
Driving force balance—the “identity card” of supramolecules in a self-sorting multicomponent assembly system†
Si
Chen‡
 ,
Likang
Zhou‡
,
Zhihang
An
,
Huiwen
He
,
Meng
Ma
,
Yanqin
Shi
and
Xu
Wang
*
,
Likang
Zhou‡
,
Zhihang
An
,
Huiwen
He
,
Meng
Ma
,
Yanqin
Shi
and
Xu
Wang
*
College of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: wangxu@zjut.edu.cn; chensi@zjut.edu.cn; Fax: +86-0571-88320855; Tel: +86-0571-88320855
First published on 23rd October 2020
Abstract
Contrary to the popular belief that multicomponent assembly systems will theoretically co-assemble under the same type of driving forces, two distinct assembly modes from a system composed of two chemically similar supramolecules were demonstrated in this work. Although with exactly the same driving forces, molecule-level self-sorting unexpectedly occurred in this two-component system made of polyhedral oligomeric silsesquioxane (POSS) core-based supramolecules with one and eight lysine derivative arms. From the experiments, it was concluded that instead of driving force types, driving force counterpoise plays a vital role here, which we called “identity card hypothesis”. The hypothesis suggests that two highly similar components show high affinity for the same molecules through the differentiated “identity card”-like balance of driving forces induced by the difference in the molecular spatial shape, which has never been reported before.
Introduction
A self-sorting assembly, which with high-fidelity distinguishes “self” from “nonself”, is capable of self-assembling simultaneously and orthogonally in the multicomponent system.1–9 It can be used to prepare multifunctional materials for various fields, such as drug-delivery systems,10 cell adhesives,11 and photoelectric devices.12 Depending on the process of system formation, self-sorting systems could be divided into thermodynamic self-sorting and kinetic self-sorting.13 Thermodynamic self-sorting occurs because of the structural differences between the components. When multiple components have diverse types of driving forces due to different structures, each component displays a high affinity for itself and the self-sorting assembly occurs. On the contrary, when the components have similar chemical structures, they are no longer able to distinguish themselves under the same kind of driving forces, then co-assembly is facilitated.14 D. K. Smith et al. have reported that the self-sorting assembly can be constructed by two dendritic molecules under different types of driving forces provided by diverse peripheral groups.15 However, self-sorting systems consisted of the components with similar structures that could be obtained by kinetic external stimuli such as pH changes16 and ultraviolet light,17,18 while the disadvantage of kinetic self-sorting systems is that it is difficult to control accurately. Therefore, developing a simple method of self-sorting assembly in multicomponent systems with similar structures is crucial to extend the application of multifunctional self-sorting materials.Supramolecular assembly systems can utilize a variety of non-covalent bond interactions to make molecules self-assemble into ordered and complex structures, which are ideal candidates for constructing self-sorting assemblies.19–31 Among them, polyhedral oligomeric silsesquioxane (POSS) core-based supramolecules have attracted wide attention due to the unique three-dimensional (3D) cage-like POSS cores and a multi-branched outer “arm”, which endows them with highly controllable multiple peripheral groups and stereoscopic structures.32–37 However, previous related studies have focused on peripheral groups of POSS core-based molecules, while the study of the contribution of POSS cores to the self-assembly is limited.38–46 According to the studies reported in the literature, when the core is low-dimensional and non-stereometric, there is almost no stereo change in the molecular spatial shape, and the length of the arm and the number of peripheral groups have limited effects on the self-assembly of the molecules.47,48 However, the POSS core is 3D cage-shaped with a strong stacking capacity; previous studies have not fully utilized the spatial structure and assembly capability of POSS cores for self-assembly studies. Thus, understanding and control of the influence of high-dimensional cores in a molecular shape and further in the self-assembly process is of great significance to complement the mechanism of the supramolecular assembly.
