plots for both NiO/C (blue line) and ID-NiO/CNT (green line) over 100 cycles at a 1C rate. Each experiment was repeated on three cells to confirm the repeatability of the electrochemical performance. In addition, the reference point for the initial capacity (Qo) was assumed to be the 2nd cycle to account for the irreversible capacity loss from SEI formation. The overall electrochemical cycling performance of NiO/C and ID-NiO/CNT showed similar capacity fade over 100 cycles – showing that the type of carbon is not an intrinsically determining factor when considering the onset of degradation. For NiO/C, the irreversible capacity loss in the first cycle was 23%. From the 2nd cycle to the 15th cycle, the rate of capacity fade was very high, and the NiO/C anode lost 41% of its capacity during this time. The average coulombic efficiency (CE) over the first 15 cycles was also low, only 97.4%. After cycle 15, the degradation rate for NiO/C was slowed, though still relatively severe, and over 100 cycles NiO/C lost 69.4% of its capacity and the average CE of a single cycle was 97.1%.
As an anode, ID-NiO/CNT (Fig. 1f green line) experienced a similar trend in capacity fade to NiO/C. The SEI-related irreversible capacity loss for ID-NiO/CNT was 35.4%, which was higher than that of NiO/C, most likely due to the higher surface area of the smaller NiO crystallites in ID-NiO/CNT (16.6 nm) compared to NiO/C (24.1 nm). Thermo-gravimetric analysis (TGA) of ID-NiO/CNT found that the total NiO mass in the active material was 95.9% (Fig. S8a in the ESI† file). The total capacity loss of ID-NiO/CNT was 73.1% over 100 cycles whereas NiO/C was 69.4%, which shows a high degree of similarity in the onset of degradation and overall capacity loss. An important point related to the capacity retention plots in Fig. 1f is that the data for both NiO/C and ID-NiO/CNT had seemingly large scatter, particularly at low cycle numbers. What this suggests is that MO degradation by metal (e.g. Li, M) trapping, SEI (re)formation, and/or transition to higher oxidation states is a complex series of events that can be triggered over a distribution of cycle numbers – though typically the onset of degradation will occur within the first 30 cycles, and the rate of initial degradation is rapid. Finally, the average CE for ID-NiO/CNT was also low, 95.8%, with a notable trend of CE loss over 50 cycles followed by a gradual recovery. The correlation between capacity loss and CE loss indicates that material is being trapped in the charged state repetitively after each cycle. This would result in larger Ni agglomerates, which was investigated by collecting TEM images of post-cycled materials and comparing them to Fig. 1b and e. Also, the low measured CE without catastrophic failure and loss of capacity is direct evidence of the conversion of the NiO to higher oxidation states during charge/discharge,21 which was further investigated by XPS before and after cycling.
Fig. 2 shows post-cycle TEM images and pre/post-cycled XPS of NiO/C and ID-NiO/CNT. It is important to observe the evolution of the MO particle size, as this impacts the electrochemical behavior. As observed in Fig. 2a for NiO/C, even after 10 cycles the interfacial boundaries between each NiO nanoparticle appear to merge. After 100 cycles, the NiO agglomerates in NiO/C are so large that individual particles are almost indistinguishable (Fig. S1 in the ESI† file). The repetitive restructuring and collapse of the MO active material during the conversion reaction leads to larger and larger phases each time it is reformed, causing a gradual decrease in the Ni/Li2O interfacial contact area during discharge (oxidation) and trapping of material in the charged state. In other words, the NiO/C undergoes significant metal trapping during discharge, and is likely the primary cause for capacity fade during cycling.
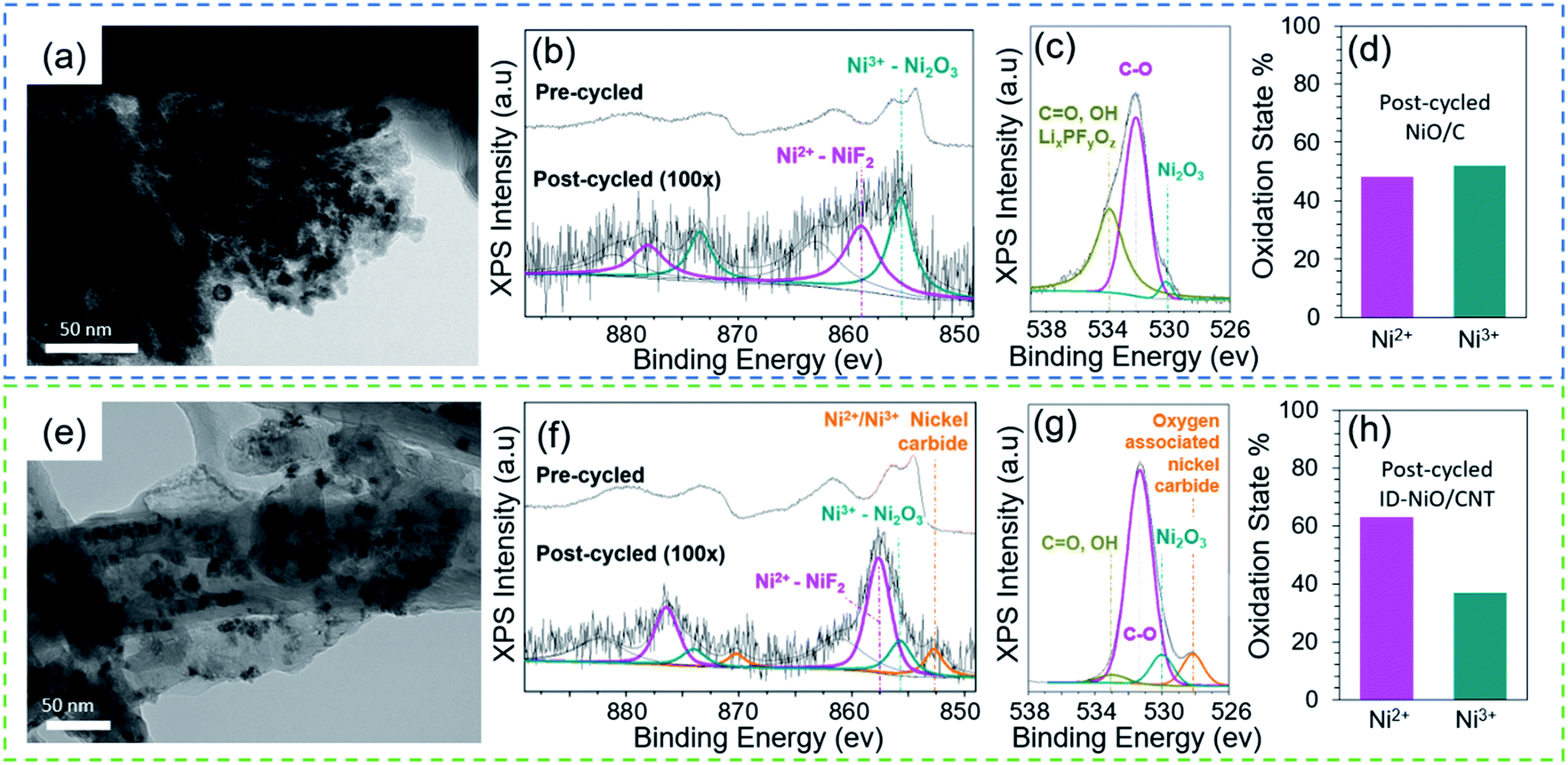 | ||
| Fig. 2 Evaluation of physical and chemical transformations. (a) TEM image of NiO/C after 10 electrochemical cycles. XPS spectra of NiO/C including (b) Ni2p, (c), O1s, and (d) oxidation state percentages for NiO/C. (e) TEM images of ID-NiO/CNT after 10 electrochemical cycles. XPS spectra of ID-NiO/CNT including (f) Ni2p, (g) O1s, and (h) oxidation state percentages for ID-NiO/CNT. | ||
Fig. 2b–d presents the pre- and post-cycle XPS analysis for NiO/C. Fig. 2b and c shows the Ni2p and O1s post-cycled XPS spectra for electrodes both before and after 100 cycles (the spectra were collected for materials in the discharged state). The high resolution Ni2p XPS spectrum before cycling (Fig. 2b) shows characteristic Ni2p1/2 (872.5 eV) and Ni2p3/2 (854 eV) with a multiplet-split and displacement of 18.5 eV (from Ni2p1/2 to Ni2p3/2), which is characteristic of NiO spin-orbital levels.21 After 100 cycles, a peak at 859 eV emerged, which could be attributed to the formation of surface NiF2 (a likely SEI product). In addition, a shoulder peak was found at a binding energy of 855.4 eV, which is characteristic of higher oxidation state Ni2O3, and indicates a pre-to-post cycle material transformation from (Ni2+ → Ni3+). The O1s XPS spectrum (Fig. 2c) for the post-cycled NiO/C shows the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, OH bonds at a binding energy of 533.0 eV and C–O bonds at 531.3 eV. Evidence for lithium alkyl carbonates, R–CH2O(CO)OLi was also found at O1s binding energies of 532.5 eV and 533.5 eV.64 A less pronounced peak for the oxygen associated to metal oxides was observed from the peak shoulder at 530.1 eV. From the XPS results, the portion of the Ni in the active material in the +2 (NiO) and +3 (Ni2O3) oxidation states are summarized in Fig. 2d. During cycling, the unconstrained NiO/C underwent a material transformation from 100% Ni2+ to a mixed oxidation state of 52% Ni3+ and 48% Ni2+, accounting for the low CE observed in Fig. 1f. As stated in the introduction, the most likely cause for the transition of Ni2+ to Ni3+ is the reaction of NiO with oxygen-containing species in the SEI and/or the electrolyte. However, because there are several oxygen-containing species in the system, it can be difficult to determine which one is responsible for the transformation of NiO to Ni2O3 over time. Though there are many possible reactions, the O1s XPS spectra does provide evidence for at least one possible reaction, shown in eqn (2).
