
Positively charged Pt-based cocatalysts: an orientation for achieving efficient photocatalytic water splitting
Jinze
Liu
a,
Yuhang
Li
 *b,
Xiaodong
Zhou
a,
Hao
Jiang
ab,
Hua Gui
Yang
b and
Chunzhong
Li
*ab
*b,
Xiaodong
Zhou
a,
Hao
Jiang
ab,
Hua Gui
Yang
b and
Chunzhong
Li
*ab
aShanghai Engineering Research Center of Hierarchical Nanomaterials, School of Chemical Engineering, East China University of Science & Technology, Shanghai 200237, China. E-mail: czli@ecust.edu.cn
bKey Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science & Technology, Shanghai 200237, China. E-mail: yuhangli@ecust.edu.cn
First published on 22nd November 2019
Abstract
Considerable effort has been made to develop efficient water-splitting photocatalysts, which are generally composed of light-absorbing semiconductors and so-called cocatalysts that play a key role in accelerating the reaction kinetics of water splitting. Platinum (Pt)-based catalysts have been widely regarded as excellent cocatalysts for photocatalytic water splitting, but the relationship between their valence state and reaction activity for photocatalytic water splitting had never been summarized. This review presents positively charged Pt nanoparticles, clusters and even single atoms that can boost the reaction activity dramatically, and summarises several strategies and techniques to tailor the positive valence states of Pt-based cocatalysts such as by using oxygen atoms. By tailoring the valence states of Pt-based cocatalysts, the capacities of these cocatalysts for water splitting can be significantly enhanced. Moreover, the operando analyses that can determine the real structures of this type of cocatalyst under working conditions are also discussed. These developments suggest that positively charged Pt-based cocatalysts could determine the direction and efficiency of photocatalytic water splitting, which may provide an ideal orientation to design state-of-the-art catalysts for achieving efficient photocatalytic water splitting.
 Jinze Liu | Jinze Liu received his Bachelor's degree in Chemical Engineering and Technology from the East China University of Science and Technology (ECUST) in 2017. He is currently a Master’s candidate in Material Chemical Engineering under the supervision of Prof. Chunzhong Li and Prof. Yuhang Li at the ECUST. His current research is focused on the design and fabrication of novel photo/electro-catalysts for water splitting and CO2 reduction. |
 Yuhang Li | Yuhang Li received his PhD degree in Materials Science and Engineering from the East China University of Science and Technology (ECUST) in 2016. He joined the Key Laboratory for Ultrafine Materials of Ministry of Education as a postdoctoral fellow in 2016–2018 at the ECUST. In September 2019, he joined the Sargent group, University of Toronto, as a visiting professor. He is currently an associate professor of the School of Materials Science and Engineering at the ECUST. His research interests are focused on the design and fabrication of novel photo/electro-catalysts for water splitting and CO2 reduction. |
 Xiaodong Zhou | Xiaodong Zhou is a Professor at the East China University of Science and Technology. He received his Bachelor's degree in Polymer Materials Science and Engineering (1990) and PhD degree in Polymer Materials Science from Nanjing University of Science and Technology (1996). He was a post-doctoral researcher (1996–1998) at the East China University of Science and Technology and has been working there since 1998. His current research interests include interface design and control of multiphase multicomponent polymer materials, preparation and molding technology of thermoplastic polymer composites and high performance and practical technology of biological resource polymer materials. |
 Hao Jiang | Hao Jiang received his PhD degree in Materials Science and Engineering from the ECUST, China, in 2009. He joined Temasek Laboratories, Nanyang Technological University (NTU) in Singapore, as a research scientist in 2009–2011. Currently he is a full professor in the Key Laboratory for Ultrafine Materials of Ministry of Education at the ECUST, and was the winner of the National Science Fund for Excellent Young Scholars in 2015. His current research interests focus on the design and preparation of novel hierarchical nanomaterials and their application in electrochemical energy storage and conversion. |
 Hua Gui Yang | Hua Gui Yang completed his PhD degree at the National University of Singapore in 2005. He joined the University of Queensland in 2007 as a Postdoctoral Research Fellow. After finishing his postdoctoral training, he came back to China and took a professor position at the East China University of Science and Technology at the end of 2008. His main research interests are focused on the rational design and fabrication of functional materials for solar-energy conversion. |
 Chunzhong Li | Chunzhong Li received his BS (1989), MS (1992) and PhD (1997) degrees from the ECUST. He became a full professor at the School of Materials Science and Engineering in 1998, and now he is the dean of the School of Chemical Engineering at the ECUST. He was elected as a Fellow of the Royal Society of Chemistry in 2015, became the Cheung Kong Distinguished Professor in 2014, and the winner of the National Science Fund for Distinguished Young Scholars in 2009. His research interests include functionalization and fabrication of nanomaterials for new energy, environment and relevant applications. |
1. Introduction
Photocatalytic water splitting to produce hydrogen (H2) as a solar fuel is considered a promising way to address environmental issues.1 This reaction is an uphill reaction, for which the ΔG can be as high as +237 kJ mol−1.2 Thus, to promote the production of H2, photocatalysts are usually involved to lower the reaction barriers.3–6 Since Fujishima and Honda demonstrated the capacity of TiO2 coupled with a platinum (Pt) electrode for photoelectrocatalytic water splitting in the early 1970s, extensive research has been conducted to fabricate high-efficiency photocatalytic water-splitting systems, which can be illustrated as a photoelectrocatalytic-like cell without applying bias.7–10The major steps involved in the photocatalytic water-splitting reaction are as follows: (I) semiconductor photocatalysts convert light energy to electron–hole carriers, (II) carriers separate and migrate to the sites of cocatalysts on photocatalysts, and (III) water reduction and/or oxidation occurs on cocatalysts. Photocatalytic water splitting can occur when these three steps are completed, and numerous strategies have been proposed to promote these processes.11 For example, semiconductors with narrow bandgaps absorb longer wavelengths of solar light, employing nanosized materials accelerates photogenerated carrier migration, and loading suitable cocatalysts reduces and/or oxidizes water molecules. Notably, it is necessary to study the half reactions of H2 and O2 evolution using sacrificial agents to identify possible photocatalysts for overall water-splitting systems.3 Moreover, investigations of the half reactions could provide insight into detailed mechanisms during water splitting. In water-splitting systems, semiconductor photocatalysts usually require the help of appropriate cocatalysts to increase the rate of the water-splitting reaction because cocatalysts that trap electrons from the bulk of the photocatalyst are key to complete proton reduction (splitting water into H2).12–15 Therefore, metal nanoparticles or clusters are widely used as cocatalysts to facilitate electron transfer and catalytic processes at the interfaces between photocatalysts and solutions.