It is well known that the assembly mode of the same molecule can be tuned by affecting the competitive relationship between thermodynamic and kinetic processes.49 The essence of this method is to affect the driving forces generated by thermodynamics or kinetics, so that the assembly mode is controlled by the dominant one. Similarly, for POSS core-based supramolecules with “cores” and peripheral “arms”, we hope that the ratio of cores to arms will be adjusted by modifying the molecular spatial shape. Furthermore, the proportion of the driving forces provided by cores and arms will change accordingly, and we will finally regulate the assembly mode of the molecule. Thus, PSS-Lys (shown in Fig. 1a and Scheme S1, ESI†) was designed and synthesized to obtain a typical tailored example to study the relationship between the molecular spatial shape and the assembly mode. Compared with the reported POSS core-based POSS-Lys (shown as Fig. 1b and Scheme S2, ESI†) with eight spatial symmetry lysine derivative arms,38PSS-Lys has exactly the same POSS core and only one lysine arm, and the remaining seven outer arms are isobutyl groups. Compared with the peripheral groups of POSS-Lys, the isobutyl groups are small in volume and similar to lysine terminal groups, and the influence of steric hindrance and hydrophobicity on the assembly process can be neglected. Thus, the exposure degree of POSS cores is greatly increased, which could gain unneglectable solvophobic interactions by its 3D structure. Therefore, the modification of the molecular spatial shape caused significant alterations of the proportion of two types of driving forces provided by cores and arms. As a result, the entire driving forces counterpoise changes, and a new balance will be achieved, which is distinct from the one formed by POSS-Lys. By transforming the molecular spatial shape, 3D cage-like POSS cores can effectively amplify the difference in the counterpoise of driving forces between PSS-Lys and POSS-Lys, and two discrepant assembly modes were obtained. Under the same type of driving forces, the two-component assembly system composed of PSS-Lys and POSS-Lys was further studied, which should theoretically co-assemble. Surprisingly, each individual component maintained its assembly mode through the unique balance of driving forces, which acted like an “identity card” of molecules, and this system performed self-sorting without any external stimuli. Herein, a two-component assembly system specifically designed to study the effect of differences in driving forces balance on self-sorting is reported.
 | ||
| Fig. 1 Chemical structure of (a) PSS-Lys and (b) POSS-Lys. | ||
Results and discussion
Assembly mode of PSS-Lys
In our previous research, the relationship between the driving forces and assembly modes was studied by performing coarse-grained molecular dynamics simulations45 (Fig. S1, ESI†). It suggested that the strength of these two driving forces was affected by the variation in the solvent polarity, which further led to two diverse assembly behaviours. In this paper, the proportion of two driving forces was adjusted to affect the supramolecular assembly mode by modifying the molecular spatial shape. PSS-Lys was synthesized to be compared with POSS-Lys (Fig. S2–S6, ESI†), and the correlation of molecular spatial shape and assembly behaviours was obtained. In terms of chemical structures, the two supramolecules have the same driving forces for self-assembly: the POSS cores provide the solvophobic effect; the arms provide hydrogen bonding of carboxyl and amino groups. However, via modifying the molecular spatial shape of PSS-Lys, the ratio of cores and arms changed. Thus, the equilibrium between these two driving forces of PSS-Lys will be completely discrepant from that of POSS-Lys. Based on this assumption, the assembly mode of PSS-Lys at the distinct balance of driving forces was studied through the following experiments.First, the gelation ability of PSS-Lys was examined through the tube inversion method in various organic solvents (Table 1). The results indicated that PSS-Lys was able to dissolve in various organic solvents of different polarities, resulting in a clear, transparent, non-viscous solution. In addition, it could not form a gel in these solvents even at a high concentration (80 mg mL−1). By contrast, POSS-Lys only needed an extremely low concentration (0.5 mg mL−1) to form a gel in the same organic solvents. It preliminarily indicated that the assembly behaviours of PSS-Lys were different from that of POSS-Lys. According to previous studies,38–46 the hydrogen bonding of arms tended to self-assemble into fibers,50,51 while the POSS cores tended to aggregate. Therefore, POSS-Lys with eight arms and one POSS core surrounded by arms had a higher proportion of hydrogen bonding than PSS-Lys, and the assembly behaviours tended to form a gel. While PSS-Lys had only a single lysine arm, the proportion of solvophobic effects generated by POSS cores increased, resulting in a decrease in the proportion of hydrogen bonds, so PSS-Lys failed to form a gel. It can be inferred that the driving force balance between the solvophobic effect and hydrogen bonding of PSS-Lys was inconsistent with POSS-Lys, resulting in the different assembly modes of PSS-Lys and POSS-Lys.