O, OH bonds at a binding energy of 533.0 eV and C–O bonds at 531.3 eV. Evidence for lithium alkyl carbonates, R–CH2O(CO)OLi was also found at O1s binding energies of 532.5 eV and 533.5 eV.64 A less pronounced peak for the oxygen associated to metal oxides was observed from the peak shoulder at 530.1 eV. From the XPS results, the portion of the Ni in the active material in the +2 (NiO) and +3 (Ni2O3) oxidation states are summarized in Fig. 2d. During cycling, the unconstrained NiO/C underwent a material transformation from 100% Ni2+ to a mixed oxidation state of 52% Ni3+ and 48% Ni2+, accounting for the low CE observed in Fig. 1f. As stated in the introduction, the most likely cause for the transition of Ni2+ to Ni3+ is the reaction of NiO with oxygen-containing species in the SEI and/or the electrolyte. However, because there are several oxygen-containing species in the system, it can be difficult to determine which one is responsible for the transformation of NiO to Ni2O3 over time. Though there are many possible reactions, the O1s XPS spectra does provide evidence for at least one possible reaction, shown in eqn (2).
| 2NiO + 2RCH2OCOOLi ⇌ Ni2O3 + Li2O + RCH2COCOCH2R | (2) |
O bonds than C–O bonds, which suggests that the alkyl carbonate SEI decomposition product R–CH2O(CO)OLi likely undergoes further decomposition. It is also noted that the asymmetry of this peak coincides well with the emergence of the Ni2O3 species, suggesting that these events are related; however, it is acknowledged that additional reactions are certainly possible, even likely.
Fig. 2e shows that after 10 cycles ID-NiO/CNT was able to retain its pre-cycled architecture with no observable deformation to the CNT. However, it was clearly observed that some NiO agglomeration occurs and that some of the active material particles were detached and isolated from the CNTs as they were engulfed by the SEI. These particles remain trapped in the electronically insulating SEI and become deactivated. The SEI was found to grow similarly to that of graphite, where the SEI partially extends out of the CNT edge planes and caps. In addition, SEI-clusters were found near large MO deposits. Fig. 2f–h presents the pre- and post-cycled XPS analysis of ID-NiO/CNT and the characteristic peaks in ID-NiO/CNT shows the degradation pathway to form nickel carbides at 852.6 eV and Ni2O3 at 857.5 eV. Fig. 2f shows the high resolution Ni2p XPS spectra for pre- and post-cycled ID-NiO/CNT. Prior to cycling, the XPS spectra were very similar to NiO/C, where Ni2p1/2 (873 eV) and Ni2p3/2 (854.5 eV) displayed an electron-spin difference of 18.5 eV. Also similar to NiO/C, significant material transformation occurred after 100 cycles and the presence of nickel fluorides (857.5 eV), nickel oxide (855.6 eV), and nickel carbides (852.8 eV) were observed. The indiscriminately deposited ID-NiO/CNT post-cycled XPS analysis suggest similar chemical transformation of Ni to higher oxidation states (Ni2+ → Ni3+) as that found in NiO/C, Fig. 2h. Finally, Fig. 2g shows that the O1s spectrum for ID-NiO/CNT showed a similar presence of CO, and OH bonds (533 eV) and C–O bonds (531 eV) as NiO/C.
In total, the observations in Fig. 1 and 2 suggest that the behavior of NiO/C and ID-NiO/CNT are very similar, though there were some subtle differences. For instance, the degradation rate of ID-NiO/CNT seems slightly slower and the transition to higher oxidation states is not quite as severe. It is possible that these differences are due to the partial confinement of NiO inside the CNT host, which might be able to minimize degradation of those particles because they are mostly isolated from the bulk electrolyte. Therefore, it was thought that fully encapsulating the MO nanoparticles inside of the CNTs might be a promising way to minimize the extent to which the inherent MO degradation mechanisms are active in these materials. Reducing the number and degree of unwanted reactions would extend the life of MO-based anode materials, which led to the synthesis and investigation of the nano-confined NiO (NC-NiO@CNTx) anodes that are shown and discussed extensively in the next section.
3.2 CNT-nanoconfined NiO
The proposed role of nanoconfinement to inhibit the dominant MO degradation pathways (in this case NiO) is illustrated in Fig. 3a. When the MO particles are deposited solely inside the CNT, the CNT acts as an immobilization host that isolates MOs from the bulk electrolyte – which is expected to limit the extent to which the MO active material can react with the electrolyte, minimizing degradation. Also, the finite space within the CNT provides some elasticity for volumetric expansion, but limited opportunity for continuous SEI growth (i.e. CNT confinement can minimize particle detachment, isolation, and ripening/agglomeration). In addition, the CNT holds all reactants and products in one closed packet, which is hypothesized to minimize possible parasitic degradation reactions and increase the interaction area between reactants – increasing the material-level reaction reversibility and improving coulombic efficiency. | ||
| Fig. 3 Material nanoconfinement. (a) Illustration of metal oxides spatial rearrangement during charge/discharge in the case of nanoconfinement, (b) SEM images of nano-confined NiO in NC-NiO@CNT50, (c) TEM images and EDS mapping of NC-NiO@CNT50, (c-1) interlayer spacing of CNT, (c-2) interface between MO-CNT, (c-3) lattice fringes of NiO, (d) TEM image of NiO@CNT50 after 10 electrochemical cycles. XPS spectra of NC-NiO@CNT50 including (e) Ni2P, (f) O1s, as well as (g) the percentage of active materials remaining in the +2 oxidation state after cycling. | ||
It should be noted that other groups have synthesized LIB anode materials with similar geometry. For example, Yu et al.65 investigated the behavior of Sn nanoparticles within the interior of carbon nanofibers and achieved stable capacity ∼737 mA h g−1 over 200 cycles at 0.5C. Ottmann et al.66 deposited Mn3O4 inside of CNTs, and was able to achieve a stable capacity of ∼430 mA h g−1 for 50 cycles at 100 mA g−1 (∼C/10). The low capacity was due to a very high carbon content of 70%. Other accounts of alloying materials such as SnSb nanodots embedded inside nanotubes and fibers67 were able to extend the cycle life by boosting the conductivity and preventing agglomeration, and increasing the cyclability of SnSb to 1000 cycles with an end-of-discharge capacity of 451 mA h g−1, though this is also far below the theoretical capacity of the material. Besides anodes, electrospun intercalation-based Li3V2(PO4)3 cathode fibers were synthesized by Liu et al.68 with high rate capability (5C), long-term stability (500 cycles), and discharge capacity (85 mA h g−1). Though these studies seem to confirm that nanoconfinement improves battery performance, none of these studies focus on the role of nanoconfinement on the suppression of chemical degradation processes, such as those involving changes in the oxidation states (e.g. M2+ → M3+) of conversion-based active materials.