Cocatalysts supply the active sites of water-splitting reactions because most bare photocatalysts have negligible H2 evolution activity, even with the help of sacrificial agents.5 Researchers have proposed some reasons for this negligible activity, such as the fast recombination of photogenerated carriers before they migrate to the surface for water splitting and the insufficient rate of catalytic processes compared to the recombination step. Among all cocatalysts developed to date, platinum (Pt)-based nanoparticles and clusters have been considered the most universal cocatalysts over many different kinds of semiconductors, including metal oxides, metal sulfides, metal nitrides, etc.,16–18 and all have been demonstrated to remarkably improve the H2 evolution activity. Pt-based cocatalysts can act as electron sinks and provide adsorption sites for protons, thus facilitating the proton reduction reaction. The superior trapping ability of Pt may be because its work function (that is, the lowest Fermi level) is the highest among those of many metals and is also greater than those of most semiconductors, forming a Schottky barrier which facilitates carrier separation at the interface.19 In addition, researchers have established a volcano-like curve between the activity of H2 generation and the adsorption energy of protons and have demonstrated the excellent activation energy for proton reduction by Pt.20 Thus, Pt has been regarded as the most powerful cocatalyst for photocatalytic water splitting because of its electronic and catalytic properties.
Since an increase in the activity of Pt nanoparticles with a mean size less than 5 nm was observed experimentally, various efforts have been devoted to studying the descriptors of the catalytic activity,16,21–25 such as the size and support interaction of Pt-based cocatalysts. Moreover, consideration of the undesirable back reaction of water formation by H2 and O2 is required because this reaction is spontaneous when using metallic Pt as a cocatalyst.26,27 To promote H2 and O2 evolution by water splitting, the addition of carbonate salts to the reaction solution was suggested in early studies, and the fabrication of core–shell structures was suggested in recent literature.28,29 Many researchers have discovered that Pt-based cocatalysts with positive valence states can enhance total performance in photocatalytic water splitting. And Pt-based cocatalysts with positive valence states can not only promote the reaction activity of photocatalysts but also solve the problem of the back-reaction of water formation by H2 and O2. Some literature studies also have reported the effect mechanisms of positively charged Pt-based cocatalysts on the performance of photocatalytic water splitting by the combination of theoretical simulations and operando characterization. Although a number of high-quality review articles describing Pt-based cocatalysts for water splitting have been published,2–5,13–15 there are few reviews devoted entirely to the effect of valence states on the activity of these cocatalysts. Here, we will focus on the role of the valence states of Pt-based cocatalysts in photocatalytic systems (Fig. 1) and highlight the use of operando characterization to determine the real structures of the cocatalysts during water splitting. Additionally, some interesting reaction mechanisms associated with positively charged Pt-based cocatalysts are also described and discussed.
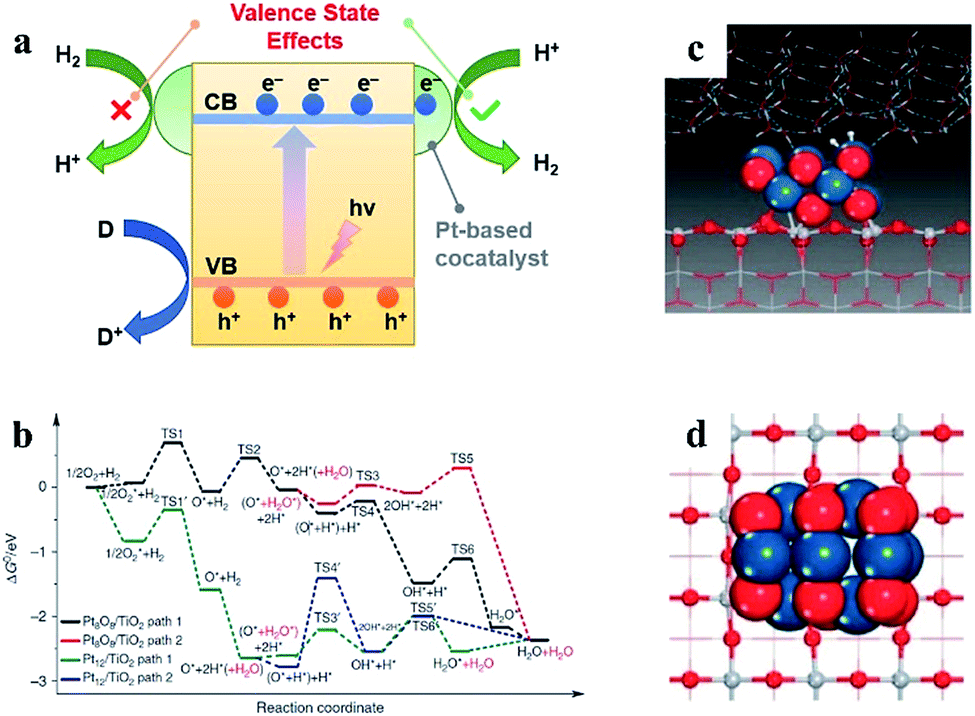 | ||
| Fig. 1 (a) Valence state effects of Pt-based cocatalysts on photocatalytic water splitting. (b) Simulations of H2 oxidation with different valence states of Pt-based cocatalysts. (c and d) Ball and stick model of a PtO cocatalyst. Reproduced with permission.30 Copyright 2013, Springer Nature. | ||
2. Valence state effects of Pt-based cocatalysts
2.1. Tailoring with oxygen atoms to suppress the HOR