The assembly mode of PSS-Lys was further investigated by measuring the water contact angle of PSS-NH2 (Fig. 2a) and PSS-Lys after grafting from a lysine derivative arm (Fig. 2b). The samples were prepared by dissolving the powders in the solvent, and the solvent was evaporated at room temperature. The water contact angle of PSS-NH2 was 119.2°, and that of PSS-Lys was 68.3°. It turned out that PSS-NH2 was hydrophobic, and PSS-Lys was hydrophilic. As the POSS inorganic core consists of Si–O, an alternately connected silicon oxide skeleton was highly hydrophobic, and PSS-NH2 tended to be hydrophobic when the arms were hydrophilic amino groups, which indicated that the amino groups were distributed inside the assembly, while the POSS cores were exposed outside. This is due to the short length of the amino groups, which have little steric effects on POSS cores. POSS cores were exposed outside to a great extent, and its strong solvophobic effect led to agglomeration, forming an assembly with the cores outside and the arms inside. For PSS-Lys, when the highly hydrophobic POSS cores were clustered inside the assembly and the hydrophilic lysine arms were at the periphery, the assemblies were generally hydrophilic. This is because the lysine arms had a stronger steric effect than amino groups, which weakened the effect of the POSS cores. In addition, the lysine arms also provided hydrogen bonding, which formed a competitive relationship with the driving forces of POSS cores. The results were the assemblies with the cores inside and arms outside. Therefore, it can be speculated that PSS-Lys will tend to form the aggregates with POSS cores inside and arms outside.
 | ||
| Fig. 2 Contact angle of the film formed by (a) PSS-NH2 and (b) PSS-Lys after grafting from Lys. | ||
The assembly mode of PSS-Lys was compared with that of POSS-Lys. In addition, the differences between the two assembly morphologies were captured by scanning electron microscopy (SEM). In the methyl methacrylate (MMA), the morphology of POSS-Lys was a unique loofah-like fiber network (Fig. S7, ESI†). However, the morphology of PSS-Lys was irregularly lumpy aggregates (Fig. 3a), indicating that PSS-Lys tended to aggregate rather than assemble into fibers, unlike POSS-Lys, which also verified the previous experimental results. In terms of chemical structures, the hydrogen bonding provided by arms is very important, but due to the change in the molecular spatial shape, the solvophobic effect of POSS cores is also critical. POSS-Lys has eight lysine derivative arms, which result in stronger hydrogen bonding. Moreover, because of the steric hindrance effect, the POSS cores cannot approach each other, and hence, the eight lysine arms provided a fixation effect. POSS-Lys riveted together in a “face-to-face” stack to form a vertical assembly, and finally, the loofah-like network was obtained (Fig. S8, ESI†). However, PSS-Lys only has a single arm, and the hydrogen bonding is greatly weakened compared to POSS-Lys. Meanwhile, the steric hindrance effect of a single arm is so weak that the POSS cores cannot be fixed, and the solvophobic effect of POSS cores is amplified. Thus, in the PSS-Lys system, the balance of the two driving forces had changed significantly when compared to POSS-Lys, resulting in two completely different assembly modes. The XRD results showed that the 2θ value corresponding to the characteristic peaks of PSS-Lys was larger than that of POSS-Lys (Fig. 3b). According to Bragg's law, when the value of 2θ became larger, the crystal plane distance became smaller, which indicated that the distance between the POSS cores in the PSS-Lys assembly was closer than that in POSS-Lys, and PSS-Lys tended to agglomerate. In addition, because the strong solvophobic effect of POSS cores towards crystallization was dominant, compared to POSS-Lys, the characteristic peaks of PSS-Lys were much sharper, manifesting that the assembly of PSS-Lys was more orderly, which further validated the assembly of PSS-Lys. In conclusion, by changing the molecular spatial shape, the 3D POSS cores amplified the difference in the balance of driving forces between PSS-Lys and POSS-Lys, and two distinct assembly modes of the two molecules with similar chemical structures were obtained.
 | ||
| Fig. 3 (a) TEM images of PSS-Lys. (b) The XRD figure of PSS-Lys and POSS-Lys. | ||
Assembly mode of the system composed of PSS-Lys and POSS-Lys
The next level of complexity involved investigating the assembly behaviours of the two-component assembly system composed of PSS-Lys and POSS-Lys. The concentration of POSS-Lys in this system was set to its minimal gel concentration (0.5 mg mL−1). On the premise that POSS-Lys was assembled into the fiber network, PSS-Lys was introduced into the system at different molar ratios (PSS-Lys![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :POSS-Lys = 1:1, 4:1, and 8:1).
:POSS-Lys = 1:1, 4:1, and 8:1).
