
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConductive hydrogel composites with autonomous self-healing properties†
Xiaohui
Li
ab,
Xia
Huang
ab,
Hatice
Mutlu
 b,
Sharali
Malik
c and
Patrick
Theato
*ab
b,
Sharali
Malik
c and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 18, D-76131 Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bSoft Matter Synthesis Laboratory, Institute for Biological Interfaces III (IBG 3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Quantum Materials and Technology, Karlsruhe Institute of Technology (KIT), Hermann von Helmholtz Platz 1, D-76131 Karlsruhe, Germany
First published on 15th October 2020
Abstract
Conventional conductive hydrogels usually lack self-healing properties, but might be favorable for smart electronic applications. Therefore, we present the fabrication of conductive self-healing hydrogels that merge the merits of electrical conductivity and self-healing properties. The conductive self-healing hydrogel composite was prepared by using single-walled carbon nanotubes (SWCNTs), poly(vinyl alcohol) (PVA), and a poly(N,N-dimethyl acrylamide) copolymer derivative modified with pyrene and borate functional moieties. While the tethered pyrene groups of the copolymer facilitated an even dispersion of the conductive components, i.e., SWCNTs, in aqueous solution via π–π stacking, the hydrogel system was formed via covalent dynamic cross-linking through tetrahedral borate ion interaction with the –OH group of PVA. The hydrogel composites exhibited bulk conductivity (1.27 S m−1 with 8 mg mL−1 SWCNTs) with a fast and autonomous self-healing ability that restored 95% of the original conductivity within 10 s under ambient conditions. Accordingly, due to their outstanding properties, we postulate that these composites may have potential in biomedical applications, such as tissue engineering, wound healing or electronic skins.
Introduction
Hydrogels are wet and soft materials with a three-dimensional network structure, in which hydrophilic polymer chains are physically or chemically cross-linked with each other, hence enabling swelling in water. Therefore, hydrogels have emerged as suitable materials in biomedical fields because of their distinctive features, such as good biocompatibility, adjustable stiffness and porous structure.1–3Conductive hydrogels, with the combination of electrical conductivity and a human tissue-like nature, have been extensively employed in bioelectric fields during the past decades, such as in biosensors,4,5 bio-actuators6,7 and artificial e-skins.8,9 Indeed, conductive hydrogels could be fabricated directly by blending conducting fillers into the hydrogel network, like metallic nanoparticles,10 single and multi-walled carbon nanotubes (CNTs)11 and graphene nanosheets.12 Nevertheless, aggregation of the fillers usually occurs in the complex systems, leading to poor conductivity and inhomogeneous mechanical properties at the same time.13–15 For CNTs, a common approach to tackle this problem is to manipulate the surface of CNTs, which involves functionalization and direct attachment of chemical moieties (such as carbenes, nitrenes, and thiolethers) to the sidewalls of CNTs. However, π-conjugation of nanotubes is disrupted during the chemical treatment process, which results inevitably in changes in their electronic properties.16 Alternatively, noncovalent modification, which involves physical adsorption of functional moieties on the CNT surface via π–π stacking, or hydrophobic, electrostatic or van der Waals forces, has received interest.17 Among the broad toolbox of functional groups, pyrene moieties, either as small molecules or polymer derivatives, are commonly used due to their strong interaction with the sidewalls of the CNTs through π–π stacking. Accordingly, the latter process facilitates dispersion of CNTs in organic solvents, as well as in aqueous solution.18
Despite being endowed with electrical conductivity, conventional conductive hydrogels still fail to completely mimic human skin, particularly due to their lack of self-healing ability. Thus, it still remains a challenge to fabricate functional hydrogels with electrical conductivity and inherent self-healing ability, which could pave the way to more suitable and durable materials for biomedical applications, such as tissue engineering, wound healing or electronic skins.
