
Discordant nature of Cd in GeTe enhances phonon scattering and improves band convergence for high thermoelectric performance†

Abstract
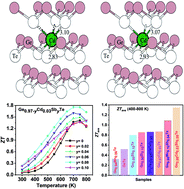
GeTe-based thermoelectric materials tend to be naturally heavily hole doped because of Ge lattice vacancies, and this complicates efforts to improve the figure of merit ZT. Herein, we report that partial substitution of Cd for Ge and doping with Sb can synergistically reduce the density of holes and improve the thermoelectric transport properties of GeTe. Density functional theory electronic structure calculations suggest that when Cd partially substitutes for Ge and forms CdxGe1−xTe, the energy offset between the light and heavy valence bands in both rhombohedral and cubic structures is reduced, promoting multi-band conduction that increases the hole carrier effective mass from 1.44m0 for pristine GeTe to 2.32m0 for Cd0.05Ge0.95Te. The ensuing valence band convergence enhances the Seebeck coefficient and yields a high power factor of 44.5 μW cm−1 K−2 at 800 K. Moreover, DFT calculations show a downward energy profile, indicating that as Cd enters the GeTe lattice it tends to move towards a hypothetical octahedral center to make six equal Cd–Te bonds. Such a local distortion in combination with the large mass and size difference between Ge and Cd strongly intensifies phonon scattering by point defects, lowering the lattice thermal conductivity in CdxGe1−xTe. Subsequent Sb doping in Cd0.03Ge0.97−ySbyTe reduces the inherent high carrier concentration in GeTe, leading to a remarkable enhancement of the Seebeck coefficient and the power factor over a wide temperature range. Furthermore, doping with Sb in Cd0.03Ge0.97−ySbyTe introduces additional point defect phonon scattering that results in an extremely low lattice thermal conductivity ∼ 0.71 W m−1 K−1 at 700 K for the Cd0.03Ge0.89Sb0.08Te sample. All those factors lead to a high thermoelectric figure of merit, ZT ∼ 1.8 at 700 K, and a high average ZT of 1.3 in the temperature interval of 400–800 K attained in Cd0.03Ge0.89Sb0.08Te.
- This article is part of the themed collection: 2020 Journal of Materials Chemistry A most popular articles
 Please wait while we load your content...
Please wait while we load your content...