In order to study the assembly mode of this two-component assembly system, the gelation ability of this system in different proportions was tested by the tube inversion method in major gelation solvents (Table 2). The mixture systems were still capable of forming a transparent gel in gelation solvents, the gelation ability of POSS-Lys did not change significantly with the addition of PSS-Lys. In addition, the gel–sol transition temperature (Tgel) of the POSS-Lys gel and PSS-Lys/POSS-Lys mixed gel was assessed at different molar ratios (Fig. S9 and S10, ESI†). It can be seen that the Tgel values of the POSS-Lys and PSS-Lys/POSS-Lys gels at different concentrations were almost the same (40 ± 1 °C, 58 ± 1 °C, 70 ± 1 °C, 76 ± 1 °C, and 78 ± 1 °C). Clearly, PSS-Lys, which could not form a gel, did not appear to disrupt the gelation ability of POSS-Lys. This phenomenon is consistent with the fact that the gelator with a weaker gelation ability in the self-sorting assembly cannot affect another gelator with strong gelation ability.15 Furthermore, microscopic observation is used to verify the assembly mode of this system, which is the most intuitionistic and effective way for assembly research. Interestingly, regardless of the PSS-Lys:POSS-Lys ratio, the morphology always turned out to be a mixture of two microstructures (irregular lumpy aggregates and loofah-like fibers) without the distinguishable difference in the SEM and TEM images (Fig. 4a–f). Each component maintained their individual assembly mode in this system. Moreover, as the concentration of PSS-Lys increased, the number and size of aggregates of PSS-Lys were getting larger, from 0.7 μm (PSS-Lys:POSS-Lys = 1:1, mol:mol) to 1.4 μm (PSS-Lys:POSS-Lys = 4:1, mol:mol), and then to 1.8 μm (PSS-Lys:POSS-Lys = 8:1, mol:mol). The surplus PSS-Lys gel still shows high affinity for itself instead of co-assembling with POSS-Lys. Therefore, the microscopic images shown in Fig. 4 can be considered a self-sorting assembly. Moreover, by analysing the energy-dispersive X-ray spectra of white lumpy aggregates and loofah-like fiber networks in this mixture system consisting of PSS-Lys:POSS-Lys = 1:1 (mol:mol) shown in Fig. 4a, the results indicated that PSS-Lys contains 51.6% C,4.9% N, 24.0% O, and 19.5% Si and POSS-Lys contains 57.0% C, 10.7% N, 25.2% O, and 7.1% Si (Fig. S11, ESI†). The element content of white lumpy aggregates was basically consistent with the theoretical content of PSS-Lys (C: 47.3%; N: 4.5%; O: 22.0%; Si: 18.0%; and H: 8.4%), and the element content of loofah-like fiber networks was consistent with the theoretical content of POSS-Lys (C: 52.0%; N: 9.6%; O: 23.7%; Si: 6.4%; and H: 8.3%). It is obvious that there was a significant difference in the content of nitrogen and silicon, which proved that the aggregation of PSS-Lys dispersed individually in the loofah-like fiber networks of POSS-Lys. These phenomena further proved intuitively that self-sorting occurred under the same type of driving forces in the two-component assembly system.
 | ||
| Fig. 4 SEM images and TEM images of the PSS-Lys/POSS-Lys two-component system. (a, c and e) SEM images of the PSS-Lys:POSS-Lys = 1:1, 4:1, and 8:1 (mol:mol) xerogel, 5 mg mL−1. (b, d and f) TEM images of the PSS-Lys:POSS-Lys = 1:1, 4:1, and 8:1 (mol:mol) xerogel, 5 mg mL−1. | ||
The assembly mode of this system was also studied at the molecular level by Fourier-transform infrared (FT-IR) spectroscopy.52 The FT-IR spectra can provide insights into the molecular interactions such as hydrogen bonding between carboxyl and amino groups in this complex system, which is one of the major driving forces in this system. It has been reported that, if a system were self-sorted, then we would expect no change in the absorption of the individual components and the combined spectrum should be similar to the overlay of the two spectra for the individual components.53 However, when the co-assembly occurs, this could be seen directly in the FT-IR spectra, with the appearance of new peaks and the shift of original peaks.54 Herein, the IR spectra of PSS-Lys, POSS-Lys xerogel and xerogel of PSS-Lys:POSS-Lys = 1:1, 4:1, and 8:1 (mol:mol) were obtained (Fig. 5). Since the hydrogen bonding effect is one of the major driving forces of this system, we focused on the peak shift for carboxyl and amino groups, which can form hydrogen bonding, and the appearance of the new peaks to probe the packing mode of the two-component gel system. As shown in Fig. 5a, there was almost no shifting in the stretching vibration peaks of N–H from amino groups (νN–H = 3349 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from carboxyl groups (νCO = 1710 cm−1). If the co-assembly occurs, there will be a hydrogen bond interaction between the two components, then the infrared absorption peaks of N–H and CO will inevitably shift to a lower wavenumber. In addition, there was no new peak in this spectra, for example, in Fig. 5b, both the single stretching vibration peak of C–H of PSS-Lys (νC–H = 2955 cm−1) and the double peaks of POSS-Lys (νC–H = 2977 cm−1 and νC–H = 2932 cm−1) still existed in the two-component gel system without any shifting, regardless of the ratio of PSS-Lys and POSS-Lys. In the two-component system, the stretching vibration peaks of C–H were only the superposition of the peaks of PSS-Lys and POSS-Lys. Therefore, the spectra of the mixed system were simply an overlay of the spectra for the two components. Based on the results of FT-IR, the assembly mode of this two-component system was self-sorting.