A facile way to achieve autonomous and inherent self-healing properties is to introduce dynamic and reversible bonds to a hydrogel. In fact, diverse noncovalent dynamic bonds, like host–guest interaction,19 hydrogen bonding,20 hydrophobic interaction21 and electrostatic interaction,22 as well as covalent bonds, such as the disulfide bond,23 imine bond,24 metal–ligand coordination25 and boronate ester bond,26,27 amongst others, have been investigated in-depth in this regard. Among the plethora of dynamic and reversible systems, the formation of boronate esters could be easily achieved in aqueous solutions through reversible complexation between boronic acids and diols (either 1,2- or 1,3-), imparting a hydrogel with self-healing characteristics upon damage. While several self-healable hydrogels based on dynamic boronate ester bonds have been developed in recent years,28–31 the toolbox of hydrogels decorated with boronate ester bonds has not been expanded yet to conductive hydrogels.
Accordingly, herein, we propose a SWCNT-based conductive hydrogel composite with autonomous self-healing properties. To achieve this, a functional copolymer decorated with pendent pyrene and phenylboronic acid moieties is synthesized via sequential reversible addition–fragmentation chain transfer (RAFT) polymerization and post-polymerization modification. Essentially, pyrene moieties are included to facilitate the homogenous dispersion of SWCNTs in aqueous media, thus alleviating SWCNT aggregation, and endowing the hydrogels with electrical conductive properties. Furthermore, phenylboronic acid moieties were able to provide dynamic boronate ester linkages with diol groups of PVA chains. Thereby, the reversible association/dissociation process between boronic acid and diol groups is expected to contribute to the self-healing ability upon damage. Last but not least, the conductivity and self-recovery ability in electrical conductivity will be investigated.
Experimental section
Materials
N,N-Dimethyl acrylamide (DMA, 99%, Sigma-Aldrich) was purified by passing through a basic aluminium column before polymerization, and 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%, Sigma-Aldrich) was recrystallized from methanol before polymerization. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (CDTPA, 97%, TCI), 1-pyrenebutyric acid (97%, Sigma-Aldrich), pentafluorophenyl trifluoroacetate (98%, Sigma-Aldrich), 1,6-hexamethylenediamine (98%, Sigma-Aldrich), (4-aminomethylphenyl)boronic acid hydrochloride (98%, abcr), poly(vinyl alcohol) (PVA, 98%, TCI, Mw ∼ 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), trimethylamine (TEA) and other solvents were used as received. Ultra-pure water was obtained from GenPure Pro™ (Thermo scientific). Pentafluorophenyl acrylate was synthesized according to the published literature.32 SWCNTs were supplied from OCSiAl Europe and heated in air at 500 °C for 30 minutes to remove amorphous carbon material before use.
000 g mol−1), trimethylamine (TEA) and other solvents were used as received. Ultra-pure water was obtained from GenPure Pro™ (Thermo scientific). Pentafluorophenyl acrylate was synthesized according to the published literature.32 SWCNTs were supplied from OCSiAl Europe and heated in air at 500 °C for 30 minutes to remove amorphous carbon material before use.
Synthesis of perfluorophenyl 4-(pyren-1-yl)butanoate
1-Pyrenebutyric acid (1.2 g, 4.2 mmol, 1.0 eq.) and trimethylamine (1.72 mL, 12.6 mmol, 3.0 eq.) were dissolved in 10 mL of dry THF, and then pentafluorophenyl trifluoroacetate (2.2 mL, 12.6 mmol, 3.0 eq.) was added dropwise. The solution was stirred overnight at ambient temperature. Afterwards, THF was removed under reduced pressure. The residue was re-dissolved in 100 mL of DCM and washed with water (3 × 20 mL). The combined organic solvent was dried with anhydrous MgSO4 and then condensed. The final product was obtained as a yellow powder after being purified by column chromatography (silica, DCM) and dried under vacuum at 45 °C (1.4 g, yield 73.7%).1H-NMR (400 Hz, CDCl3, δ in ppm): 8.32 (d, J = 9.2 Hz, 1H), 8.24–8.13 (m, 4H), 8.12–7.99 (m, 3H), 7.91 (d, J = 7.7 Hz, 1H), 3.58–3.46 (m, 2H), 2.81 (t, J = 7.2 Hz, 2H), 2.42–2.31 (m, 2H).
19F-NMR (377 MHz, CDCl3, δ in ppm): −152.71 (ortho), −158.03 (para), −162.28 (meta).