Therefore, to explore the realized effect of confinement on long-term reversibility and stability, NC-NiO@CNT50 was synthesized. The SEM images in Fig. 3b show that the synthesized material was nearly completely free of surface MO particles compared to ID-NiO/CNT in Fig. 1d. Not only is this expected to yield the advantages discussed above, it should also be noted that the resulting electrode would primarily consist of a highly conductive, interconnected carbon matrix that is able to freely transport electrons throughout the active layer.69,70Fig. 3c further shows the successful synthesis of the nanoconfinement concept presented in Fig. 3a, providing a high-resolution view of the structure alongside EDS elemental mapping, which showed the presence and distribution of Ni, O, and C. In addition, the NiO particles appeared to be tethered to the inner walls of the CNT, which physically restricts the MOs and their charge products (M and Li2O) to a finite space, thereby suppressing movement. The high-resolution TEM images (HR-TEM) in Fig. 3(c-1 to c-3) show the lattice fringes of the NiO nanocrystallites with a d-spacing of 2.4 Å, which is ascribed to (111) plane of NiO. The interlayer separation of the CNT was determined to be 3.4 Å. Finally, XRD patterns for NC-NiO@CNT50 (purple line) are shown in Fig. 1e. The same characteristic peaks for NiO (at 37°, 44°, 63°, 75°, and 80°) and carbon (at 26° and 44°) were observed in NC-NiO@CNT50 that were observed for NiO/C and ID-NiO/CNT. Application of the Scherrer equation yielded a calculated average crystallite domain size from all planes for NC-NiO@CNT50 of 18.5 nm, similar to ID-NiO/CNT (18.6 nm).
After synthesis, the electrochemical response of NC-NiO@CNT50 was investigated and its normalized capacity retention (Q/Qo) over 100 cycles is shown in Fig. 1f, alongside the data for NiO/C and ID-NiO/CNT for comparison. Unlike NiO/C and ID-NiO/CNT, the NC-NiO@CNT50 retained essentially all of its capacity over the first 100 cycles. Also positive, NC-NiO@CNT50 had a much lower irreversible capacity loss than the other two NiO materials, only 12.2%. Because of this excellent initial performance, the NC-NiO@CNT50 was allowed to cycle for an additional 300 cycles. Over the 400 total deep charge/discharge cycles at 1C, the NC-NiO@CNT50 retained 96.3% of its capacity with very little scatter over three repeated cells (Fig. S2 and S3 in the ESI† file). In addition, the average coulombic efficiency of NC-NiO@CNT50 was 99.2%. However, one aspect that could be improved in future iterations of these materials is the NiO content, which was estimated by TGA to be 87.4% (Fig. S8b in the ESI† file). The theoretical minimum carbon content for this specific CNT was calculated to be 3.4% based on the volume of the Ni + Li2O phase after charging. This suggests that future versions of the NiO@CNT anodes could have a carbon content of around 5%, and no other additive carbon, which is comparable to modern batteries.
Fig. 3d shows post-cycle TEM images of NC-NiO@CNT50, which shows that minimal structural changes occurred during charge/discharge, in stark contrast to NiO/C and ID-NiO/CNT (Fig. 2). The NiO particle size distribution of confined MOs was very similar pre- and post-cycling. Also, there was no evidence of either particle detachment or migration that lead to the formation of large M/MO agglomerates. Furthermore, the SEI appeared to remain confined within the interior of the CNT, which can effectively limit SEI growth during long-term cycling. Finally, XPS analysis showed that confinement also inhibited the transition of the nickel to higher oxidation states. Fig. 3e shows a similar pre-cycled high resolution Ni2p XPS spectrum for NC-NiO@CNT to that of NiO/C and ID-NiO/CNT. Post-cycling (100 cycles), NC-NiO@CNT only showed the presence of NiF2 (from SEI formation) and NiO, which suggests that after repeated charge/discharge cycles, Ni2+ → Ni2+ retention was achieved, and that no measurable transformation to Ni3+ was observed (Fig. 3g). Therefore, the possible reaction pathways for a constrained material appear to be limited to the reversible electrochemical reaction pathways – which should allow for longer term operation.
To better understand the electrochemical behavior of the NC-NiO@CNT50, Fig. 4a and b shows charge/discharge and differential capacity analysis (dQ/dV) curves, respectively, during the 1st, 100th, 200th, 300th, and 400th cycle. The conversion reaction and the SEI formation is shown in the 1st cycle at a mixed potential of 0.350 V. The subsequent reduction plateau occurs at 1.15 V and the oxidation plateau at 2.15 V. Cycles 100, 200, 300, and 400 show minimal deviations in both the charge and discharge voltage plateaus/peaks, which indicates excellent reaction reversibility and active material utilization over time. Subsequent cycles result in a decoupled reduction plateau that is characteristic of the reaction from eqn (1). It is noted that the CNT is a non-dilutive conductive carbon additive that also allows for some Li ion intercalation, which may contribute to a little to the total capacity. During the oxidation, a Li de-intercalation peak was observed at 0.2 V and a metallic Ni reconversion back to NiO at 1.5 V and 2.15 V.
