
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersible porous classical polymer photocatalysts for visible light-mediated production of pharmaceutically relevant compounds in multiple solvents†
Calum T. J.
Ferguson
a,
Niklas
Huber
a,
Thomas
Kuckhoff
a,
Kai A. I.
Zhang
 *ab and
Katharina
Landfester
*a
*ab and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de
bDepartment of Materials Science, Fudan University, 200433 Shanghai, P. R. China. E-mail: kai_zhang@fudan.edu.cn.
First published on 12th December 2019
Abstract
The dispersibility of photocatalytic polymeric materials is currently a limiting factor for wide scale use. Indeed, the dispersibility of photocatalytic material in solvents dictates its use as heterogenous catalysts, due in part to reagent solubility. Here, we describe a new platform for producing porous polymer photocatalytic nanoparticles that are easily dispersible in a broad range of solvents. Photocatalytic porous classical polymer nanoparticles have been designed, based on classical porous nanoparticles copolymerised with photocatalytic moieties. The combination of both classical and photocatalytic monomers results in an affordable highly photocatalytically effective material, that can be easily dispersed in a wide range of solvents. The versatility and useability of this new material has been demonstrated by photocatalysing a number of pharmaceutically relevant compounds in solvents with varying polarity.
The employment of heterogeneous polymer photocatalysts under visible light irradiation is of high research interest with applications in energy, environmental, and biological systems. These applications require a broad range of dispersing solvents due to the desired application and solubility. For example there is need for the use of “green” aqueous based solvents for water splitting,1–6 anti-bacterial treatment,7 photodynamic therapy,8–10 enzyme regeneration11,12 and waste water treatment.13–15 Conversely, organic systems are required for the majority of redox reactions such as: C–C bond formation,16–19 C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage,20,21 oxidative coupling of amines,22,23 and hydroxylations24 for example. However, the dispersibility of photocatalytic polymeric materials in a broad range of solvents is currently a limiting factor. Indeed, the dispersibility of the heterogeneous photocatalytic material in solvents dictates what it may be used for, due in part to reagent solubility. As a result, polymer photocatalysts are restricted to work in either an organic or aqueous continuous phase.
C bond cleavage,20,21 oxidative coupling of amines,22,23 and hydroxylations24 for example. However, the dispersibility of photocatalytic polymeric materials in a broad range of solvents is currently a limiting factor. Indeed, the dispersibility of the heterogeneous photocatalytic material in solvents dictates what it may be used for, due in part to reagent solubility. As a result, polymer photocatalysts are restricted to work in either an organic or aqueous continuous phase.
The study of colloidally stable materials in a range of solvents has been extensively studied in classical polymer chemistry. Careful selection of steric stabilisers can enable the dispersibility in one or more continuous phases. Notably, polyethylene glycol (PEG) based steric stabilisers have been utilised in to stabilise colloidal materials in a range of both aqueous and organic continuous phases.25–28 These polymeric materials tend to be non-porous, which, if used for catalytic purposes, would lead to a low efficiency due to their limited active surface. Recently, however, classical microporous organic polymeric materials have been developed which have high surface areas.29–31 This enhanced surface area may be utilised in conjunction with the recently developed organic small molecule photocatalytic monomers to produce a photocatalytically active porous classical polymeric material.32 Moreover, this new material may provide a platform to produce a new class of polymer photocatalysts that are colloidally stable in a wide range of solvents. Easy control of photocatalyst conjugation length, highest occupied molecular orbital (HOMO) and lowest occupied molecular orbital (LUMO) levels may also be engineered into these new materials by varying the copolymerised photocatalytic moiety.
Ideally, we would be able to construct a photocatalytic material that can be easily dispersed in a broad range of solvents providing versatility. Photocatalytic porous polymer particles stabilised by PEG chains extending from the surface may enable the dispersibility in a range of solvents. RAFT-mediated polymerisation induced self-assembly (PISA) may be utilised to ensure that the stabilising polymer chains extend from the surface which ensure the easy dispersibility of the photocatalytic material.29,33 Further, to enhance the potential of the photocatalytic material its surface area should be maximised. Porous classical polymer particles have previously been reported through the combination of divinyl benzene (DVB) and fumaronitrile (FN).29,34 This produces a highly crosslinked polymeric material with a microporous structure. Photocatalytic units with controllable donor acceptor units as well as conjugation length may be incorporated into this porous material.