O from carboxyl groups (νCO = 1710 cm−1). If the co-assembly occurs, there will be a hydrogen bond interaction between the two components, then the infrared absorption peaks of N–H and CO will inevitably shift to a lower wavenumber. In addition, there was no new peak in this spectra, for example, in Fig. 5b, both the single stretching vibration peak of C–H of PSS-Lys (νC–H = 2955 cm−1) and the double peaks of POSS-Lys (νC–H = 2977 cm−1 and νC–H = 2932 cm−1) still existed in the two-component gel system without any shifting, regardless of the ratio of PSS-Lys and POSS-Lys. In the two-component system, the stretching vibration peaks of C–H were only the superposition of the peaks of PSS-Lys and POSS-Lys. Therefore, the spectra of the mixed system were simply an overlay of the spectra for the two components. Based on the results of FT-IR, the assembly mode of this two-component system was self-sorting.
 | ||
| Fig. 5 (a) FT-IR spectra of PSS-Lys, POSS-Lys and PSS-Lys:POSS-Lys = 1:1, 4:1, and 8:1 (mol:mol) xerogel obtained from MMA. (b) Amplified image of the dashed box in (a). | ||
The rheological measurements are also crucial to understand the self-sorting assembly system, when the concentration of PSS-Lys remained unchanged (Fig. 6). It was found that with the increase in the concentration of POSS-Lys, the storage modulus (G′) and loss modulus (G′′) gradually increased, which meant that POSS-Lys self-assembled into a denser and stabler nanofiber network with the increase in concentration. This proved that the assemblies of POSS-Lys were dominant in this two-component system.
 | ||
| Fig. 6 Frequency dependence of G′ and G′′ for PSS-Lys/POSS-Lys system, measured at 25 °C with a frequency from 0.1 to 100 rad s−1 and strain g = 0.1%. PSS-Lys/POSS-Lys with 1.4%:0.5% and 1.4% were prepared in MMA. | ||
Similarly, when the concentration of POSS-Lys was constant (Fig. 7a and b). The data showed that the mechanical strength of this system was almost unchanged with the increase in concentration of PSS-Lys. As shown in SEM images (Fig. 4), the size of PSS-Lys assemblies was much smaller than that of POSS-Lys. In addition, the assembly mode of this system was self-sorting, which meant that the two components self-assembled separately. Therefore, the increase in the PSS-Lys aggregates had almost no effect on the mechanical strength of the entire system. Hence, the rheological results proved that the self-sorting assembly occurred under the same kind of driving forces.
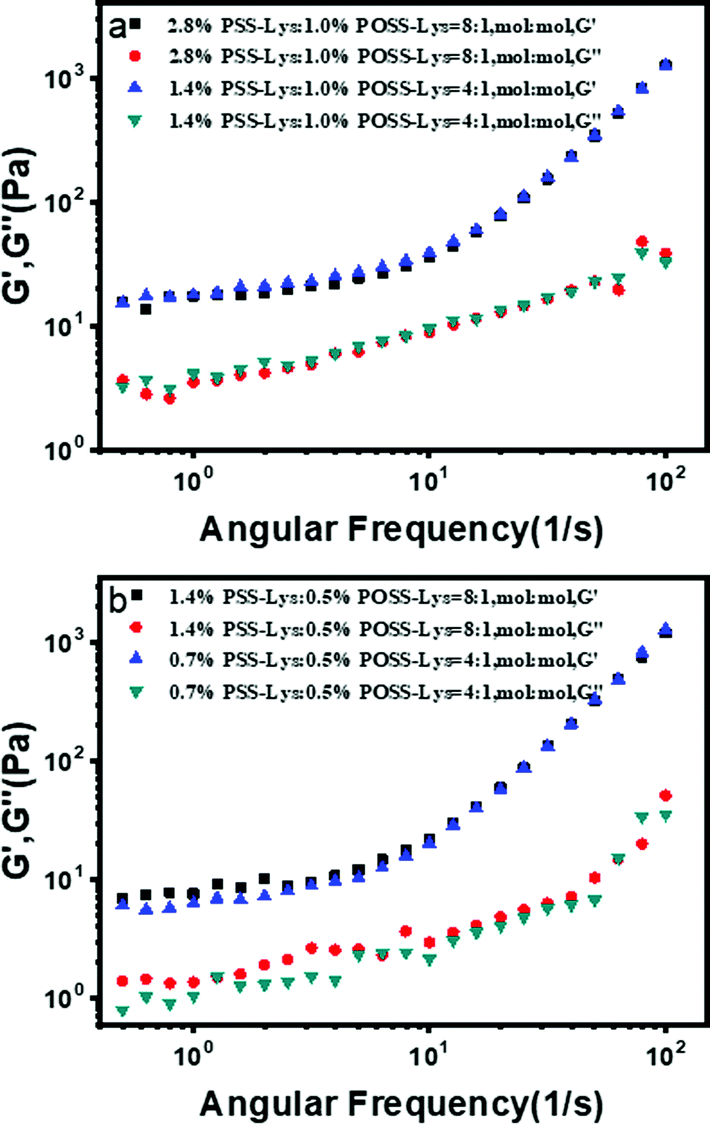 | ||