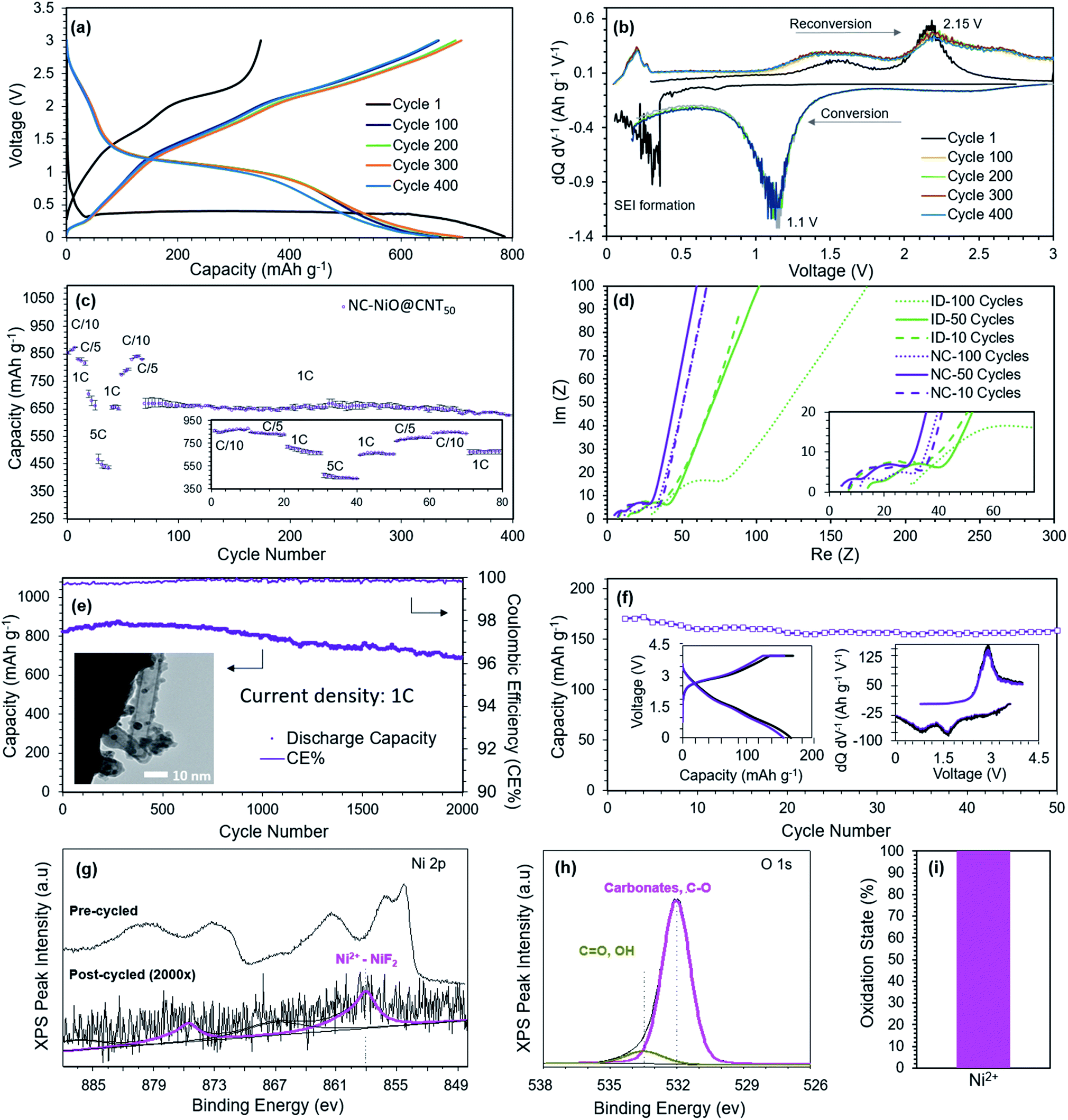 | ||
| Fig. 4 Electrochemical performance (a) the charge-discharge curves and (b) differential capacity analysis (dQ/dV) of NC-NiO@CNT50. (c) Rate capability evaluation for charge reversibility and rate-specific side reactions at different current densities for NC-NiO@CNT50. (d) EIS spectra after 10, 50 and 100 charge/discharge cycles for ID-NiO/CNT and NC-NiO@CNT50, showing the evolution of chemical and physical processes (e.g. diffusion, charge transfer, electrolyte conductivity, SEI resistance) during cycling. (e) Capacity retention over 2000 cycles for NC-NiO@CNT10 at 1C (718 mA h g−1). (f) Capacity retention, charge-discharge curves, and differential capacity analysis for high loading full cells made from NC-NiO@CNT10 anodes and Li(Ni0.5Mn0.3Co0.2)O2 cathodes (anode loading: 5.16 mg cm−2; cathode loading: 18.23 mg cm−2) full cell. XPS spectra for NC-NiO@CNT10 before and after cycling including (g) Ni2P, (h) O1s, and (i) the percentage of NiO remaining in the +2 oxidation post-cycling. | ||
Electrodes made of well-embedded MOs in conductive carbon matrices will exhibit less contact resistance at the MO/Carbon interface. Therefore, it might also be possible that these confined, nano-sized MOs will exhibit enhanced reaction kinetics and rapid diffusion of Li ions compared to other structures. This was explored through experiments at several charge/discharge rates. Fig. 4c shows the rate capability of NC-NiO@CNT50 and shows a capacity of 460 mA h g−1 at 5C (refer to Fig. S4 in the ESI† file for 1000 stable cycles at 5C). Even after alternating among various C-rates (based on NiO), the cell capacity retention after 400 cycles was at 627 mA h g−1 at 1C, which was 94% of the original capacity. EIS measurements (Fig. 4d) can be used to explain the evolution of the physical and chemical processes because each of the characteristic phenomena (diffusion, reaction, etc.) have their own time constants and relaxation times, which hence can be probed at different frequencies. It was observed that unlike ID-NiO/CNT, where the evolution of the Rohmic increased over 100 cycles, the nano-confined NC-NiO@CNT50 showed a negligible increase in charge transport impedance. NC-NiO@CNT50 also had a lower charge transfer resistance than ID-NiO/CNT and the charge transfer resistance was unaffected by cycling.
The combination of cycling performance, pre-/post-cycled TEM/XPS, and EIS analysis suggest that the degree of confinement and isolation from the bulk electrolyte might play a pivotal role in concomitantly increasing the MO reaction reversibility and minimizing the size reactions responsible for the change in Ni oxidation state and long-term SEI growth (refer to Fig. S8 in the ESI† file for pre/post-cycled XRD). Therefore, a version of the NC-NiO/CNTx anode was synthesized with the specific goal of providing even less void space (i.e. less volume for excess electrolyte exposure) and more rapid Li+ diffusion. This was accomplished by using CNTs with a small diameter, 10 nm versus 50 nm. The total active loading of NiO was determined to be 89.9% through thermogravimetric analysis (TGA) (Fig. S7 in the ESI† file). Fig. 4e shows the capacity retention of NC-NiO@CNT10 over 2000 charge/discharge cycles at 1C. It was found that this material was highly reversible, showing an achievable capacity 686 mA h g−1 after 2000 cycles (initial capacity = 824 mA h g−1, retention = 83.3%). Also, a coulombic efficiency greater than 99.9% was measured (to be more quantitative would require high precision coulometry measurements, which is beyond the scope of this study). Fig. 4f shows the capacity and capacity retention for full cells that paired NC-NiO@CNT10 anodes and Li(Ni0.5Mn0.3Co0.2)O2 cathodes at a C/2 rate. The anode and cathode active material loading were 5.16 mg cm−2 and 18.23 mg cm−2, respectively, resulting in a N/P ratio of 1.13. To be consistent with the literature, the full cell capacity is reported based on the cathode capacity. The achievable cell capacity was very good during the 50-cycle experiment, with a stable reversible capacity of 159 mA h g−1 (based on the total cathode mass – suggesting that nearly all of the cathode is fully utilized during cycling). By deconvoluting the charge–discharge curves into differential capacity plots (dQ/dV), inset of Fig. 4f, the full metal oxide cell displays a charge peak at 2.9 V and 2 discharge peaks at 1.7 V and 0.9 V, which correspond well with the half-cell peaks at ∼1.5 V and 2.15 V. Additionally, Fig. 4g–i shows the pre- and post-cycle XPS spectra after 2000 cycles for the highly-constrained NC-NiO@CNT10 in half cells. In short, it was found that all of the nickel after 2000 cycles was in the +2 oxidation state, suggesting that the confined MO in NC-NiO@CNT50 is highly stable, highly reversible, and capable of enabling full metal oxide Li ion cells.
