
In situ exsolution of Ni particles on the PrBaMn2O5 SOFC electrode material monitored by high temperature neutron powder diffraction under hydrogen†
 *a
Praveen B.
Managutti,
a
Vincent
Dorcet,
a
Annie
Le Gal La Salle,
b
Thomas C.
Hansen
c
*a
Praveen B.
Managutti,
a
Vincent
Dorcet,
a
Annie
Le Gal La Salle,
b
Thomas C.
Hansen
c
Abstract
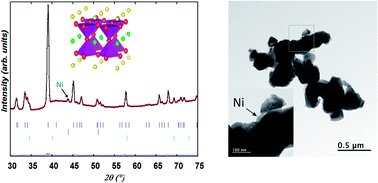
NiO has been incorporated into the Pr0.5Ba0.5MnO3−δ perovskite to produce, upon heating under a hydrogen atmosphere, in situ exsolved Ni-catalysts supported on the PrBaMn2O5 anode material. Transmission electron microscopy (TEM) and neutron powder diffraction (NPD) showed that the initial composition obtained by annealing in air at 950 °C consists of two perovskite phases: orthorhombic Pr0.65Ba0.35Mn0.975Ni0.025O3 (S.G. Ibmm, ∼75 wt%) and 2H-hexagonal BaMnO3−δ (S.G. P63/mcm, ∼25 wt%). On heating the two-phase sample under wet hydrogen, MnO particles exsolve at T ∼ 500 °C meanwhile the orthorhombic phase transforms to tetragonal (S.G. I4/mcm) then to cubic (S.G. Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) at T ∼ 665 °C. When the temperature approaches 900 °C, the emergence of Ni metal particles was detected in the neutron diffraction patterns meanwhile the two perovskite phases start to transform into a Ni-free layered double perovskite, PrBaMn2O5. In situ real time observation of the structural changes under hydrogen atmosphere provided evidence of the simultaneity of Ni exsolution and phase transformation within our timescale resolution. From quantitative Rietveld analysis, the fraction of exsolved nickel represents the whole amount of Ni introduced in the synthesis. Impedance spectroscopy measurements in a 5% H2/Ar atmosphere show promising electrochemical performance for the Ni-exsolved layered perovskite electrode with a polarization resistance of 0.4 Ω cm2 at 800 °C (0.135 Ω cm2 at 850 °C) without any optimization.
m) at T ∼ 665 °C. When the temperature approaches 900 °C, the emergence of Ni metal particles was detected in the neutron diffraction patterns meanwhile the two perovskite phases start to transform into a Ni-free layered double perovskite, PrBaMn2O5. In situ real time observation of the structural changes under hydrogen atmosphere provided evidence of the simultaneity of Ni exsolution and phase transformation within our timescale resolution. From quantitative Rietveld analysis, the fraction of exsolved nickel represents the whole amount of Ni introduced in the synthesis. Impedance spectroscopy measurements in a 5% H2/Ar atmosphere show promising electrochemical performance for the Ni-exsolved layered perovskite electrode with a polarization resistance of 0.4 Ω cm2 at 800 °C (0.135 Ω cm2 at 850 °C) without any optimization.
 Please wait while we load your content...
Please wait while we load your content...