In 2013, Li et al. synthesized a PtO/TiO2 photocatalyst that exhibits enhanced performance and reported that the valence state of Pt can influence photocatalytic water splitting by controlling unidirectional H2 oxidation (Fig. 2). According to the STEM characterization in Fig. 2b, the mean size of a PtO-cluster is found to be around 1 nm, and further from the Fourier-transformed spectrum of the Pt L3-edge extended X-ray absorption fine structure, the Pt species of PtO/TiO2 is indicated to be the PtO phase (Fig. 2c). In addition to the core–shell structures used for inhibiting the back reaction, Pt in a high valence state can suppress H2 back-oxidation. In this study, the abilities of both Pt and PtO as cocatalysts to suppress the H2 oxidation reaction were studied in an aqueous methanol solution. In the light reaction, the photocatalytic H2 production rate of the Pt cocatalyst is 1/4 that of PtO. Interestingly, the H2 pressure on the Pt cocatalyst decreases faster than that on PtO in the dark reaction (Fig. 2d). The photocatalytic water splitting benefited from the suppressing effect of the PtO clusters on the H2 oxidation reaction. In this photocatalyst system, there are two roles for the PtO cocatalyst: (i) providing efficient H2 evolution active sites and (ii) suppressing reverse H2 oxidation. The X-ray photoelectron spectral analysis results indicate that the Pt species in the cocatalysts are in the form of metallic Pt and PtO.30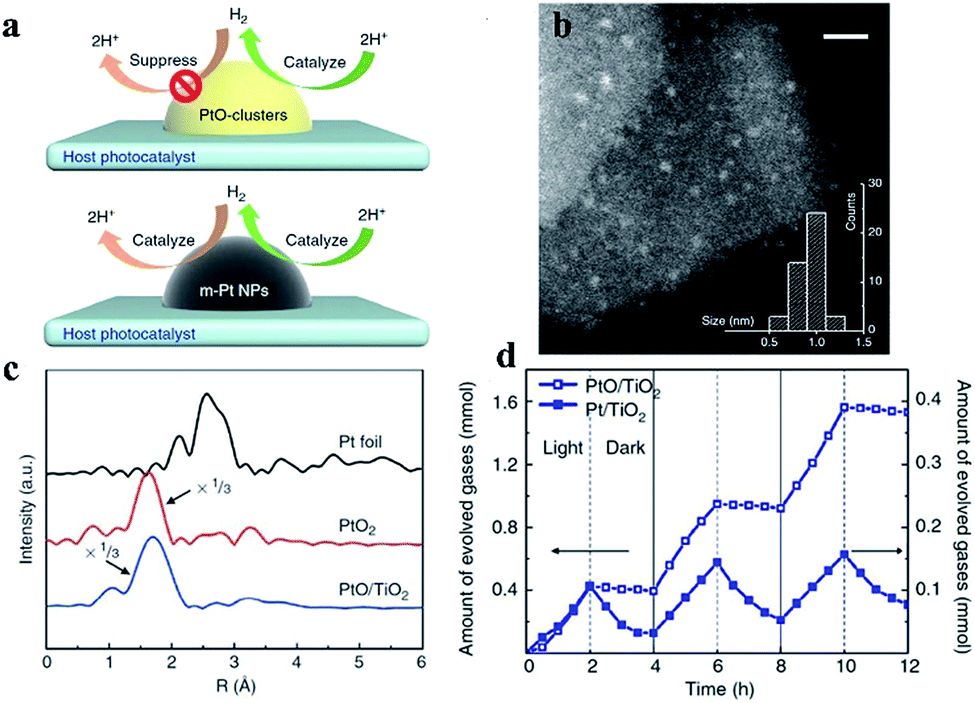 | ||
| Fig. 2 (a) H2 oxidation can be suppressed and catalyzed by PtO and metallic Pt cocatalysts. (b) Scanning transmission electron microscopy image of a PtO cocatalyst loaded on TiO2. The inset shows the size distribution of PtO. Scale bar, 5 nm. (c) The PtO cocatalyst contains only Pt–O bonds, as detected using X-ray absorption fine structure spectra. (d) Photocatalytic H2 generation and H2 oxidation rates using methanol as a sacrificial agent with alternating light on and off in 2 h intervals. Reproduced with permission.30 Copyright 2013, Springer Nature. | ||
Moreover, by using PtO cocatalysts and by utilizing advanced electron microscopy and simulation calculations, Li et al. demonstrated in 2015 that TiO2 crystal facets do not play a critical role in photocatalytic reactions (Fig. 3). The possible electron transfer between the cocatalyst and host catalyst is illustrated in Fig. 3a. They have systematically investigated the true photoreactivity order of the {001} and {101} facets of TiO2 by means of loading PtO clusters as a cocatalyst (Fig. 3c and d). The valence states of the Pt-based cocatalyst are the main reason why the photoactivity of the PtO-loaded photocatalyst is higher than that of the Pt-loaded photocatalyst,31 which could be inferred from Fig. 3b. Furthermore, to obtain structural information about the PtO catalyst during water splitting, the structural changes of the PtO cocatalyst were detected by operando X-ray absorption fine structure spectroscopy during water splitting (Fig. 4a–d), in which a Pt–O bond length of 2.13 Å and a coordination number of 2.5 were quantitatively determined under working conditions (Fig. 4f). The PtO cocatalyst can be observed by STEM with an average size of 1.5 nm, and the PtO cocatalyst is uniformly coated on the surface (Fig. 4e). The results show that the structure of PtO during the reaction is quite different from that of PtO with fewer O atoms and longer bond lengths, and even these longer bonds are shorter than the Pt–Pt bond in metallic Pt, consistent with the higher valence state of the PtO cocatalyst than that of metallic Pt. These results indicate the important role of valence states of the PtO cocatalyst in the unidirectional inhibition of the H2 oxidation reaction.32
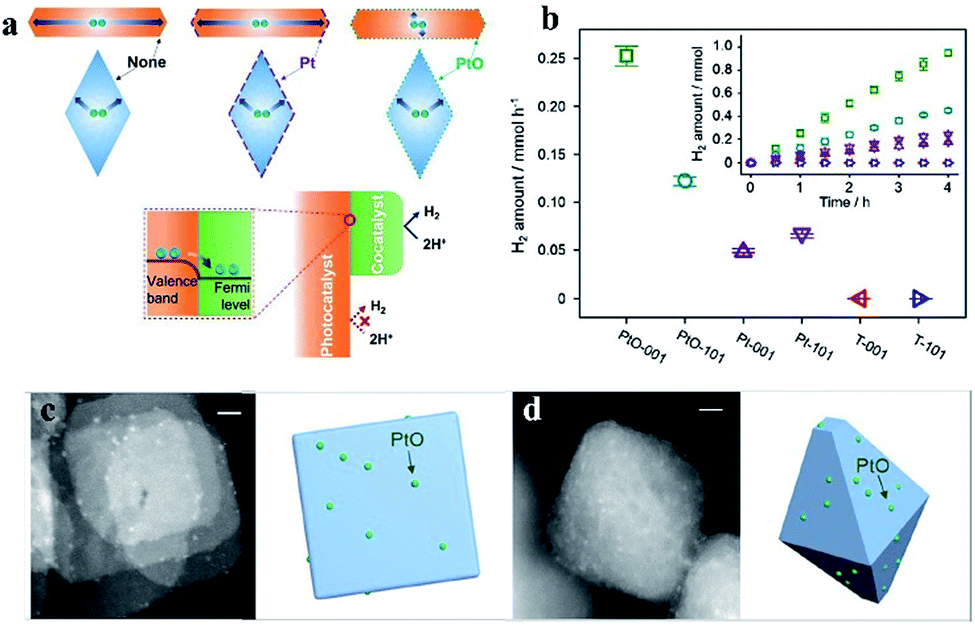 | ||
| Fig. 3 (a) Possible photogenerated electron migration when loading different valence states of Pt-based cocatalysts on TiO2. (b) Photocatalytic H2 generation rates of TiO2 photocatalysts loaded with different valence states of Pt-based cocatalysts. Scanning transmission electron microscopy images of PtO cocatalysts on sheet-like (c) and octahedron-like (d) photocatalysts. Reproduced with permission.31 Copyright 2015, Elsevier. | ||