3.3 Non-confined vs. confined MO nanoparticles
As discussed above, the main descriptor for whether high capacity and capacity retention was achieved was whether or not the MO was exposed to (NiO/C and ID-NiO/CNT) or isolated from (NC-NiO@CNTx) the electrolyte. As summarized in Fig. 5, MOs undergo four types of physical processes that influence their behavior in operating LIBs: uncontrolled growth of the SEI, transition to higher oxidation states, metal agglomeration, and metal trapping. These processes directly impact the coulombic efficiency, capacity retention, and onset of degradation. Randomly dispersed MOs on conductive carbon (whether it is carbon black or advanced carbon) are susceptible to continuous uncontrolled growth of the SEI, and this phenomenon is primarily found to be caused by the repetitive expansion/contraction of MOs during phase separation/recombination that exposes the highly reactive surfaces of the MO nanoparticles. Without a proper barrier, the SEI grows onto the highly-exposed surface and uncontrollably dislodges particles from the conductive medium in the electrode. These electronically isolated particles become electrochemically deactivated, and with the loss of active material, results in capacity fade. In addition, the destabilized oxygen balance and local overpotentials due to elevated internal resistance, drives the evolution to higher oxidation states. The reactions that guide the transformation of the MO to higher oxidation states do not consume Li, but they do consume oxygen in the system and change the number of electrons transferred per metal atom. This results in lower coulombic efficiencies, but does not result in capacity loss. The SEI reactions and the reactions associated with coulombic efficiency loss in operating cells are summarized in Table 1.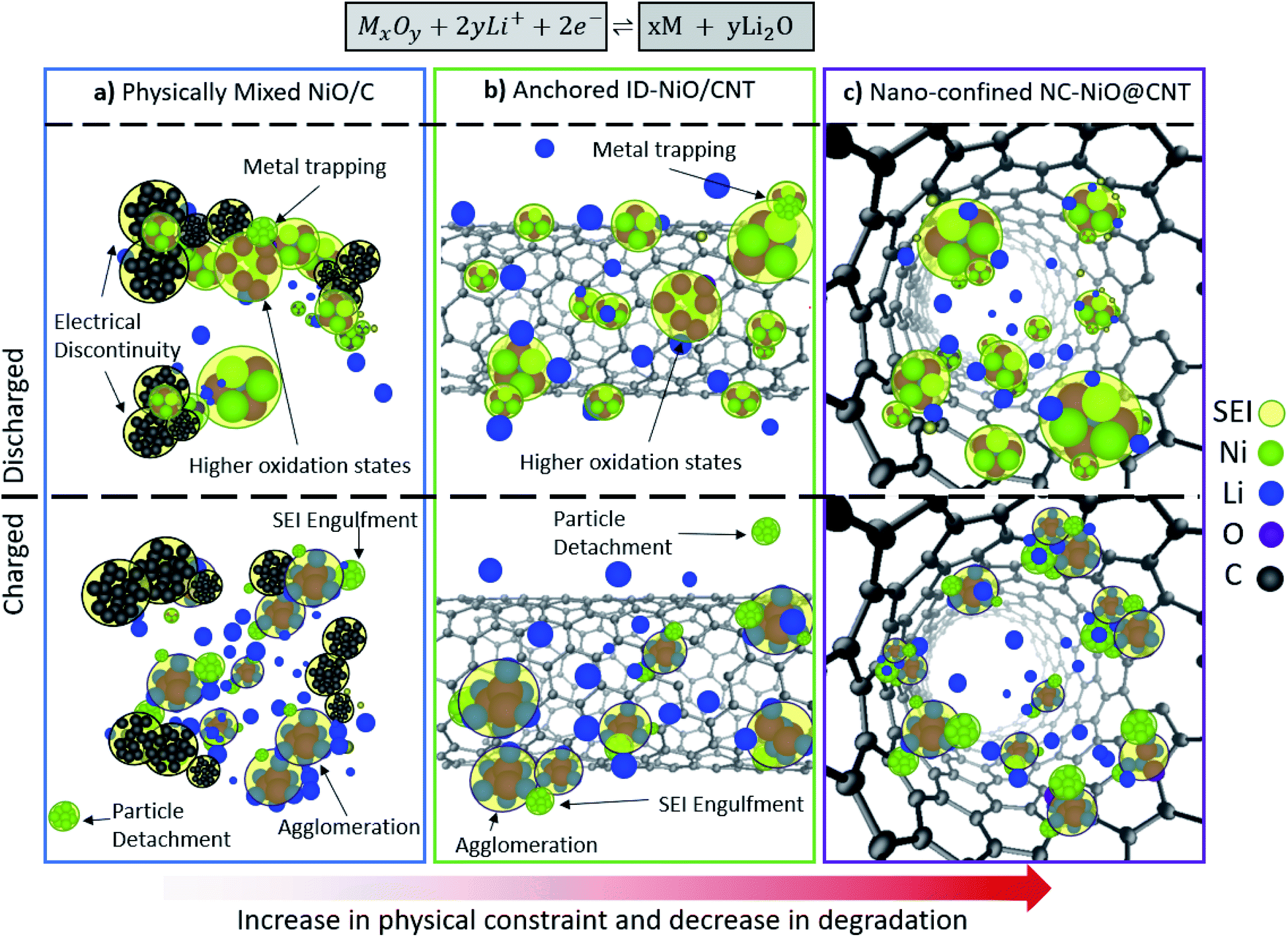 | ||
| Fig. 5 Phase separation and degradation pathway. Identifying the degradation pathways in conversion MOs (e.g. M/MO agglomeration, metal trapping, electrical detachment, SEI engulfment of MO, particle delamination) caused by fluctuations in the local e−, Li+, oxygen, and metal balance during phase separation for (a) physically mixed NiO/C, (b) anchored ID-NiO/CNT, and (c) nano-confined NC-NiO@CNT. | ||
| Case study | Pre-cycled | Post-cycle | Possible reaction pathway | Results |
|---|---|---|---|---|
| Control (NiO/C) 100 cycles | 100% Ni2+ | 48% Ni2+ | 2LiF + Ni ⇌ NiF2 + 2Li+ + 2e− (SEI) | Resulted in the formation of higher oxidation states (HOS) +3 oxidation state |
| 52% Ni3+ | 2NiO + 2RCH2OCOOLi ⇌ Ni2O3 + Li2O + RCH2COCOCH2R (HOS) | |||
| Indiscriminately deposited (ID-NiO/CNT) 100 cycles | 100% Ni2+ | 63% Ni2+ | 2LiF + Ni ⇌ NiF2 + 2Li+ + 2e− (SEI) | Similar results to control formation of higher oxidation states (HOS) and NiCyOzspecies |
| 37% Ni3+ | 2NiO + 2RCH2OCOOLi ⇌ Ni2O3 + Li2O + RCH2COCOCH2R (HOS), LixCyOz + Ni ⇌ NiCyOz + xLi+ + xe− (SEI) | |||
| Nanoconfined (NC-NiO@CNTx) 2000 cycles | 100% Ni2+ | 100% Ni2+ | 2LiF + Ni ⇌ NiF2 + 2Li+ + 2e− (SEI) | NiO transformed to NiF 2 and remained in the +2 oxidation state |
In comparison, well confined materials where the MO was mostly isolated from the electrolyte and confined inside of the CNT the number of size reactions was reduced, the SEI was physically restricted in its growth and the MO particles were always contained within a highly conducting matrix. Also, the Ni atoms in the discharged state were in the +2 oxidation state. NiO was extremely stable over a large number of cycles and high coulombic efficiency was achieved. Therefore, it appears that constraint-based synthesis routes are able to minimize the degradation pathways associated MOs and such nano-confinement represents a very promising strategy to increase the adoption and practicality of MO-based anodes for Li-ion batteries.