Here, porous classical polymers containing copolymerised photocatalytic moieties are produced in the form of photocatalytic polymeric material, as illustrated in Fig. 1. These photocatalytic porous particles are easily dispersible in a broad range of solvents, ranging from water to toluene. As a result, the reactions solvent is no longer limited by the photocatalytic material and the optimum solvent for the reagents can be selected. The porosity and resulting high surface area of the designed material leads to a highly efficient photocatalytic material by maximising the photocatalytic units incorporated. We have demonstrated the ability of this material to catalyse a range of highly desired reactions providing functionality that has importance in pharmaceutical molecules. Specifically, we have used this material to synthesise organosulfones by two different methods in both aqueous and organic conditions. Moreover, nitrogen based benzimidazole derivatives have been synthesised.
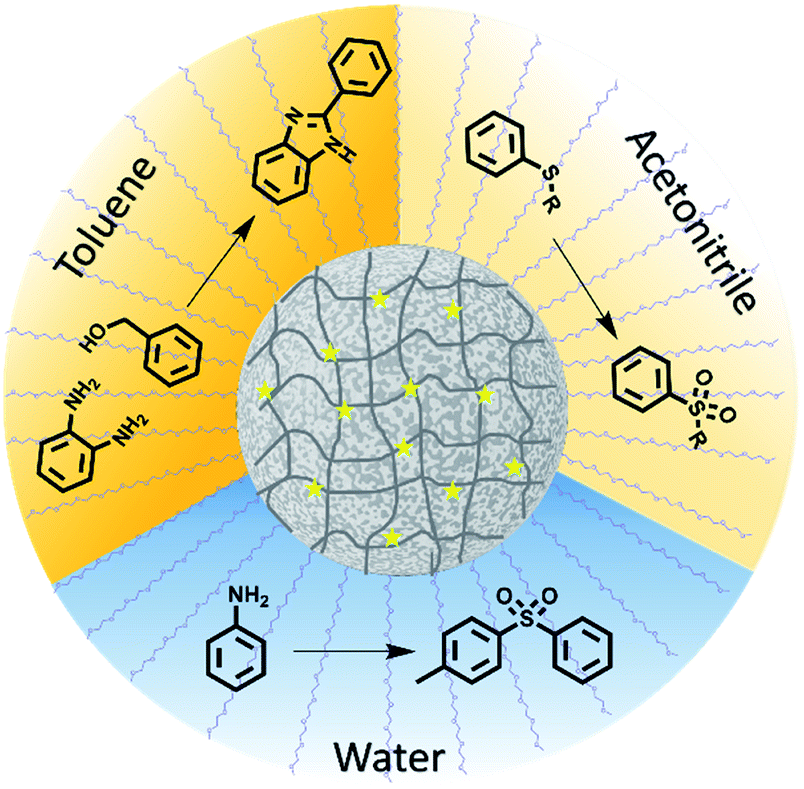 | ||
| Fig. 1 Porous polymer photocatalysts produced by combining classical polymer chemistry and conjugated polymer photocatalysts. The designed material is easily dispersible in a spectrum of solvents enabling a range of reactions to be catalysed. | ||
Photocatalytic porous classical polymers were synthesised using RAFT mediated PISA similarly to previously reported, where a PEG macro chain transfer agent was chain extended with DVB, FN and the photocatalytic crosslinker 4,7-bis(4-vinylphenyl)benzo[c][1,2,5]thiadiazole (Fig. 2a, S1 and S2).29 This creates a highly crosslinked particle core containing a small portion of photocatalytic units (0.33 mol%). This core is stabilised by easily solvated PEG chains that extend from the surface into the continuous phase. Transmission electron microscopy (TEM) was utilised to examine the morphology of the designed material (Fig. 2b). The aggregated particle morphology observed is not typically observed for particles produced by PISA; however, similar structures have previously been reported for hypercrosslinked systems.29,35 This morphology is attributed to the crosslinking of the small particles by DVB. The photocatalytic porous material could be easily dispersed in a spectrum of solvents with polarity ranging from 0.099 to 1 (water to toluene; Fig. S3†). The diameter of the photocatalytic material upon dispersion in water was initially centred around 1700 nm with a large polydispersity. Sonication of the sample lead to a large reduction in the average diameter to 375 nm (Fig. S4†).