| Fig. 7 Frequency dependence of G′ and G′′ for the PSS-Lys/POSS-Lys system, measured at 25 °C with a frequency from 0.1 to 100 rad s−1 and strain g = 0.1%. (a) PSS-Lys/POSS-Lys with 2.8%:1.0% and 1.4% were prepared in MMA. (b) PSS-Lys/POSS-Lys with 1.4%:0.5% and 0.7% were prepared in MMA. | ||
Based on this anomalous phenomenon of self-sorting in this assembly system composed of two similar chemical structural components, PSS-Lys and POSS-Lys, one hypothesis was proposed: under multiple driving forces, each supramolecule will have a unique balance of driving forces in the assembly process. This balance of driving forces can be considered as a molecular “identity card”. In this system, the “identity card” was the balance of the solvophobic effect provided by POSS cores and the hydrogen bonding provided by lysine arms, the counterpoise of which plays a key role instead of each one individually. As shown in Fig. 8, due to the difference in the molecular spatial shape, the proportions of driving forces in the two molecules changed greatly, which made the equilibrium of driving forces unique to each component. The distinction of driving force balance between PSS-Lys and POSS-Lys led to their very differentiated assembly mode. It is this “identity card” that enabled them to show high affinity for the same molecules without any interference with other molecules and eventually forming the self-sorting assembly in the complex two-component system.
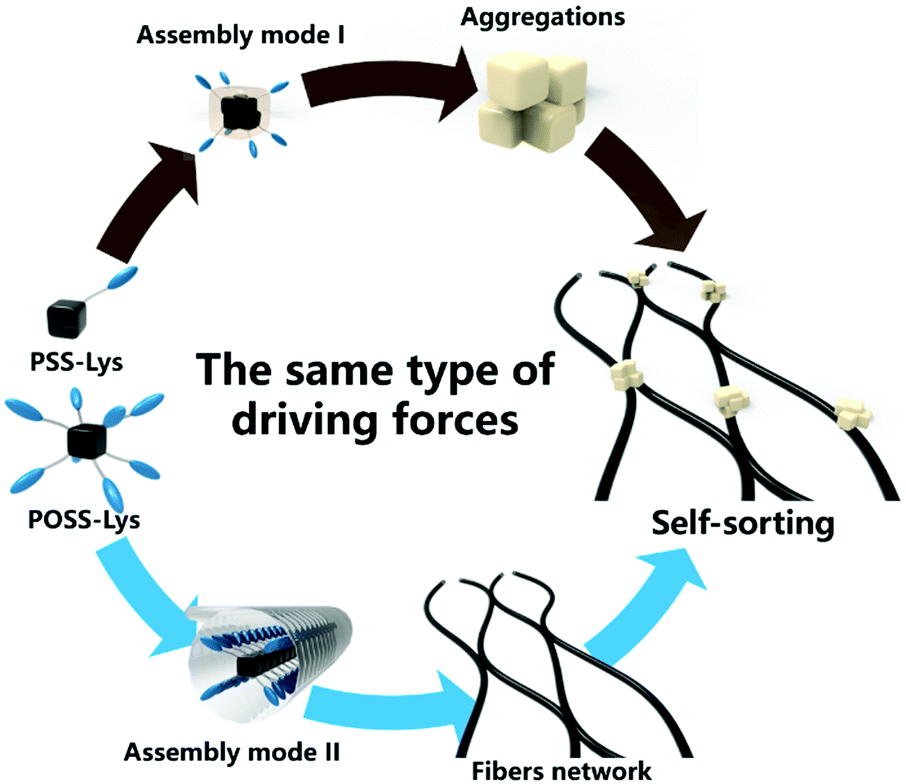 | ||
| Fig. 8 Schematic of the PSS-Lys/POSS-Lys two-component assembly system. | ||
Conclusions
In this paper, the assembly behaviours of PSS-Lys with one lysine derivative arm and POSS-Lys with eight arms were studied. By changing the molecular spatial shape, the 3D cage-like POSS cores magnified the difference in the balance of driving forces provided by the POSS cores and lysine arms. Through a series of tests, it was proved that two distinct assembly modes of PSS-Lys and POSS-Lys with similar chemical structures can be obtained. Furthermore, a two-component assembly system was constructed from these two supramolecules with similar chemical structures to study the assembly mode of this system under the same type of driving forces. Unexpectedly, a series of studies had shown that this two-component system self-sorted, which theoretically tended to form a co-assembly. Moreover, two highly similar components showed high affinity for the same molecules through the differentiated “identity card”-like equilibrium relationship between two kinds of driving forces, and hence, the precise self-sorting occurred in the complicated two-component system. It provides a new strategy for the precise self-sorting mechanism without any external stimuli, which helps develop multifunctional self-sorting supramolecular materials.