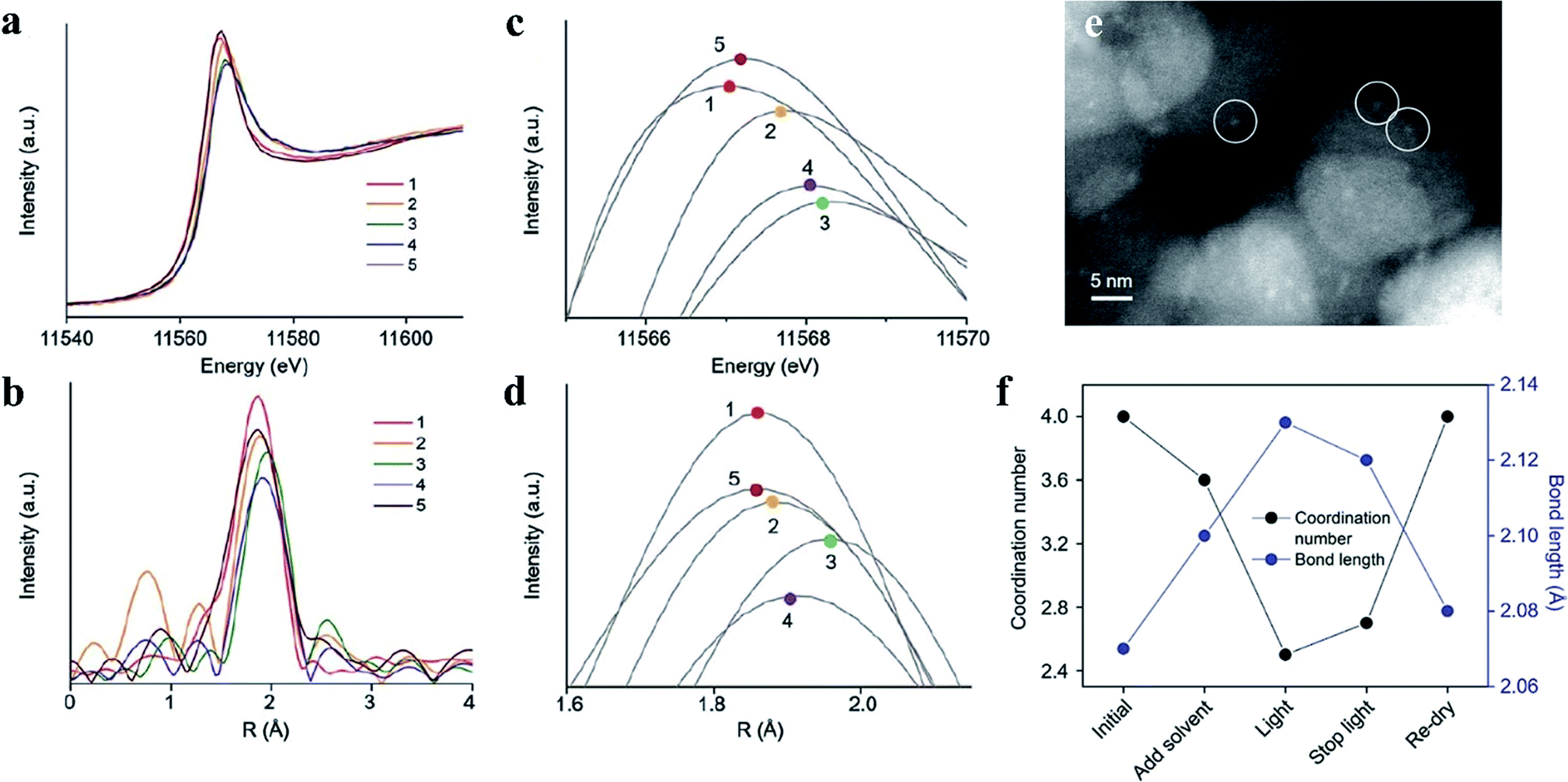 | ||
| Fig. 4 (a–d) Spectra of PtO as a cocatalyst detected by operando X-ray absorption fine structure spectroscopy. The numbers 1–5 indicate the initial time, addition of solvent, light on, light off and recycled sample, respectively. (e) Scanning transmission electron microscopy image of a PtO cocatalyst (in white circles) loaded on TiO2. (f) Parameters such as Pt–O bond length and coordination number under different conditions. Reproduced with permission.32 Copyright 2017, The Royal Society of Chemistry. | ||
Ren et al. reported an enhanced photocatalytic H2 evolution performance of Pt/PtO heterojunction nanodots on TiO2. In this report, a NaOH modification method is utilized to anchor the Pt/PtO nanodots onto porous TiO2 with interparticle mesopores, resulting in enhanced photochemical H2 production.
The formation of Pt2+ species is related to the influence of NaOH on the reduction rate of H2PtCl6. Metallic Pt can effectively separate the photogenerated carriers, and PtO can serve as a reaction site for H2 evolution while inhibiting the H2 oxidation reaction.33 Manwar et al. introduced Pt/CeO2 for donor-assisted photocatalytic H2 evolution reactions, where the photocatalyst is prepared via the wet impregnation method. X-ray photoelectron spectra of the Pt component have been reported, indicating that the Pt species are in the form of Pt, Pt2+ and Pt4+. The existence of Pt and PtO together can promote H2 evolution and suppress H2 oxidation.34
2.2. Tailoring with oxygen atoms to boost the HER and OER
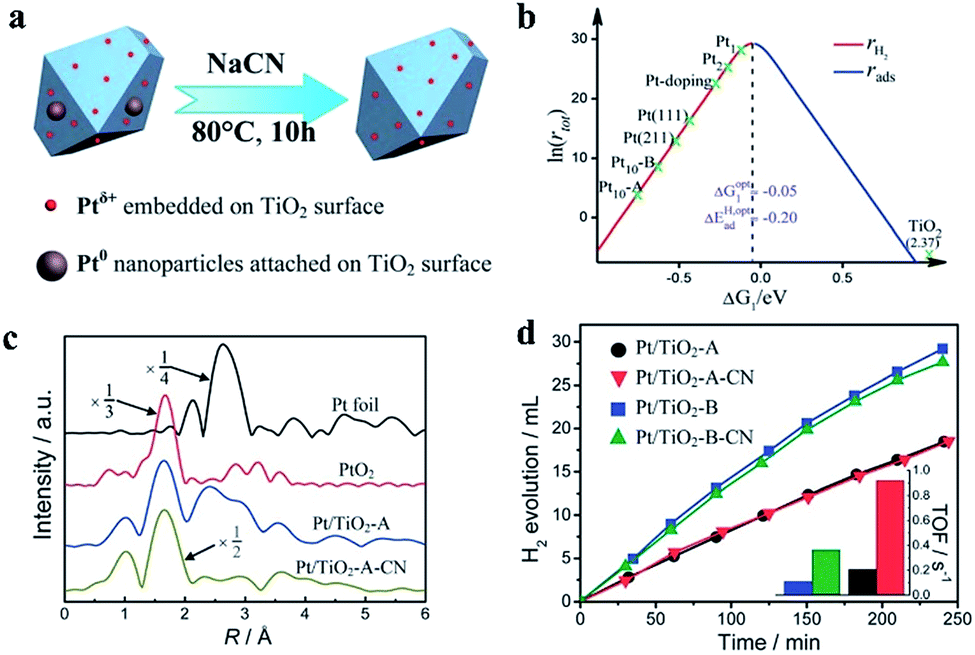 | ||
| Fig. 5 (a) Schematic preparation process of a Ptδ+ cocatalyst. (b) Relationship between the catalytic rate and ΔG of H adsorption with different Pt-based cocatalysts. (c) The Ptδ+ cocatalyst has only Pt–O bonds, as determined from the X-ray absorption fine structure spectra. (d) Photocatalytic H2 generation rates and turnover frequencies of different valence states of Pt-based cocatalysts. Reproduced with permission.36 Copyright 2013, The Royal Society of Chemistry. | ||
In 2017, Sui et al. reported a Pt/TiO2 photocatalyst fabricated by a facile hydrothermal method. In this photocatalyst, atomic Pt is loaded on TiO2. The photocatalyst composition is 72% single atoms, 16% clusters of 0.4–1 nm, and 12% particles of 1–2 nm, as demonstrated by the size distribution frequency. The X-ray photoelectron spectral analysis results suggest that more deficient-state Ptδ+ is present, while the Pt species in commonly used photocatalysts are mainly in the form of metallic Pt. The H2 generation rate of the photocatalyst can reach 84.5 μmol h−1, which is higher than that of the control sample due to the valence state effects of Pt-based cocatalysts.39 Recently, Talas et al. modified PtOx–TiO2 photocatalysts by introducing tin. In this article, the valence state of Pt in the initial and recycled samples following high-temperature H2 reduction and annealing was studied. Pt exists in the form of Pt0 and Pt2+ in the initial sample after calcination, while mainly Pt0 can be detected after the photocatalytic tests, indicating that the reduction of Ptδ+ occurs during water splitting. The H2 production can be hindered by reducing the Pt nanoparticles at the start of the H2 evolution reaction in the case of the calcined samples, which has a negative effect on activating the photocatalyst by high-temperature hydrogenation. PtOx not only prevents carrier recombination but also acts as the reaction site for the photocatalytic process.40