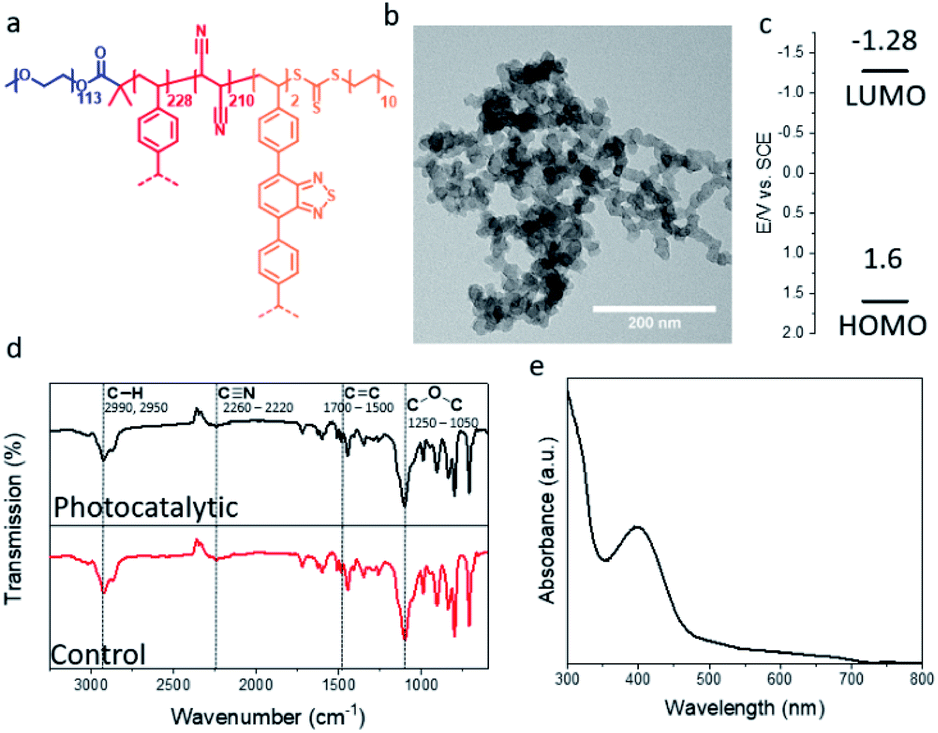 | ||
| Fig. 2 (a) Structure; (b) TEM image; (c) HOMO and LUMO levels; FTIR spectrum and UV/vis absorption spectrum of the photocatalytic porous polymer nanoparticles. | ||
The porosity of the material was investigated using nitrogen gas sorption at 77 K (Fig. S5†). The BET (Brunauer–Emmett–Teller) surface area of the photocatalytic was determined over a relative pressure range of 0.01–0.3 P/P0 (Fig. S5a† insert). The surface area of the photocatalytic material was found to be around 290 m2 g−1, comparable to previous reports of non-photocatalytic material (Fig. S5a†).29 The pore size of the material was calculated using NLDFT pore size analysis, the major peak of the pore size distribution for the photocatalytic material was centred on 1.7 nm (Fig. S5b†). Along with pores in the microporous region, meso- and macrospores were also observed, which are due the highly aggregated morphology of the material (Fig. S5b†). The HOMO and LUMO of the photocatalytic unit were calculated to be 1.6 and −1.28 EV vs. SCE as previously reported (Fig. 2c).36
Solid state NMR spectroscopy was used to determine the composition of the polymer particles (Fig. S6†). As expected signals were detected for the quaternary aromatic carbon (145 ppm), the unreacted vinyl group (138 ppm), the aromatic C–H (128 ppm), the nitrile group (114 ppm), the PEG CH2 group (71 ppm) and the polymer backbone (42 & 30 ppm) (Fig. S6†). Fourier transform infrared (FTIR) spectroscopy characterisation was undertaken to determine the chemical composition of the porous polymeric particles with and without the photocatalytic moiety. The FTIR spectrum (Fig. 2d) showed typical signals at 2992 and 2950 cm−1, which are characteristic for the C–H bond stretch vibrations of the –CH3 and –CH2– groups, respectively. The signal at 2260–2220 cm−1 is attributed to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch vibration originating from the FN. Signals from 1700–1500 cm−1 are due to aromatic CC bending from the DVB and photocatalytic monomer. The signal at 1250–1050 cm−1 result from the C–O–C stretch in the PEG chains. The effect of the photocatalytic unit on the FTIR spectrum is limited. As the photocatalytic unit only constitutes around 0.33 mol% of the material its effect is easily masked. The UV/vis spectra of the photocatalytic polymer particles have a broad absorption peak in the visible light region up to 700 nm (Fig. 2e).