4. Conclusion
In operating Li-ion batteries, MO-based anode materials undergo four primary degradation pathways that limit their achievable capacity, coulombic efficiency and long-term stability. When the MO particles are fully exposed to the electrolyte in networks with insufficient electronic conductivity, all of the degradation pathways are active and the MO anode is able to undergo rapid material degradation. However, this study shows that nano-confinement of MO nanoparticles can significantly reduce the rate of some degradation mechanisms and completely eliminate others – extending the achievable lifetime of MO anodes significantly. In fact, the top-performing material in this study, NC-NiO@CNT10, was able to cycle for more than 2000 deep charge/discharge cycles at 1C while retaining >80% of its initial capacity (824 mA h g−1) with a coulombic efficiency >99.9%. Nanoconfined NiO was also able to support a very high capacity of 460 mA h g−1 at 5C, a rate that is very relevant for fast-charging and high-power applications. In addition, high loading full metal oxide Li-ion cells (prelithiated NC-NiO@CNT10/Li (Ni(Ni0.5Mn0.3Co0.2)O2) were assembled and tested. The MO/LMO full cell confirmed the hypothesis that confinement inhibits anode degradation (i.e. >99.9 coulombic efficiency = less side reactions) and the long-range conductive CNT networks can help electrons percolate through thick high loading electrodes (i.e. boost material utilization).Finally, the discovery that nanoconfinement can improve the reaction reversibility and longevity of conversion metal oxides can likely also be applied to other conversion-based chemistries as well (e.g. metal hydrides, nitrides, oxides, fluorides, phosphides, and sulfides) whose reaction can be generalized below:
| Mαμ+Xβπ− + βπ(Li+ + e−) ⇌ αM + βLiπX | (3) |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded by the Ford Motor Company through the University Research Program (URP). The authors would like to specifically thank Bob Kudla and Andy Drews at Ford and Stavros G. Karakalos at the University of South Carolina XPS facility for their guidance and help throughout this work.References
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Electrical energy storage for transportation – Approaching the limits of, and going beyond, lithium-ion batteries, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- M. A. Hannan, M. S. H. Lipu, P. J. Ker, R. A. Begum, V. G. Agelidis and F. Blaabjerg, Power electronics contribution to renewable energy conversion addressing emission reduction: Applications, issues, and recommendations, Appl. Energy, 2019, 251, 113404 CrossRef.
- G. L. Zhu, C. Z. Zhao, J. Q. Huang, C. He, J. Zhang, S. Chen, L. Xu, H. Yuan and Q. Zhang, Fast Charging Lithium Batteries: Recent Progress and Future Prospects, Small, 2019, 15, 1–14 Search PubMed.
- C. P. Grey and J. M. Tarascon, Sustainability and in situ monitoring in battery development, Nat. Mater., 2016, 16, 45–56 CrossRef CAS PubMed.
- T. Kim, W. Song, D. Y. Son, L. K. Ono and Y. Qi, Lithium-ion batteries: outlook on present, future, and hybridized technologies, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Materials for lithium-ion battery safety, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- X. Lin, M. Salari, L. M. R. Arava, P. M. Ajayan and M. W. Grinstaff, High temperature electrical energy storage: Advances, challenges, and frontiers, Chem. Soc. Rev., 2016, 45, 5848–5887 RSC.
- S. Gao, L. Lu, M. Ouyang, Y. Duan, X. Zhu, C. Xu, B. Ng, N. Kamyab, R. E. White and P. T. Coman, Experimental Study on Module-to-Module Thermal Runaway-Propagation in a Battery Pack, J. Electrochem. Soc., 2019, 166, A2065–A2073 CrossRef CAS.
- A. Palmieri, S. Yazdani, R. Kashfi-Sadabad, S. G. Karakalos, B. Ng, A. Oliveira, X. Peng, M. T. Pettes and W. E. Mustain, Improved Capacity Retention of Metal Oxide Anodes in Li-Ion Batteries: Increasing Intraparticle Electronic Conductivity through Na Inclusion in Mn3O4, ChemElectroChem, 2018, 5, 2059–2063 CrossRef CAS.
- S. Tippmann, D. Walper, L. Balboa, B. Spier and W. G. Bessler, Low-temperature charging of lithium-ion cells part I: Electrochemical modeling and experimental investigation of degradation behavior, J. Power Sources, 2014, 252, 305–316 CrossRef CAS.
- N. Kim, S. Chae, J. Ma, M. Ko and J. Cho, Fast-charging high-energy lithium-ion batteries via implantation of amorphous silicon nanolayer in edge-plane activated graphite anodes, Nat. Commun., 2017, 8, 812 CrossRef PubMed.
- Q. Li, G. Zhu, Y. Zhao, K. Pei and R. Che, NixMnyCozO Nanowire/CNT Composite Microspheres with 3D Interconnected Conductive Network Structure via Spray-Drying Method: A High-Capacity and Long-Cycle-Life Anode Material for Lithium-Ion Batteries, Small, 2019, 15, 1–12 Search PubMed.
- H. Huang, X. Wang, E. Tervoort, G. Zeng, T. Liu, X. Chen, A. Sologubenko and M. Niederberger, Nano-Sized Structurally Disordered Metal Oxide Composite Aerogels as High-Power Anodes in Hybrid Supercapacitors, ACS Nano, 2018, 12, 2753–2763 CrossRef CAS PubMed.
- N. S. Spinner, A. Palmieri, N. Beauregard, L. Zhang, J. Campanella and W. E. Mustain, Influence of conductivity on the capacity retention of NiO anodes in Li-ion batteries, J. Power Sources, 2015, 276, 46–53 CrossRef CAS.
- Y. Dong, B. Wang, K. Zhao, Y. Yu, X. Wang, L. Mai and S. Jin, Air-Stable Porous Fe2N Encapsulated in Carbon Microboxes with High Volumetric Lithium Storage Capacity and a Long Cycle Life, Nano Lett., 2017, 17, 5740–5746 CrossRef CAS PubMed.
- H. Yuan, M. Wu, J. Zheng, Z. G. Chen, W. Zhang, J. Luo, C. Jin, O. Sheng, C. Liang, Y. Gan, Y. Xia, J. Zhang, H. Huang, Y. Liu, J. Nai and X. Tao, Empowering Metal Phosphides Anode with Catalytic Attribute toward Superior Cyclability for Lithium-Ion Storage, Adv. Funct. Mater., 2019, 29, 1–8 Search PubMed.
- Z. Liu, S. Yang, B. Sun, X. Chang, J. Zheng and X. Li, A Peapod-like CoP@C Nanostructure from Phosphorization in a Low-Temperature Molten Salt for High-Performance Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2018, 57, 10187–10191 CrossRef CAS PubMed.
- L. Li, R. Jacobs, P. Gao, L. Gan, F. Wang, D. Morgan and S. Jin, Origins of Large Voltage Hysteresis in High-Energy-Density Metal Fluoride Lithium-Ion Battery Conversion Electrodes, J. Am. Chem. Soc., 2016, 138, 2838–2848 CrossRef CAS PubMed.
- C. Chen, X. Xie, B. Anasori, A. Sarycheva, T. Makaryan, M. Zhao, P. Urbankowski, L. Miao, J. Jiang and Y. Gogotsi, MoS2-on-MXene Heterostructures as Highly Reversible Anode Materials for Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2018, 57, 1846–1850 CrossRef CAS PubMed.
- J. Zhan, K. Wu, X. Yu, M. Yang, X. Cao, B. Lei, D. Pan, H. Jiang and M. Wu, α-Fe2O3 Nanoparticles Decorated C@MoS2 Nanosheet Arrays with Expanded Spacing of (002) Plane for Ultrafast and High Li/Na-Ion Storage, Small, 2019, 15, 1–10 Search PubMed.
- A. Palmieri, N. Spinner, S. Zhao and W. E. Mustain, Explaining the role and mechanism of carbon matrices in enhancing reaction reversibility of metal oxide anodes for high performance Li ion batteries, Carbon, 2018, 130, 515–524 CrossRef CAS.