Jiang et al. reported g-C3N4 modified with PtO, which is synthesized via a typical photodeposition method, for the H2 evolution reaction. When a PtO cocatalyst was utilized, the H2 generation rate increased from 1.6 to 11.5 μmol h−1, which demonstrated that PtO has an excellent ability to serve as an electron sink and provide active sites to lower the reaction barrier of H2 evolution (Fig. 6).52 Due to the surface plasmon resonance of Au and the synergetic action of PtO and Au, the HER activity of g-C3N4 is enhanced dramatically (Fig. 6a). The HRTEM image confirms the existence of Au and PtO nanoparticles on the g-C3N4 (Fig. 6b). The rate of H2 evolution in Fig. 6c indicates that the PtO lowers the overpotential for hydrogen evolution by acting as an active site. Wang et al. deposited PtO nanodots with high crystallinity on g-C3N4. A series of electronic tests revealed that PtO has enhanced conductivity on g-C3N4 compared with metallic Pt. The H2 production rate is more than 260 μmol h−1, which is approximately 50 times higher than that of metallic Pt as a cocatalyst.53 In addition, Pt oxide cocatalysts can also be loaded onto other photocatalysts, such as KCa2Nb3O10,54 BiVO4,55 and TaON,41,56,57 to promote the water-splitting reaction. These cocatalysts can even act as efficient electrocatalysts on carbon substrates for H2 generation.58,59
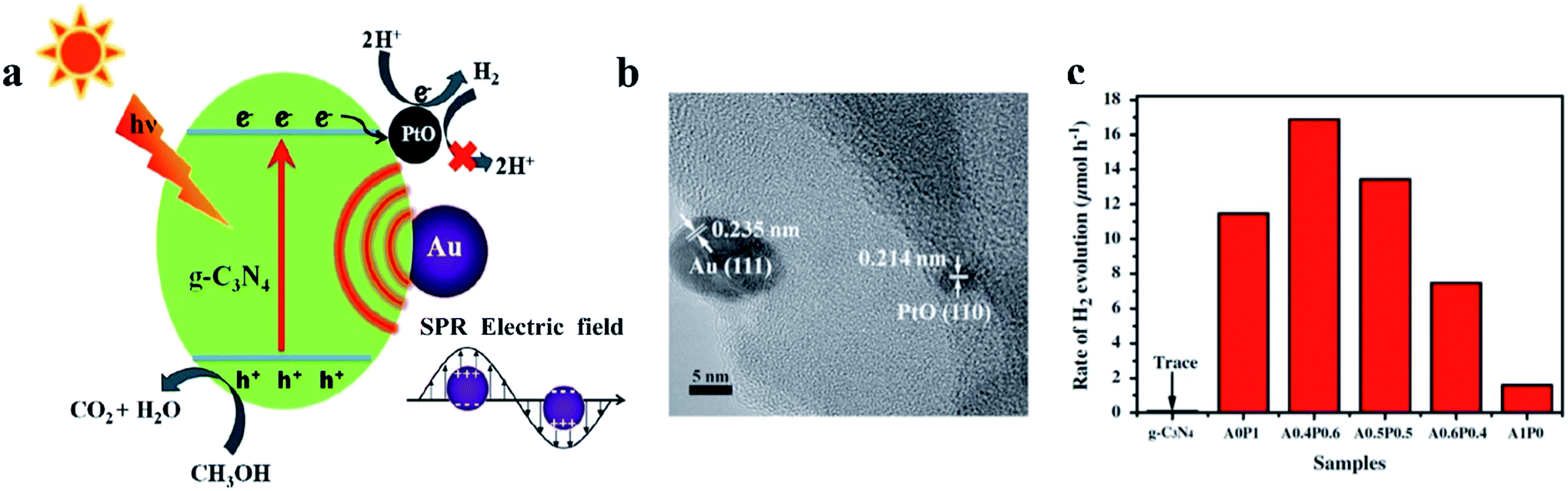 | ||
| Fig. 6 (a) Schematic mechanism of a PtO cocatalyst as a H2 evolution site, (b) high-resolution transmission electron microscopy image and (c) photocatalytic H2 generation rates of PtO cocatalysts loaded on a g-C3N4 photocatalyst. Reproduced with permission.52 Copyright 2016, Elsevier. | ||
2.3. Tailoring with halogen atoms or substrates
To suppress this undesired back reaction, Wang has proven the significant ability of Pt modified with fluorine ions to inhibit the back reaction. The H2 generation rate of fluorine-modified Pt cocatalysts can be 2.3 times that of unmodified Pt cocatalysts in photocatalytic reactions because fluorine occupies the adsorption sites to prevent adsorption of H2 on metallic Pt cocatalysts.60 In addition, Wang and coworkers have further studied the suppression of H2 combination with O2 by halogen elements on Pt-based cocatalysts for photocatalytic water splitting by detailed theoretical and experimental analysis (Fig. 7a–e). Remarkably, simulations of the adsorption energy of H2 on modified Pt-based cocatalysts were carried out, and the results show the following order of H2 adsorption energy: F-modified < Cl-modified < I-modified < Br-modified < unmodified cocatalysts. Then they synthesized halogen/Pt–TiO2 and confirmed the existence of halogen in photocatalysts via high-angle annular dark field scanning transmission electron microscopy (Fig. 7f–i). The suppression of H2 oxidation follows the same trend (Fig. 7j).61 Recently, Li et al. successfully inhibited the recombination of O2 and H2 by the addition of hemin chloride; the hemin captures O2 generated from water and transfers it away from the surface of the photocatalyst, and the Cl− ions further inhibit the back-recombination of O2 and H2.62
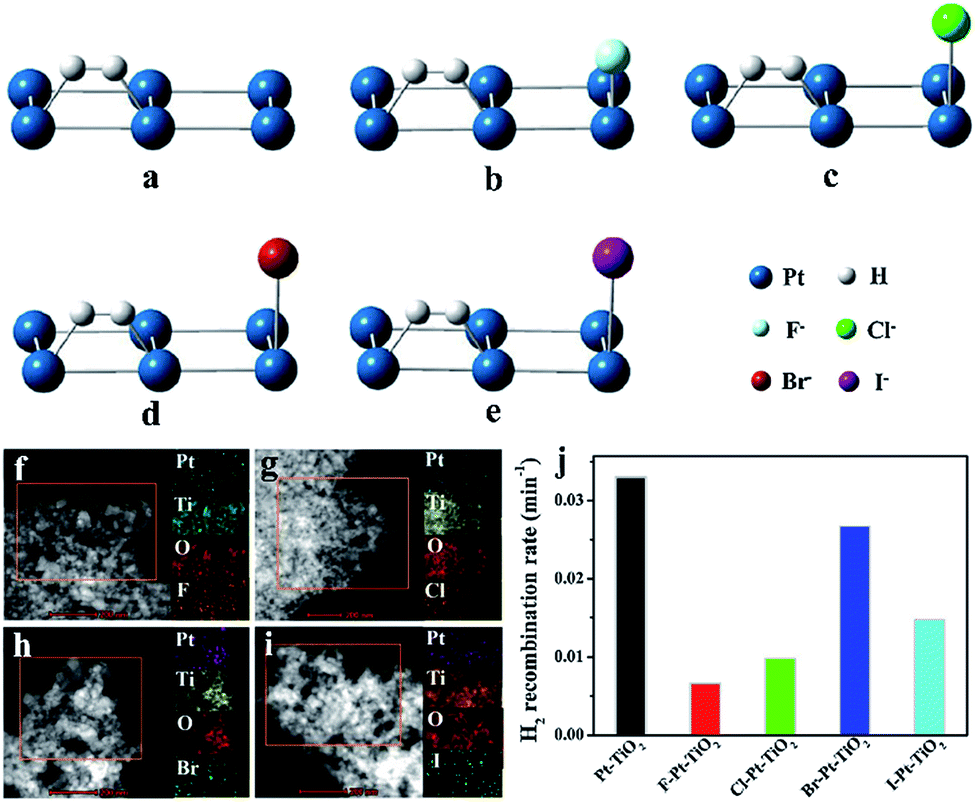 | ||
| Fig. 7 H2 adsorption (a–e), scanning transmission electron microscopy images (f–i) and photocatalytic H2 generation rates (j) of Pt cocatalysts modified using different halogen elements. Reproduced with permission.61 Copyright 2018, Elsevier. | ||
In a supported metal catalyst, the metal atoms can be adsorbed on the surface of the support and undergo charge transfer with the support. During this process, the electronic structures of the metal catalysts will be tuned, presenting enhanced catalytic activity. In particular, Pt atoms as catalysts tend to adsorb onto the bridge formed by two oxygen atoms in anatase TiO2, and according to the simulations, this composite structure could increase the activity of the adsorption surface.63,64 Xing and coworkers prepared a Ptδ+ cocatalyst on a TiO2 photocatalyst using a leaching method. In this study, these authors investigated the valence state effects of Pt-based cocatalysts on H2 generation and demonstrated that the photocatalytic H2 generation rates can be sensitive to the valence states of Pt but that the size effects are negligible at several nanometers. According to simulations of the H adsorption energy of Pt with different valence states, the poor