N stretch vibration originating from the FN. Signals from 1700–1500 cm−1 are due to aromatic CC bending from the DVB and photocatalytic monomer. The signal at 1250–1050 cm−1 result from the C–O–C stretch in the PEG chains. The effect of the photocatalytic unit on the FTIR spectrum is limited. As the photocatalytic unit only constitutes around 0.33 mol% of the material its effect is easily masked. The UV/vis spectra of the photocatalytic polymer particles have a broad absorption peak in the visible light region up to 700 nm (Fig. 2e).
The versatility of the newly designed porous classical polymer photocatalyst to synthesise pharmaceutically relevant compounds was investigated. Moreover, we have investigated the ability of this new material to synthesise compounds in three very different solvents water, acetonitrile and toluene. This range of solvents would insure reagent solubility. Furthermore, we would expect the lifetime of the photocatalytically produced radicals to differ as a result of solvent.37 Initially, we investigated the production of benzimidazole with functional groups which are of significant importance due to their prevalence in pharmaceutically and biologically active molecules.38,39 Typically, the synthesis of benzimidazole containing compounds requires harsh conditions in strongly acidic conditions with temperatures in excess of 200 °C.40 Alternatively, oxidation of benzimidazole intermediates has been undertaken, however, this method often requires a large excess of strong oxidants.41
2-Substituted benzimidazoles have been synthesised using TiO2 loaded with platinum as a photocatalyst in anaerobic conditions.38 Here, porous classical polymer photocatalysts have been utilised to produce benzimidazole containing compounds under ambient conditions using visible light in air in various solvents. 2-Phenyl-1H-benzo[d]imidazole was synthesised in this study in water, acetonitrile and toluene with conversions of 96, 50 and 75% conversion respectively (Table 1 entry 1–3 and Fig S7–S9†). Control reactions in dark conditions and in the absence of photocatalyst did not result in measurable conversion (Table 1 entry 4 & 5, Fig S10 & S11†). Interestingly, when the experiment was repeated with the photocatalytic monomer only in aqueous conditions were the same conversions obtained. Poorer photocatalytic performance was observed in acetonitrile and a higher yield in toluene (Table 1 entry 12–14). Control experiments with scavengers showed that 1O2, ˙OH and h+ were integral for the photocatalytic reaction (Fig. S15†). The photocatalytic moiety concentration used in this study was substantially lower than previously reported and a much lower excess of the alcohol is required to form the benzimidazole.38 Interestingly, in previous studies the reaction was undertaken in acetonitrile, which was the poorest performing solvent in this study.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst | Light | Conversion |
| a Reaction conditions: 1,2 benzenediamine (50 mM), benzyl alcohol (200 mM), photocatalytic particles (2.5 mg mL−1 nanogel, 27 μg mL−1 photocatalytic units), 10 mL solvent, RT, 16 h, conversion determined by GC-MS. | ||||
| 1 | H2O | + | + | 96% |
| 2 | CH3CN | + | + | 50% |
| 3 | Tol | + | + | 16% |
| 4 | H2O | + | — | 0% |
| 5 | H2O | — | + | 0% |
| 6 | H2O | Monomer | + | 98% |
| 7 | CH3CN | Monomer | + | 10% |
| 8 | Tol | Monomer | + | 41% |
The synthesis of sulfonyl containing molecules is of importance due to the biological properties they produce.42,43 Furthermore, sulfonyls can be used as important reagents for a number of chemical transformations including Julia olefination,44,45 Ramberg–Bäcklund reaction46 and Smiles rearrangement.47 The photocatalytic synthesis of sulfonyl typically proceeds by the oxidation of a sulfide containing molecule. Sulfide oxidation has been catalysed by several different photocatalysts,48–50 where the monoxide product is formed in high yields. Conversely, Zhang et al.51 have reported the selective formation of the dioxide product using mesoporous graphitic carbon nitride and an a large excess of aldehyde oxidant. Initially, a high selectivity for the monoxide was observed with this product transforming into the dioxide over time.