FT-IR: ν (cm−1): 1774 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester bond), 1520 (aromatic CC from PFP).
O ester bond), 1520 (aromatic CC from PFP).
Synthesis of N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB)
In a round flask, 1,6-hexamethylenediamine (2.04 g, 17.5 mmol, 20 eq.) and TEA (0.24 mL, 1.75 mmol, 2 eq.) were dissolved into 10 mL of THF, and then perfluorophenyl 4-(pyren-1-yl)butanoate (0.4 g, 0.88 mmol, 1.0 eq.) was dissolved in 2 mL of THF and added dropwise. The mixture was stirred overnight at ambient temperature, and then THF was removed under reduced pressure. The residue was re-dissolved in 50 mL of DCM and washed with water (3 × 10 mL). The combined organic solvent was dried with anhydrous MgSO4 and then condensed under reduced pressure. The final product was obtained as a yellow powder after being dried under vacuum at 45 °C (240 mg, yield 70.6%).1H-NMR (400 MHz, DMSO, δ in ppm): 8.48–7.88 (m, 9H), 3.33–3.29 (m, 2H), 3.06 (dd, J = 12.8, 6.6 Hz, 2H), 2.50–2.47 (m, 2H), 2.23 (t, J = 7.2 Hz, 2H), 2.08–1.96 (m, 2H), 1.44–1.37 (m, 2H), 1.35–1.20 (m, 6H).
FT-IR: ν (cm−1): 3300 (N–H stretching vibration), 1650 (amide band I), 1550 (amide band II).
Synthesis of poly(N,N-dimethyl acrylamide-co-pentafluorophenyl acrylate) (P(DMA-co-PFPA)) (P1)
Pentafluorophenyl acrylate (PFPA) was synthesized according to a previously published procedure.27 Accordingly, the hydrophilic copolymer P(DMA-co-PFPA) was synthesized via typical RAFT polymerization. Briefly, N,N-Dimethyl acrylamide (20 mmol, 2.0 g), pentafluorophenyl acrylate (2.2 mmol, 0.534 g), the RAFT agent CDTPA (0.075 mmol, 30 mg), AIBN (0.0075 mmol, 1.2 mg) and 6 mL of dried 1,4-dioxane were added into a pre-dried Schlenk tube. The mixture was degassed three times through freeze–thaw cycles. Subsequently, the Schlenk tube was transferred into a pre-heated oil bath at 70 °C and stirred for an additional 16 h. The reaction was terminated by cooling down in liquid nitrogen and then exposure to air. The crude copolymer was purified via precipitation from THF into ice cold n-hexane three times. The final product was dried under vacuum at 45 °C overnight (2.3 g, yield 92%).1H-NMR (400 Hz, CDCl3, δ in ppm): 3.18–2.76 (d,7H, –CH2–CH– from the backbone of PPFPA and –N(CH3)2 from the side chain of PDMA), 2.75–2.54 (s, 1H, CH2–CH in the backbone of PDMA), 2.0–1.0 (m, 2H, –CH2–CH– in the backbone of PPFPA and PDMA), 0.88 (t, 3H, –CH3 from CTA).
19F-NMR (377 MHz, CDCl3, δ in ppm): −152.54 (ortho), −157.44 (para), −162.05 (meta) from PPFPA.
GPC: Mn,GPC = 2.32 × 104 g mol−1, Mw/Mn = 1.44.