performance of metallic Pt as a cocatalyst may be due to its too strong H adsorption energy, thus decreasing the H2 generation activity.65 Further, Xing synthesized isolated Pt atoms stabilized by anchoring onto TiO2, and X-ray absorption fine structure spectra were utilized to observe the valence states of Pt in different samples in this study (Fig. 8). The Fourier transformed spectra of Pt L3-edge EXAFS of 0.2 Pt/TiO2 reflect that the Pt atoms in the photocatalyst exist as Ptδ+, in which the isolated Pt atom is combined with oxygen atoms in the TiO2 substrate (Fig. 8a and b). The H2 production rate of the sample was measured to be 169.6 mmol h−1, which is the best performance among the controlled samples66 (Fig. 8c). The theoretical calculations demonstrate that the performance of photocatalytic H2 evolution on the TiO2 photocatalyst can be optimized through introducing single-atom Pt, and due to the decreased H adsorption energy, the catalytic activity of the isolated Pt atom could be even better than that of the metallic Pt deposited on TiO2 (Fig. 8d–f).
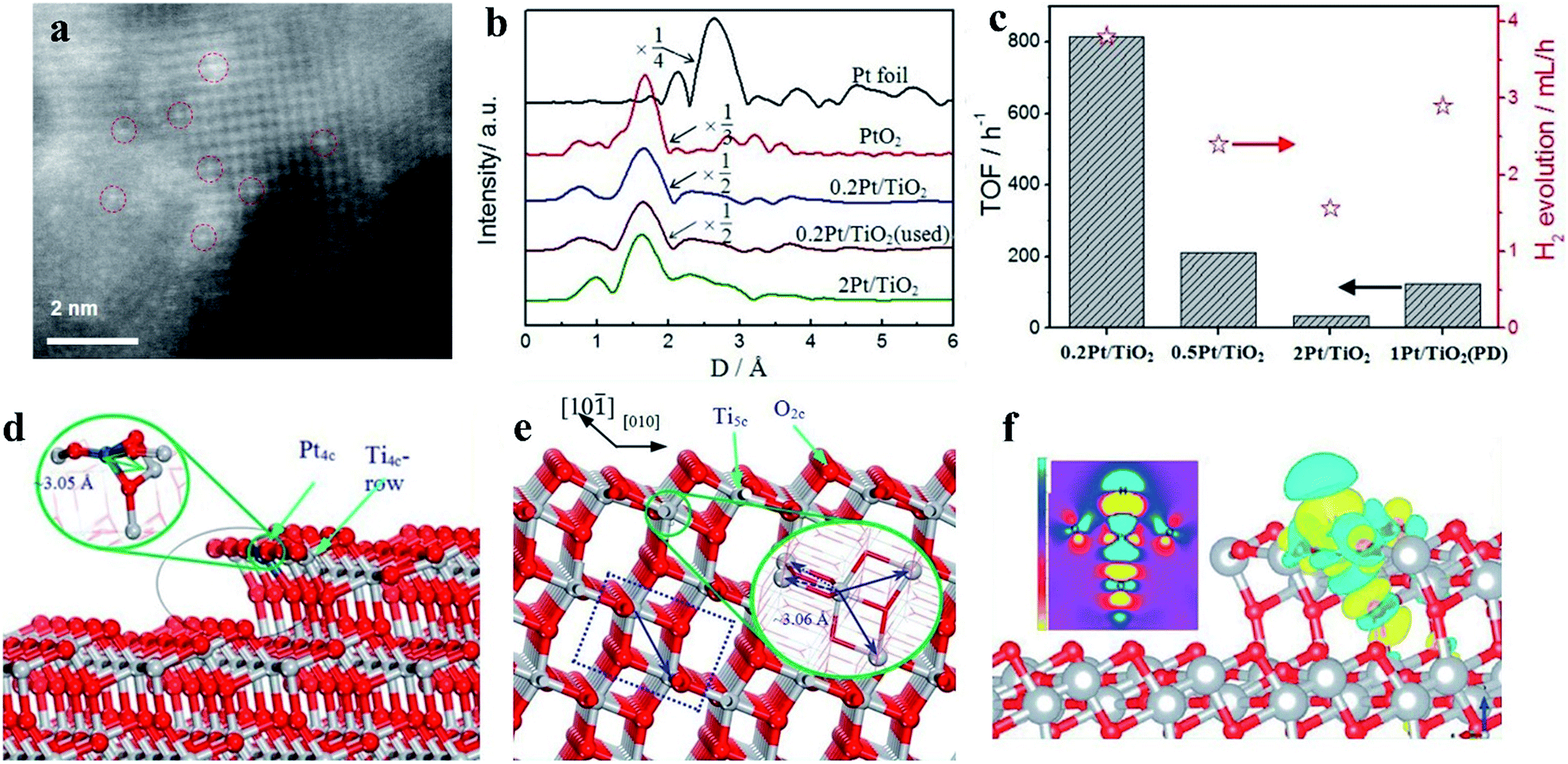 | ||
| Fig. 8 (a) Scanning transmission electron microscopy image of a single Pt atom as a cocatalyst. (b) The single Pt atom is combined with oxygen atoms in a TiO2 substrate as detected by X-ray absorption fine structure spectroscopy. (c) Photocatalytic H2 generation rates and turnover frequencies of different valence states of Pt-based cocatalysts. (d–f) Simulations of the density of states of bare TiO2 and single Pt atom-modified TiO2. Reproduced with permission.66 Copyright 2014, Wiley-VCH. | ||
In 2017, Jiang et al. synthesized a photocatalyst comprising Pt loaded on anatase TiO2 with postmodification by a hydrothermal method. The modified in situ preparation process to load Pt cocatalysts shows an enhanced rate compared to other conventional processes and is a favored method to form positively charged Pt cocatalysts by wet impregnation of TiO2 photocatalysts in a H2PtCl6 solution. The enhanced performance for photocatalytic H2 generation can be attributed to the existence of Ptδ+ as a cocatalyst in this system.67 In the same year, Ou et al. successfully prepared g-C3N4 nanosheets with single Pt atoms loaded through a wet impregnation method. These nanosheets present the best performance for photocatalytic H2 generation. The X-ray photoelectron spectra reveal that the valence state of Pt is Ptδ+. Regarding the location of the single Pt atoms in g-C3N4, the conduction band of the host photocatalyst can be downshifted, and the photogenerated carriers can migrate faster. The Pt species in the form of Ptδ+ show the capacity to improve the H2 generation performance.68 Recently, Xue and coworkers reported a highly active and durable single Pt atom as a cocatalyst supported on g-C3N4 for photocatalytic H2 generation. The X-ray photoelectron spectral analysis shows that there exists more deficient-state Ptδ+ in the reported sample than in compared systems, leading to the most excellent activity of 458.1 mmol h−1 mgPt−1.69
3. Summary and outlook
Suitable cocatalysts are of great importance for obtaining high-efficiency photocatalysts for water splitting. Pt-based cocatalysts can trap photogenerated carriers and act as active sites to lower the activation barriers of water-splitting reactions, thus serving as the most efficient cocatalysts for photocatalytic H2 evolution. In this review, the positively charged Pt-based cocatalysts for photocatalytic water splitting that have been proposed in recent years have been discussed. Benefitting from the unique characteristics of different valence states of Pt, such as the optimized adsorption of H atoms and suppression capacity for the H2 oxidation reaction, Pt-based cocatalysts with a positive valence state have been considered ideal reaction sites on semiconductor-based photocatalysts, including metal oxides, sulfides, nitrides and their composites. The valence states of Pt-based cocatalysts can be tailored using oxygen atoms, halogen