Here, we have utilised the porous classical polymer photocatalyst to induce the sulfide oxidation of methyl phenyl sulfide and diphenyl sulfide in water, acetonitrile and toluene. For both reactions high conversion of over 99% was observed when using water and acetonitrile (Table 2 entry 1, 2, 5 & 6, Fig. S16, S17, S19 & S20†). Reactions undertaken in toluene had reduced conversion of less than 70% (Table 2 entry 3 & 7, Fig. S18 & 21†). Differences in the selectivity were observed between water and acetonitrile, where reactions in water favoured the formation of the monoxide whereas reactions in acetonitrile favoured the formation of the dioxide.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Cat. | Light | R | Conversion | Selectivity (b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :c) :c) |
| a Reaction conditions: methyl phenyl sulfide (50 mM), isobutylaldehyde (100 mM) photocatalytic particles (2.5 mg mL−1 nanogel, 27 μg mL−1 photocatalytic units), 10 mL solvent, RT, 18 h, conversion determined by GC-MS. b Reaction conditions: diphenyl sulfide (50 mM), photocatalytic particles (2.5 mg mL−1 nanogel, 27 μg mL−1 photocatalytic units), 10 mL solvent, RT, 20 h, conversion determined by GC-MS. | ||||||
| 1a | H2O | + | + | Me | >99% | 93:7 |
| 2a | CH3CN | + | + | Me | >99% | 40:60 |
| 3a | Tol | + | + | Me | 59% | 98:2 |
| 4b | H2O | + | + | Ph | >99% | 68:32 |
| 5b | CH3CN | + | + | Ph | >99% | 19:81 |
| 6b | Tol | + | + | Ph | 68% | 94:6 |
| 7a | CH3CN | + | — | Me | Trace | — |
| 9a | CH3CN | — | + | Me | 7% | 100:0 |
To test if this selectivity was due to a faster reaction in acetonitrile compared to water kinetic testing was undertaken. It was found that indeed the reaction rate is faster in acetonitrile and this results in a higher portion of the dioxide (Fig. S22†). Control reactions without photocatalyst or in the dark did not proceeded (Fig. S23 and S24†). Reactions in the presence of scavengers revealed that 1O2, h+, e− and ˙O2− are all integral for the sulfonyl formation (Fig. S25†). This method does, however, require the addition of isobutylaldehyde as an oxidant as previously reported.51
Recently, a new strategy for the photocatalytic production of dioxide sulfonyl through the reaction of diazonium salts with sulfonate salts has been reported. However, our attempts showed that the reaction proceeded without photocatalyst (Fig. S28†). The photocatalytic in situ formation of the diazonium moiety from aniline and its subsequent reaction has been reported.52 We have utilised our porous classical polymer photocatalysts to undertake similar reactions in a heterogeneous metal free nature. The reaction proceeded with the highest conversion in acetonitrile (84%; Table 3 entry 2 and S30†) with significantly lower conversions in water (42%; Table 3 entry 1 and S29†) and toluene (68%; Table 3 entry 3 and S31†). Scavenger experiments showed that 1O2, ˙OH, h+, e− and ˙O2− were required for the experiment (Fig. S32†). This synthetic route selectively produces the dioxide sulfonyl over the monoxide formed by sulfide oxidation. However, byproducts of the sulfonamide and the direct C–C coupling between phenyl rings was observed (Fig. S29–S31†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Light | Conversion | Selectivity |
| a Reaction conditions: sodium benzenesulfinate (75 mM), Analine (50 mM), photocatalytic particles (5 mg mL−1 nanogel, 54 μg mL−1 photocatalytic units), MsOH (20 mol%), t-BuONO (75 mM), 10 mL solvent, RT, 20 h, conversion determined by GC-MS. | |||||
| 1 | H2O | + | + | 42% | 50% |
| 2 | CH3CN | + | + | 84% | 75% |
| 3 | Tol | + | + | 68% | 32% |
| 4 | CH3CN | + | — | 0% | — |
| 5 | CH3CN | — | + | 10% | 100% |
Conclusions
In summary, we present here a new class of porous photocatalytic polymeric materials comprised mainly of inexpensive classical monomers. In addition, specifically designed divinyl photocatalytic monomers have been incorporated into the porous material to produce novel highly efficient photocatalysts. This material can easily be dispersed in a range of solvents with low to high polarity. The designed material has been utilised to catalyses pharmaceutically relevant reactions in water, acetonitrile and toluene. We have demonstrated that the reaction can be conducted in the optimum solvent for the synthesis, not based on the photocatalyst. This new material and synthetic route produces highly effective polymeric porous photocatalytic material in a range of solvents with greater efficiency.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
N. H. acknowledges the Kekulé fellowship of Fonds der chemischen Industrie (FCI) and the Gutenberg-Akademie of the Johannes Gutenberg University of Mainz. K. A. I. Z. acknowledges the Deutsche Forschungsgemeinschaft (DFG) for funding. This work is part of the research conducted by the MaxSynBio consortium that is jointly funded by the Federal Ministry of Education and Research of Germany (BMBF) and the Max Planck Society (MPG).References
- S. Bi, Z. A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang and X. Zhang, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- Z.-a. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 479–483 CrossRef.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-h. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180 CrossRef CAS PubMed.
- B. C. Ma, S. Ghasimi, K. Landfester and K. A. Zhang, J. Mater. Chem. B, 2016, 4, 5112–5118 RSC.
- L. Xu, L. Cheng, C. Wang, R. Peng and Z. Liu, Polym. Chem., 2014, 5, 1573–1580 RSC.
- K. Yang, H. Xu, L. Cheng, C. Sun, J. Wang and Z. Liu, Adv. Mater., 2012, 24, 5586–5592 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- B. C. Ma, L. Caire da Silva, S.-M. Jo, F. R. Wurm, M. Bannwarth, K. A. Zhang, K. Sundmacher and K. Landfester, ChemBioChem, 2019, 20, 2593–2596 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- C. Lu, P. Zhang, S. Jiang, X. Wu, S. Song, M. Zhu, Z. Lou, Z. Li, F. Liu, Y. Liu, Y. Wang and Z. Le, Appl. Catal., B, 2017, 200, 378–385 CrossRef CAS.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14549–14553 CrossRef CAS PubMed.
- L. Wang, I. Rörich, C. Ramanan, P. W. Blom, W. Huang, R. Li and K. A. Zhang, Catal. Sci. Technol., 2018, 8, 3539–3547 RSC.
- L. Wang, J. Byun, R. Li, W. Huang and K. A. Zhang, Adv. Synth. Catal., 2018, 360, 4312–4318 CrossRef CAS.
- X.-H. Li, M. Baar, S. Blechert and M. Antonietti, Sci. Rep., 2013, 3, 1743 CrossRef.
- A. Mills and C. O'Rourke, Catal. Today, 2014, 230, 256–264 CrossRef CAS.
- C. Ayed, L. Caire Da Silva, D. Wang and K. A. I. Zhang, J. Mater. Chem. A, 2018, 6, 22145–22151 RSC.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2015, 56, 653–656 CrossRef CAS.
- C. Ayed, W. Huang, R. Li, L. C. da Silva, D. Wang, O. Suraeva, W. Najjar and K. A. Zhang, Part. Part. Syst. Charact., 2018, 35, 1700234 CrossRef.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS.
- Z. J. Wang, R. Li, K. Landfester and K. A. Zhang, Polymers, 2017, 126, 291–295 CrossRef CAS.