- Z. Li, H. Zhao, P. Lv, Z. Zhang, Y. Zhang, Z. Du, Y. Teng, L. Zhao and Z. Zhu, Watermelon-Like Structured SiOx–TiO2 @C Nanocomposite as a High-Performance Lithium-Ion Battery Anode, Adv. Funct. Mater., 2018, 28, 1–11 Search PubMed.
- J. Luo, B. Ma, J. Peng, Z. Wu, Z. Luo and X. Wang, Modified Chestnut-Like Structure Silicon Carbon Composite as Anode Material for Lithium-Ion Batteries, ACS Sustainable Chem. Eng., 2019, 7, 10415–10424 CrossRef CAS.
- P. Li, J. Y. Hwang and Y. K. Sun, Nano/microstructured silicon-graphite composite anode for high-energy-density Li-ion battery, ACS Nano, 2019, 13, 2624–2633 CAS.
- X. Cai, W. Liu, S. Yang, S. Zhang, Q. Gao, X. Yu, J. Li, H. Wang and Y. Fang, Dual-Confined SiO Embedded in TiO2 Shell and 3D Carbon Nanofiber Web as Stable Anode Material for Superior Lithium Storage, Adv. Mater. Interfaces, 2019, 6, 1–11 Search PubMed.
- W. Wang, B. Jiang, W. Xiong, H. Sun, Z. Lin, L. Hu, J. Tu, J. Hou, H. Zhu and S. Jiao, A new cathode material for super-valent battery based on aluminium ion intercalation and deintercalation, Sci. Rep., 2013, 3, 3383 CrossRef PubMed.
- N. Liu, Z. Lu, J. Zhao, M. T. Mcdowell, H. W. Lee, W. Zhao and Y. Cui, A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. Mcdowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Stable cycling of double-walled silicon nanotube battery anodes through solid-electrolyte interphase control, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- M. Zheng, H. Tang, L. Li, Q. Hu, L. Zhang, H. Xue and H. Pang, Hierarchically Nanostructured Transition Metal Oxides for Lithium-Ion Batteries, Adv. Sci., 2018, 5, 1700592 CrossRef PubMed.
- J. Zhou, H. Song, X. Chen, L. Zhi, S. Yang, J. Huo and W. Yang, Carbon-encapsulated metal oxide hollow nanoparticles and metal oxide hollow nanoparticles: A general synthesis strategy and its application to lithium-ion batteries, Chem. Mater., 2009, 21, 2935–2940 CrossRef CAS.
- J. Ming, J. B. Park and Y. K. Sun, Encapsulation of metal oxide nanocrystals into porous carbon with ultrahigh performances in lithium-ion battery, ACS Appl. Mater. Interfaces, 2013, 5, 2133–2136 CrossRef CAS PubMed.
- L. Luo, J. Wu, J. Xu and V. P. Dravid, Atomic resolution study of reversible conversion reaction in metal oxide electrodes for lithium-ion battery, ACS Nano, 2014, 8, 11560–11566 CrossRef CAS PubMed.
- K. Karki, L. Wu, Y. Ma, M. J. Armstrong, J. D. Holmes, S. H. Garofalini, Y. Zhu, E. A. Stach and F. Wang, Revisiting Conversion Reaction Mechanisms in Lithium Batteries: Lithiation-Driven Topotactic Transformation in FeF2, J. Am. Chem. Soc., 2018, 140, 17915–17922 CrossRef CAS PubMed.
- G. Evmenenko, R. E. Warburton, H. Yildirim, J. P. Greeley, M. K. Y. Chan, D. B. Buchholz, P. Fenter, M. J. Bedzyk and T. T. Fister, Understanding the Role of Overpotentials in Lithium Ion Conversion Reactions: Visualizing the Interface, ACS Nano, 2019, 13, 7825–7832 CrossRef CAS PubMed.
- S. H. Choi, J. H. Lee and Y. C. Kang, Perforated Metal Oxide-Carbon Nanotube Composite Microspheres with Enhanced Lithium-Ion Storage Properties, ACS Nano, 2015, 9, 10173–10185 CrossRef CAS PubMed.
- J. Sun, C. Lv, F. Lv, S. Chen, D. Li, Z. Guo, W. Han, D. Yang and S. Guo, Tuning the Shell Number of Multishelled Metal Oxide Hollow Fibers for Optimized Lithium-Ion Storage, ACS Nano, 2017, 11, 6186–6193 CrossRef CAS PubMed.
- N. Du, H. Zhang, J. Chen, J. Sun, B. Chen and D. Yang, Metal oxide and sulfide hollow spheres: Layer-by-layer synthesis and their application in lithium-ion battery, J. Phys. Chem. B, 2008, 112, 14836–14842 CrossRef CAS PubMed.
- S. Tao, W. Yue, M. Zhong, Z. Chen and Y. Ren, Fabrication of graphene-encapsulated porous carbon-metal oxide composites as anode materials for lithium-ion batteries, ACS Appl. Mater. Interfaces, 2014, 6, 6332–6339 CrossRef CAS PubMed.
- D. Lee, M. Wu, D. H. Kim, C. Chae, M. K. Cho, J. Y. Kim, S. S. Lee, S. Choi, Y. Choi, T. J. Shin, K. Y. Chung, S. Jeong and J. Moon, Understanding the Critical Role of the Ag Nanophase in Boosting the Initial Reversibility of Transition Metal Oxide Anodes for Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2017, 9, 21715–21722 CrossRef CAS PubMed.
- S. Müller, P. Pietsch, B. E. Brandt, P. Baade, J. Eller, M. Ebner, C. Burns, D. Westhoff, J. Feinauer, V. De Andrade, F. Marone, M. Stampanoni, J. Dahn, V. Schmidt, F. De Carlo and V. Wood, Nanoscale imaging, quantification, and modeling of mechanical degradation in lithium-ion batteries, Chem. Mater. Lead-Based Batter. Symp. 2018, Held AABC Eur. 2018, 2018, vol. 9, pp. 389–390 Search PubMed.
- T. Nordh, R. Younesi, M. Hahlin, R. F. Duarte, C. Tengstedt, D. Brandell and K. Edström, Manganese in the SEI Layer of Li4Ti5O12 Studied by Combined NEXAFS and HAXPES Techniques, J. Phys. Chem. C, 2016, 120, 3206–3213 CrossRef CAS.
- O. C. Harris and M. H. Tang, Molecular Probes Reveal Chemical Selectivity of the Solid-Electrolyte Interphase, J. Phys. Chem. C, 2018, 122, 20632–20641 CrossRef CAS.
- J. A. Gilbert, J. Bareño, T. Spila, S. E. Trask, D. J. Miller, B. J. Polzin, A. N. Jansen and D. P. Abraham, Cycling behavior of NCM523/Graphite lithium-ion cells in the 3-4.4 V range: Diagnostic studies of full cells and harvested electrodes, J. Electrochem. Soc., 2017, 164, A6054–A6065 CrossRef CAS.
- J. Deng, X. Yu, X. Qin, D. Zhou, L. Zhang, H. Duan, F. Kang, B. Li and G. Wang, Co–B Nanoflakes as Multifunctional Bridges in ZnCo2O4 Micro-/Nanospheres for Superior Lithium Storage with Boosted Kinetics and Stability, Adv. Energy Mater., 2019, 9, 1–11 Search PubMed.
- S. Yan, H. Song, S. Lin, H. Wu, Y. Shi and J. Yao, GeO2 Encapsulated Ge Nanostructure with Enhanced Lithium-Storage Properties, Adv. Funct. Mater., 2019, 29, 1–7 Search PubMed.