atoms and the surface atoms of loading substrates. Moreover, the Pt valence states can be rationally designed via ligand-assisted chemical reduction, hydrothermal, template and wet impregnation methods. With tuned valence states, the back reaction of Pt-based cocatalysts can be suppressed, and the forward H2 evolution reaction can be further promoted.Although significant advances have been made in Pt-based cocatalysts in the field of water splitting, further work is urgently needed. Structural changes are always involved in small Pt-based cocatalysts during photocatalytic reactions, and a lack of precise control of atomic structures and valence states will undoubtedly decrease the water splitting performance. Therefore, the fabrication of Pt-based cocatalysts with target structures on the nanoscale or even the atomic level as well as the valence states must be achieved, and high-quality transient operando techniques to track the structural changes of cocatalysts during water splitting must be developed. The loading location of cocatalysts has shown its superiority in promoting photocatalytic activity for overall water splitting, but the detailed mechanism is not yet understood, and an in-depth investigation combining experimental studies with corresponding simulations should be carried out. Moreover, most reported Pt-based cocatalysts are aimed at the half reaction of H2 evolution, in which sacrificial reagents are commonly required. The final goal to produce H2 as a solar fuel should be realized by the fabrication of high-efficiency photocatalysts that split pure water into H2 and O2 without added sacrificial reagents. Thus, positively charged Pt-based cocatalysts that can inhibit H2 oxidation are highly desired for overall water splitting.
In this review, we provide a concise survey of the valence state effects of Pt-based cocatalysts for photocatalytically splitting water into H2 products. By precisely tailoring the atomic structures and valence states, positively charged Pt-based cocatalysts can determine the direction and efficiency of the water-splitting reaction and thus make promising contributions to photocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21808061, 21838003, 91834301), the Shanghai Scientific and Technological Innovation Project (18JC1410500, 18DZ2252400, 19JC1410400), the “Fundamental Research Funds for the Central Universities” (222201714001, 222201718002), the Shanghai Sailing Program (17YF1402900), the “Chen Guang” project supported by the Shanghai Municipal Education Commission and the Shanghai Education Development Foundation (16CG31).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- B. Mei, K. Han and G. Mul, ACS Catal., 2018, 8, 9154–9164 CrossRef CAS PubMed.
- Y. Moriya, T. Takata and K. Domen, Coord. Chem. Rev., 2013, 257, 1957–1969 CrossRef CAS.
- Y. Kuang, T. Yamada and K. Domen, Joule, 2017, 1, 290–305 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- C.-F. Fu, X. Wu and J. Yang, Adv. Mater., 2018, 30, 1802106 CrossRef PubMed.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20, 110–117 CrossRef CAS.
- J. R. Darwent and G. Porter, J. Chem. Soc., Chem. Commun., 1981, 145–146 RSC.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- D. Y. C. Leung, X. Fu, C. Wang, M. Ni, M. K. H. Leung, X. Wang and X. Fu, ChemSusChem, 2010, 3, 681–694 CrossRef CAS PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef PubMed.
- J. Xing, W. Q. Fang, H. J. Zhao and H. G. Yang, Chem.–Asian J., 2012, 7, 642–657 CrossRef CAS PubMed.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218–13219 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- W. R. Grove, The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 1839, 14, 127–130 CrossRef.
- R. Abe, K. Sayama and H. Arakawa, Chem. Phys. Lett., 2003, 371, 360–364 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- Y. Hang Li, J. Xing, Z. Jia Chen, Z. Li, F. Tian, L. Rong Zheng, H. Feng Wang, P. Hu, H. Jun Zhao and H. Gui Yang, Nat. Commun., 2013, 4, 2500 CrossRef PubMed.