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Lehn, Science, 1985, 227, 849–856 CrossRef CAS.
- J. M. Lehn, Science, 1993, 260, 1762–1763 CrossRef CAS.
- M. C. LeMieux, M. Roberts, S. Barman, Y. W. Jin, J. M. Kim and Z. Bao, Science, 2008, 321, 101–104 CrossRef CAS.
- M. M. Safont-Sempere, G. Fernandez and F. Wurthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS.
- F. Wang, C. Y. Han, C. L. He, Q. Z. Zhou, J. Q. Zhang, C. Wang, N. Li and F. H. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS.
- J. F. Ayme, J. M. Lehn, C. Bailly and L. Karmazin, J. Am. Chem. Soc., 2020, 142, 5819–5824 CrossRef CAS.
- J. Deng, D. Bezold, H. Jessen and A. Walther, Angew. Chem., Int. Ed., 2020, 132, 12182–12190 CrossRef.
- Y. Yang, H. Hu, L. Chen, H. Bai, S. Wang, J. F. Xu and X. Zhang, Mater. Chem. Front., 2019, 3, 806–811 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- K. Sugiyasu, S. I. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- A. X. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- S. Bloom, C. Liu, D. K. Kolmel, J. X. Qiao, Y. Zhang, A. M. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS.
- J. M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379 RSC.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS.
- L. He, S. C. Wang, L. T. Lin, J. Y. Cai, L. Li, T. H. Tu and Y. T. Chan, J. Am. Chem. Soc., 2020, 142, 7134–7144 CrossRef CAS.
- Q. Shi, X. Zhou, W. Yuan, X. Su, A. Neniskis, X. Wei, L. Taujenis, G. Snarskis, J. S. Ward, K. Rissanen, J. de Mendoza and E. Orentas, J. Am. Chem. Soc., 2020, 142, 3658–3670 CrossRef CAS.
- H. Barbero, N. A. Thompson and E. Masson, J. Am. Chem. Soc., 2020, 142, 867–873 CrossRef CAS.
- K. Aratsu, R. Takeya, B. R. Pauw, M. J. Hollamby, Y. Kitamoto, N. Sihmizu, H. Takagi, R. Haruki, S. I. Adachi and S. Yagai, Nat. Commun., 2020, 11, 1623 CrossRef CAS.
- Y. Li, C. Liu, X. Bai, F. Tian, G. Hu and J. Sun, Angew. Chem., Int. Ed., 2020, 59, 3486–3490 CrossRef CAS.
- Y. Wang, T. K. Piskorz, M. Lovrak, E. Mendes, X. Guo, R. Eelkema and J. H. van Esch, Adv. Sci., 2020, 7, 1902487 CrossRef CAS.
- X. Shi, X. Zhang, X. L. Ni, H. Zhang, P. Wei, J. Liu, H. Xing, H. Q. Peng, J. W. Y. Lam, P. Zhang, Z. Wang, H. Hao and B. Z. Tang, Macromolecules, 2019, 52, 8814–8825 CrossRef CAS.