Synthesis of poly(N,N-dimethyl acrylamide-co-aminohexyl pyren-butane acrylamide-co-methylphenylboronic acid acrylamide) (P(DMA-co-APB-co-PBA)) (P2)
P(DMA-co-APB-co-PBA) (P2) was synthesized via a sequential post-polymerization modification process. Briefly, P(DMA-co-PFPA) (1.0 g, containing 1 mmol of PPFPA) was dissolved into 6 mL of anhydrous DMF in a vial. To this, N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB) (115.8 mg, 0.3 mmol) in 1 mL of dry DMF was added, followed by 0.15 mL of TEA. The vial was sealed with a rubber septum and placed into a pre-heated oil bath at 45 °C. After 6 h, an excess of (4-aminomethylphenyl)boronic acid hydrochloride (PBA) (190.5 mg, 1.05 mmol) was dissolved into 1 mL of dry DMF and added into the reaction mixture. Subsequently, the mixture was stirred overnight at 45 °C. After that, the copolymer was purified via dialysis against MeOH for 24 h, and DI water, respectively, for another 24 h. The final product was isolated as a pale yellow solid after lyophilisation (850 mg, yield 85%).1H-NMR (400 Hz, CDCl3, δ in ppm): 8.30–7.86 (m, 9H, pyrene group), 7.85–7.67 (s, 2H, Ar–B(OH)2), 7.28–7.18 (s, 2H, Ar–B(OH)2), 3.37–3.21 (s, 2H, –NH–CH2-Ar), 3.18–2.76 (d, 6H, –N(CH3)2 from the side chain of PDMA), 2.75–2.0 (m, 1H, –CH2–CH in the backbone), 2.0–1.0 (m, 2H, –CH2–CH– in the backbone).
FT-IR: ν (cm−1): 3433 (broad, ν–OH– in Ar-B(OH)2), 1720 (ν–CO– in pendent N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB) moieties).
GPC: Mn,GPC = 2.69 × 104 g mol−1, Mw/Mn = 1.40.
Preparation of hydrogel composites with SWCNTs
100 mg of copolymer P2 (containing ∼11.6 mg, 0.03 mmol of N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB)) was dissolved in 0.9 mL of purified DI water in a vial with different amounts of SWCNTs (3, 6, 9 and 12 mg, respectively). The vial was sonicated for 20 min to evenly disperse CNTs in the aqueous media. Afterwards, 600 mg of PVA solution (10 wt%) was added, and the mixture was stirred heavily until gelation was observed. The final concentration of SWCNTs was 2, 4, 6, and 8 mg mL−1, respectively.Characterization
All 1H- and 19F-NMR measurements were recorded using a Bruker Avance 400 spectrometer (400 MHz for 1H and 377 MHz for 19F) with tetramethylsilane as the internal standard, and all the samples were dissolved in deuterated solvents before measurements. Gel permeation chromatography (GPC) was carried out in DMAc to determine the polymer samples’ apparent molecular weights and molecular weight distributions. FT-IR spectra were obtained using a Bruker Vertex 70 spectrometer with an ATR unit. Morphological images of freeze-dried hydrogels were obtained by scanning electron microscopy (SEM, Sigma-Zeiss). Optical micro-images were obtained using an optical microscope (VHX-950F, Keyence).UV-vis analysis
The dispersion of SWCNTs was prepared by dissolving SWCNTs and P2 in aqueous solution with the weight ratio of SWCNTs:P2 = 1:10 (corresponding weight ratio of CNTs:pyrene moieties ≈1:1), and then treating with sonication for 1 h. UV/vis absorption spectra were recorded using a PerkinElmer Lambda 35 UV/vis spectrometer with a Peltier system. The transmittances of the SWCNT dispersion with and without P2 were recorded using an Ocean optics spectrometer, and the content of SWCNTs was kept at 0.02 wt%.
Rheological analysis
Rheological measurements were performed using a HAAKE RS 150 Rheometer with a 20 mm plate-plate geometry. First, oscillation amplitude strain sweep measurements were conducted to determine the linear viscoelastic region of the hydrogels at a constant frequency of 1 Hz. Then, frequency sweep measurements were performed at a constant strain of 1% to determine the storage modulus G′ and loss modulus G′′ over the frequency region from 0.1 to 10 Hz. As for the self-healing experiments, dynamic rheology tests were performed on hydrogel samples with 4 mg mL−1 SWCNTs. A strain-sweep test was first carried out over the strain region from 1% to 200% with a constant frequency of 1 Hz at 20 °C. Afterwards, the alternate strain-sweep measurement was performed at 1 Hz and 20 °C during cyclic change from small strain (1%) to large strain (100%).Electrical conductivity measurement
To determine the conductivity of the hydrogels with different CNT concentrations, hydrogel samples were first prepared and then moulded into rectangular thin films (50 mm in length, 5 mm in width). Subsequently, electrical resistance (Ω) was monitored using an LCR meter (KEYSIGHT, E4980AL) and the four-point probe method,33 and the corresponding electrical conductivity was calculated by the following formula:where R is the measured resistance, l is the length between the adjacent probes, w is the width of the sample, and t is the thickness of the sample.