- Y. H. Li, C. Peng, S. Yang, H. F. Wang and H. G. Yang, J. Catal., 2015, 330, 120–128 CrossRef CAS.
- Y. H. Li, C. Z. Li and H. G. Yang, J. Mater. Chem. A, 2017, 5, 20631–20634 RSC.
- X. N. Ren, Z. Y. Hu, J. Jin, L. Wu, C. Wang, J. Liu, F. Liu, M. Wu, Y. Li, G. Van Tendeloo and B. L. Su, ACS Appl. Mater. Interfaces, 2017, 9, 29687–29698 CrossRef CAS PubMed.
- N. R. Manwar, A. A. Chilkalwar, K. K. Nanda, Y. S. Chaudhary, J. Subrt, S. S. Rayalu and N. K. Labhsetwar, ACS Sustainable Chem. Eng., 2016, 4, 2323–2332 CrossRef CAS.
- B. S. Huang, F. Y. Chang and M. Y. Wey, Int. J. Hydrogen Energy, 2010, 35, 7699–7705 CrossRef CAS.
- J. Xing, H. B. Jiang, J. F. Chen, Y. H. Li, L. Wu, S. Yang, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 15258–15264 RSC.
- S. K. Parayil, H. S. Kibombo, C. M. Wu, R. Peng, T. Kindle, S. Mishra, S. P. Ahrenkiel, J. Baltrusaitis, N. M. Dimitrijevic, T. Rajh and R. T. Koodali, J. Phys. Chem. C, 2013, 117, 16850–16862 CrossRef CAS.
- J. Jin, C. Wang, X. N. Ren, S. Z. Huang, M. Wu, L. H. Chen, T. Hasan, B. J. Wang, Y. Li and B. L. Su, Nano Energy, 2017, 38, 118–126 CrossRef CAS.
- Y. L. Sui, S. B. A. Liu, T. F. Li, Q. X. Liu, T. Jiang, Y. F. Guo and J. L. Luo, J. Catal., 2017, 353, 250–255 CrossRef CAS.
- E. Talas, Z. Paszti, L. Korecz, A. Domjan, P. Nemeth, G. P. Szijjarto, J. Mihaly and A. Tompos, Catal. Today, 2018, 306, 71–80 CrossRef CAS.
- R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228–237 CrossRef CAS PubMed.
- G. Ma, S. Chen, Y. Kuang, S. Akiyama, T. Hisatomi, M. Nakabayashi, N. Shibata, M. Katayama, T. Minegishi and K. Domen, J. Phys. Chem. Lett., 2016, 7, 3892–3896 CrossRef CAS PubMed.
- K. Maeda, ACS Appl. Mater. Interfaces, 2014, 6, 2167–2173 CrossRef CAS PubMed.
- K. Maeda, D. L. Lu and K. Domen, ACS Catal., 2013, 3, 1026–1033 CrossRef CAS.
- Y. Miseki, S. Fujiyoshi, T. Gunji and K. Sayama, Catal. Sci. Technol., 2013, 3, 1750–1756 RSC.
- Y. Qi, S. S. Chen, M. R. Li, Q. Ding, Z. Li, J. Y. Cui, B. B. Dong, F. X. Zhang and C. Li, Chem. Sci., 2017, 8, 437–443 RSC.
- Z. Song, T. Hisatomi, S. Chen, Q. Wang, G. Ma, S. Li, X. Zhu, S. Sun and K. Domen, ChemSusChem, 2019, 12, 1906–1910 CrossRef CAS PubMed.
- K. Tsuji, O. Tomita, M. Higashi and R. Abe, ChemSusChem, 2016, 9, 2201–2208 CrossRef CAS PubMed.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390–8397 RSC.
- K. Song, J. Yang, P. F. Jiang, W. L. Gao, R. H. Cong and T. Yang, Eur. J. Inorg. Chem., 2015, 5786–5792 CrossRef CAS.
- S. R. Fu, B. B. Zhang, H. Y. Hu, Y. J. Zhang and Y. P. Bi, Catal. Sci. Technol., 2018, 8, 2789–2793 RSC.
- J. Jiang, J. G. Yu and S. W. Cao, J. Colloid Interface Sci., 2016, 461, 56–63 CrossRef CAS PubMed.
- C. Wang, H. Q. Fan, X. H. Ren, Y. Wen and W. J. Wang, Appl. Surf. Sci., 2018, 462, 423–431 CrossRef CAS.
- H. Suzuki, O. Tomita, M. Higashi and R. Abe, Catal. Sci. Technol., 2015, 5, 2640–2648 RSC.
- J. Yan, H. Wu, H. Chen, Y. Zhang, F. Zhang and S. F. Liu, Appl. Catal., B, 2016, 191, 130–137 CrossRef CAS.
- S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. Asai, Z. Li, S. S. K. Ma, F. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS PubMed.
- H. Hagiwara, M. Nagatomo, C. Seto, S. Ida and T. Ishiahara, Catalysts, 2013, 3, 614–624 CrossRef.
- X. Cheng, Y. H. Li, L. R. Zheng, Y. Yan, Y. F. Zhang, G. Chen, S. R. Sun and J. J. Zhang, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- M. K. Kundu, T. Bhowmik, R. Mishra and S. Barman, ChemSusChem, 2018, 11, 2388–2401 CrossRef CAS PubMed.
- M. Wang, Z. Li, Y. Q. Wu, J. T. Ma and G. X. Lu, J. Catal., 2017, 353, 162–170 CrossRef CAS.
- M. Wang, W. L. Zhen, B. Tian, J. T. Ma and G. X. Lu, Appl. Catal., B, 2018, 236, 240–252 CrossRef CAS.
- Z. Li, B. Tian, W. L. Zhen, Y. Q. Wu and G. X. Lu, Appl. Catal., B, 2017, 203, 408–415 CrossRef CAS.
- C. Jin, Y. Dai, W. Wei, X. C. Ma, M. M. Li and B. B. A. Huang, Appl. Surf. Sci., 2017, 426, 639–646 CrossRef CAS.
- F. S. Han, Z. H. Zhou, X. H. Zhang, Z. X. Huang, M. T. Li and L. J. Guo, J. Phys. Chem. C, 2018, 122, 2546–2553 CrossRef CAS.
- J. Xing, Y. H. Li, H. B. Jiang, Y. Wang and H. G. Yang, Int. J. Hydrogen Energy, 2014, 39, 1237–1242 CrossRef CAS.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem.–Eur. J., 2014, 20, 2138–2144 CrossRef CAS PubMed.
- Z. Jiang, M. A. Isaacs, Z. W. Huang, W. F. Shangguan, Y. F. Deng and A. F. Lee, ChemCatChem, 2017, 9, 4268–4274 CrossRef CAS.
- M. Ou, S. P. Wan, Q. Zhong, S. L. Zhang and Y. A. Wang, Int. J. Hydrogen Energy, 2017, 42, 27043–27054 CrossRef CAS.
- Y. Xue, Y. G. Lei, X. Y. Liu, Y. N. Li, W. N. Deng, F. Wang and S. X. Min, New J. Chem., 2018, 42, 14083–14086 RSC.
| This journal is © The Royal Society of Chemistry 2020 |