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. Müller, J.-M. Verbavatz and R. Langer, Adv. Drug Delivery Rev., 1995, 16, 215–233 CrossRef CAS.
- T. Golds, P. Maliga and H.-U. Koop, Biotechnol. J., 1993, 11, 95 CAS.
- T. Nishio, K. Takahashi, T. Yoshimoto, Y. Kodera, Y. Saito and Y. Inada, Biotechnol. Lett., 1987, 9, 187–190 CrossRef CAS.
- K. Takahashi, Y. Kodera, T. Yoshimoto, A. Ajima, A. Matsushima and Y. Inada, Biochem. Biophys. Res. Commun., 1985, 131, 532–536 CrossRef CAS.
- A. M. James, M. J. Derry, J. S. Train and R. Dawson, Polym. Chem., 2019, 10, 3879–3886 RSC.
- Y. Yang, B. Tan and C. D. Wood, J. Mater. Chem. A, 2016, 4, 15072–15080 RSC.
- W. Mai, B. Sun, L. Chen, F. Xu, H. Liu, Y. Liang, R. Fu, D. Wu and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 13256–13259 CrossRef CAS PubMed.
- C. T. Ferguson, N. Huber, K. Landfester and K. A. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10567–10571 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- F. Xie, W. Hu, L. Ding, K. Tian, Z. Wu and L. Li, Polym. Chem., 2017, 8, 6106–6111 RSC.
- H. Xu, J. Wu, B. Zheng, W. Mai, F. Xu, L. Chen, H. Liu, R. Fu, D. Wu and K. Matyjaszewski, ChemComm, 2017, 53, 5294–5297 RSC.
- L. Wang, W. Huang, R. Li, D. Gehrig, P. W. Blom, K. Landfester and K. A. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9783–9787 CrossRef CAS.
- P. R. Ogilby and C. S. Foote, J. Am. Chem. Soc., 1983, 105, 3423–3430 CrossRef CAS.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS PubMed.
- R. S. Keri, A. Hiremathad, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 19–65 CrossRef PubMed.
- J. B. Wright, Chem. Rev., 1951, 48, 397–541 CrossRef CAS PubMed.
- K. Bahrami, M. M. Khodaei and A. Nejati, Green Chem., 2010, 12, 1237–1241 RSC.
- N. Neamati, A. Mazumder, H. Zhao, S. Sunder, T. Burke, R. J. Schultz and Y. Pommier, Antimicrob. Agents Chemother., 1997, 41, 385–393 CrossRef CAS PubMed.
- C. J. Dinsmore, T. M. Williams, T. J. O'Neill, D. Liu, E. Rands, J. C. Culberson, R. B. Lobell, K. S. Koblan, N. E. Kohl and J. B. Gibbs, Bioorg. Med. Chem. Lett., 1999, 9, 3301–3306 CrossRef CAS PubMed.
- P. R. Blakemore, J. Am. Chem. Soc., 2002, 2563–2585 CAS.
- M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 14, 4833–4836 CrossRef.
- R. J. Taylor, G. D. McAllister and R. W. Franck, Carbohydr. Res., 2006, 341, 1298–1311 CrossRef CAS.
- K. Plesniak, A. Zarecki and J. Wicha, in Sulfur-Mediated Rearrangements II, Springer, 2006, pp. 163–250 Search PubMed.
- C. Herrero, A. Quaranta, R. Ricoux, A. Trehoux, A. Mahammed, Z. Gross, F. Banse and J.-P. Mahy, Dalton Trans., 2016, 45, 706–710 RSC.
- Y. Huang, Z. Xin, W. Yao, Q. Hu, Z. Li, L. Xiao, B. Yang and J. Zhang, ChemComm, 2018, 54, 13587–13590 RSC.
- X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357–361 RSC.
- P. Zhang, Y. Wang, H. Li and M. Antonietti, Green Chem., 2012, 14, 1904–1908 RSC.
- R. Chawla and L. D. S. Yadav, Org. Biomol. Chem., 2019, 17, 4761–4766 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta11242a |
| This journal is © The Royal Society of Chemistry 2020 |