- J. Hao, J. Zhang, G. Xia, Y. Liu, Y. Zheng, W. Zhang, Y. Tang, W. K. Pang and Z. Guo, Heterostructure Manipulation via in Situ Localized Phase Transformation for High-Rate and Highly Durable Lithium Ion Storage, ACS Nano, 2018, 12, 10430–10438 CrossRef CAS PubMed.
- L. Ma, T. Chen, G. Zhu, Y. Hu, H. Lu, R. Chen, J. Liang, Z. Tie, Z. Jin and J. Liu, Pitaya-like microspheres derived from Prussian blue analogues as ultralong-life anodes for lithium storage, J. Mater. Chem. A, 2016, 4, 15041–15048 RSC.
- G. Zhu, L. Wang, H. Lin, L. Ma, P. Zhao, Y. Hu, T. Chen, R. Chen, Y. Wang, Z. Tie, J. Liu and Z. Jin, Walnut-Like Multicore-Shell MnO Encapsulated Nitrogen-Rich Carbon Nanocapsules as Anode Material for Long-Cycling and Soft-Packed Lithium-Ion Batteries, Adv. Funct. Mater., 2018, 28, 1800003 CrossRef.
- Z. Liu, X. Y. Yu and U. Paik, Etching-in-a-Box: A Novel Strategy to Synthesize Unique Yolk-Shelled Fe3O4@Carbon with an Ultralong Cycling Life for Lithium Storage, Adv. Energy Mater., 2016, 6, 3–7 Search PubMed.
- Y. Lu, J. Nai and X. W. D. Lou, Formation of NiCo2V2O8 Yolk–Double Shell Spheres with Enhanced Lithium Storage Properties, Angew. Chem., Int. Ed., 2018, 57, 2899–2903 CrossRef CAS PubMed.
- J. Zhang, J. Wan, J. Wang, H. Ren, R. Yu, L. Gu, Y. Liu, S. Feng and D. Wang, Hollow Multi-Shelled Structure with Metal–Organic-Framework-Derived Coatings for Enhanced Lithium Storage, Angew. Chem., Int. Ed., 2019, 58, 5266–5271 CrossRef CAS PubMed.
- Q. Xie, P. Liu, D. Zeng, W. Xu, L. Wang, Z. Z. Zhu, L. Mai and D. L. Peng, Dual Electrostatic Assembly of Graphene Encapsulated Nanosheet-Assembled ZnO–Mn–C Hollow Microspheres as a Lithium Ion Battery Anode, Adv. Funct. Mater., 2018, 28, 1–11 Search PubMed.
- T. Chen, Y. Hu, B. Cheng, R. Chen, H. Lv, L. Ma, G. Zhu, Y. Wang, C. Yan, Z. Tie, Z. Jin and J. Liu, Multi-yolk-shell copper oxide@carbon octahedra as high-stability anodes for lithium-ion batteries, Nano Energy, 2016, 20, 305–314 CrossRef CAS.
- J. Yu, Y. Wang, L. Mou, D. Fang, S. Chen and S. Zhang, Nature-Inspired 2D-Mosaic 3D-Gradient Mesoporous Framework: Bimetal Oxide Dual-Composite Strategy toward Ultrastable and High-Capacity Lithium Storage, ACS Nano, 2018, 12, 2035–2047 CrossRef CAS PubMed.
- S. Yun, S. M. Bak, S. Kim, J. S. Yeon, M. G. Kim, X. Q. Yang, P. V. Braun and H. S. Park, Rational Design of Hierarchically Open-Porous Spherical Hybrid Architectures for Lithium-Ion Batteries, Adv. Energy Mater., 2019, 9, 1–9 CAS.
- P. Xiong, R. Ma, N. Sakai and T. Sasaki, Genuine Unilamellar Metal Oxide Nanosheets Confined in a Superlattice-like Structure for Superior Energy Storage, ACS Nano, 2018, 12, 1768–1777 CrossRef CAS PubMed.
- H. Higuchi, K. Uenae and A. Kawakami, Low-crystallized carbon materials for lithium-ion secondary batteries, J. Power Sources, 1997, 68, 212–215 CrossRef CAS.
- M. D. Fang, T. H. Ho, J. P. Yen, Y. R. Lin, J. L. Hong, S. H. Wu and J. J. Jow, Preparation of advanced carbon anode materials from mesocarbon microbeads for use in high C-rate lithium ion batteries, Materials, 2015, 8, 3550–3561 CrossRef CAS.
- S. Shrestha and W. E. Mustain, Properties of nitrogen-functionalized ordered mesoporous carbon prepared using polypyrrole precursor, J. Electrochem. Soc., 2010, 157, B1665 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- N. Spinner, L. Zhang and W. E. Mustain, Investigation of metal oxide anode degradation in lithium-ion batteries via identical-location TEM, J. Mater. Chem. A, 2014, 2, 1627–1630 RSC.
- Z. Lin, C. Du, B. Yan, C. Wang and G. Yang, Two-dimensional amorphous NiO as a plasmonic photocatalyst for solar H2 evolution, Nat. Commun., 2018, 9, 4036 CrossRef PubMed.
- Y. M. Chen, X. Y. Yu, Z. Li, U. Paik and X. W. Lou, Hierarchical MoS2 tubular structures internally wired by carbon nanotubes as a highly stable anode material for lithium-ion batteries, Sci. Adv., 2016, 2, e1600021 CrossRef PubMed.
- M. Xu, W. Li and B. L. Lucht, Effect of propane sultone on elevated temperature performance of anode and cathode materials in lithium-ion batteries, J. Power Sources, 2009, 193, 804–809 CrossRef CAS.
- Y. Yu, L. Gu, C. Wang, A. Dhanabalan, P. A. Van Aken and J. Maier, Encapsulation of Sn@carbon nanoparticles in bamboo-like hollow carbon nanofibers as an anode material in lithium-based batteries, Angew. Chem., Int. Ed., 2009, 48, 6485–6489 CrossRef CAS PubMed.
- A. Ottmann, M. Scholz, M. Haft, E. Thauer, P. Schneider, M. Gellesch, C. Nowka, S. Wurmehl, S. Hampel and R. Klingeler, Electrochemical Magnetization Switching and Energy Storage in Manganese Oxide filled Carbon Nanotubes, Sci. Rep., 2017, 7, 13625 CrossRef PubMed.
- R. Chen, X. Xue, Y. Hu, W. Kong, H. Lin, T. Chen and Z. Jin, Intermetallic SnSb nanodots embedded in carbon nanotubes reinforced nanofabric electrodes with high reversibility and rate capability for flexible Li-ion batteries, Nanoscale, 2019, 11, 13282–13288 RSC.
- P. Sun, X. Zhao, R. Chen, T. Chen, L. Ma, Q. Fan, H. Lu, Y. Hu, Z. Tie, Z. Jin, Q. Xu and J. Liu, Li3V2(PO4)3 encapsulated flexible free-standing nanofabric cathodes for fast charging and long life-cycle lithium-ion batteries, Nanoscale, 2016, 8, 7408–7415 RSC.
- Y. Wang, Z. Wang, Y. Chen, H. Zhang, M. Yousaf, H. Wu, M. Zou, A. Cao and R. P. S. Han, Hyperporous Sponge Interconnected by Hierarchical Carbon Nanotubes as a High-Performance Potassium-Ion Battery Anode, Adv. Mater., 2018, 30, 1–10 CAS.
- Z. Wu, Y. Wang, X. Liu, C. Lv, Y. Li, D. Wei and Z. Liu, Carbon-Nanomaterial-Based Flexible Batteries for Wearable Electronics, Adv. Mater., 2019, 31, 1–25 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta11708c |
| This journal is © The Royal Society of Chemistry 2020 |