- L. Volbach, N. Struch, F. Bohle, F. Topic, G. Schnakenburg, A. Schneider, K. Rissanen, S. Grimme and A. Lutzen, Chemistry, 2020, 3335–3347 CrossRef CAS.
- Y. T. Tsai, G. Raffy, H. F. Liu, B. J. Peng, K. P. Tseng, L. Hirsch, A. Del Guerzo, D. M. Bassani and K. T. Wong, Mater. Chem. Front., 2020, 4, 845–850 RSC.
- M. A. Castriciano, N. Leone, P. Cardiano, S. Manickam, L. M. Scolaro and S. Lo Schiavo, J. Mater. Chem. C, 2013, 1, 4746–4753 RSC.
- M. B. Hu, Z. Y. Hou, W. Q. Hao, Y. Xiao, W. Yu, C. Ma, L. J. Ren, P. Zheng and W. Wang, Langmuir, 2013, 29, 5714–5722 CrossRef CAS.
- H. K. Shin, C. C. Hsieh, M. G. Mohamed, C. Y. Zhu and S. W. Kuo, Soft Matter, 2016, 12, 1847–1858 RSC.
- G. Tang, X. Wang, D. Li, Y. Ma and D. Wu, Polym. Chem., 2018, 9, 5377–5384 RSC.
- L. Fan, X. Wang, Q. Cao, Y. Yang and D. Wu, Biomater. Sci., 2019, 7, 1984–1994 RSC.
- N. Lu, Y. Lu, S. Liu, C. Jin, S. Fang, X. Zhou and Z. Li, Biomacromolecules, 2019, 20, 3485–3493 CrossRef CAS.
- G. Tang, S. Chen, F. Ye, X. Xu, J. Fang and X. Wang, Chem. Commun., 2014, 50, 7180–7183 RSC.
- S. Chen, X. Luo, H. He, X. Tong, B. Wu, M. Ma and X. Wang, J. Mater. Chem. C, 2015, 3, 12026–12031 RSC.
- H. He, S. Chen, J. Bai, H. Zheng, B. Wu, M. Ma, Y. Shi and X. Wang, RSC Adv., 2016, 6, 34685–34691 RSC.
- H. He, S. Chen, X. Tong, Y. Chen, B. Wu, M. Ma, X. Wang and X. Wang, Soft Matter, 2016, 12, 957–964 RSC.
- S. Chen, X. Tong, H. He, M. Ma, Y. Shi and X. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 11924–11932 CrossRef CAS.
- Z. An, S. Chen, X. Tong, H. He, J. Han, M. Ma, Y. Shi and X. Wang, Langmuir, 2019, 35, 2649–2654 CrossRef CAS.
- X. Tong, T. Shan, Q. Du, Z. An, H. He, M. Ma, Y. Shi, S. Chen and X. Wang, Adv. Electron. Mater., 2019, 5, 1900373 CrossRef.
- T. Shan, L. Gao, X. Tong, Q. Du, Z. An, H. He, J. Lin, S. Chen and X. Wang, Chem. – Eur. J., 2019, 25, 12098–12104 CrossRef CAS.
- H. He, H. Zheng, M. Ma, Y. Shi, Z. Gao, S. Chen and X. Wang, Chem. Commun., 2020, 56, 2983–2986 RSC.
- V. Percec, P. W. Chu, G. Ungar and J. P. Zhou, J. Am. Chem. Soc., 1995, 117, 11441–11454 CrossRef CAS.
- R. Zhang, X. Feng, R. Zhang, W. Shan, Z. Su, J. Mao, C. Wesdemiotis, J. Huang, X. Y. Yan, T. Liu, T. Li, M. Huang, Z. Lin, A. C. Shi and S. Z. D. Cheng, Angew. Chem., Int. Ed., 2019, 58, 11879–11885 CrossRef CAS.
- J. Matern, Y. Dorca, L. Sanchez and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS.
- D. Yan, G. R. Williams, M. Zhao, C. Li, G. Fan and H. Yang, Langmuir, 2013, 29, 15673–15681 CrossRef CAS.
- J. Puigmarti-Luis, A. Minoia, A. P. del Pino, G. Ujaque, C. Rovira, A. Lledos, R. Lazzaroni and D. B. Amabilino, Chem. – Eur. J., 2006, 12, 9161–9175 CrossRef CAS.
- Y. M. Abul-Haija, S. Roy, P. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS.
- K. Sugiyasu, S. I. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- P. Xing, X. Chu, S. Li, F. Xin, M. Ma and A. Hao, New J. Chem., 2013, 37, 3949–3955 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01405b |
| ‡ Si Chen and Likang Zhou contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |