
PIM-1-based carbon–sulfur composites for sodium–sulfur batteries that operate without the shuttle effect†

Abstract
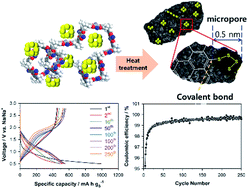
Room temperature sodium–sulfur (RT Na–S) batteries have distinct advantages over other next generation batteries because of their use of abundant and inexpensive resources with high theoretical capacities of 1166 and 1675 mA h g−1, namely for sodium and sulfur. However, problematic side reactions, called the shuttle effect, lead to low coulombic efficiency during cycling. Here, we propose a new strategy to fundamentally suppress the shuttle phenomenon by combining two widely used concepts, covalent bonds and physical confinement, through the preparation of a PIM-1-based carbon–sulfur composite. This sulfur–carbon material was prepared through one-step heat treatment of a mixture of sulfur and PIM-1. The resulting sulfur–carbon composites have characteristics of both ∼0.5 nm-sized ultra-micropores and covalent bonding in a single material, which fundamentally obstruct the dissolution of polysulfide into the electrolyte. This strategy led to long cycling stability over 250 cycles, with a capacity of 556 mA h gs−1 and a coulombic efficiency of approximately 100%.
 Please wait while we load your content...
Please wait while we load your content...