Real-time electrical self-healing measurement
Real-time monitoring of the electrical signals from the cut and self-healed hydrogels was performed using a Fluke 287 digital multimeter. The hydrogel samples were tethered to the multimeter by copper tape.Results and discussion
A functional copolymer, P(DMA-co-APB-co-PBA) (P2), as depicted in Scheme 1, was prepared via a sequential RAFT polymerization and post-polymerization modification. To accomplish the post modification functionalization, amine end-functionalized pyrene, i.e., N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB), was synthesized in a sequential two step procedure, and characterized with 1H-NMR, 19F-NMR and FT-IR spectroscopy to prove the purity of the compound (Schemes S1, S2 and Fig. S1–S4, ESI†). Afterwards, a linear random precursor copolymer, i.e., P(DMA-co-PFPA) (P1), featuring reactive pentafluorophenyl ester groups, was synthesized by RAFT polymerization. The 1H-NMR spectrum of P(DMA-co-PFPA) (Fig. 1(a)) enabled the composition of P1 to be estimated by comparing the integrals of signals (e + d) at 2.76–3.18 ppm and signals (b) at 2.54–2.75 ppm with the end methyl groups at 0.88 ppm from the chain transfer agent (CTA). Accordingly, P1 consisted of 186 DMA and 26 PFPA repeating units, respectively. The corresponding ratio of PFPA repeating units was calculated to be 12.2 mol%, which was in close proximity to the feeding molar ratio (i.e., 10 mol%). Besides, the number average molecular weight of P1 was 2.32 × 104 g mol−1, as determined by gel permeation chromatography (GPC) with DMAc as the eluent, which was in accordance with the theoretical value (2.53 × 104 g mol−1), thus suggesting a well-controlled copolymerization. Note, the hydrophilic DMA monomer was the comonomer of choice, since it imparts P1 with good solubility in water. In the next step, copolymer P2 was obtained by substituting sequentially the active ester parts of P1 with amine nucleophiles, i.e., the N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB) moiety, and then with a slight excess of (4-aminomethylphenyl) boronic acid (PBA). Compared to P1, the 1H-NMR spectrum of P2 (Fig. 1(b)) showed additional signals (k) at 8.30–7.86 ppm, which were assigned to pyrene groups from N-(6-aminohexyl)-4-(pyren-1-yl)butanamide (APB), as well as distinctive peaks (j) at 7.85–7.67 ppm, (i) at 7.28–7.18 ppm and (h) at 3.37–3.21 ppm, which could be attributed to the aromatic and methylene proton signals from the (4-aminomethylphenyl) boronic acid (PBA) groups, respectively. Besides, the integrals of protons from k and j were calculated, and apparently they were in agreement with the theoretical feeding molar ratio of APB:PBA of 30:70, therefore demonstrating the controlled post-modification process of P1 in order to deliver P2 (Fig. S5, ESI†).
 | ||
| Scheme 1 The synthetic route towards the functional copolymer P(DMA-co-APB-co-PBA), P2. First, the copolymer P(DMA-co-PFPA), P1, was synthesized via RAFT polymerization, and subsequent nucleophilic post-polymerization modification was performed to deliver copolymer P2. | ||
 | ||
| Fig. 1 1H-NMR (400 MHz) spectra of (a) P(DMA-co-PFPA) and (b) P(DMA-co-APB-co-PBA), respectively P1 and P2. | ||
The nucleophilic amine substitution process was monitored via19F-NMR and FT-IR spectroscopy. As shown in Fig. 2(a), the complete disappearance of the characteristic signals of PFP ester at (δ) −152.54, −157.44, and −162.05 ppm in the 19F-NMR spectrum revealed the thorough and successful modification. In a similar manner, the disappearance of indicative pentafluorophenyl ester bands at 1780 cm−1 (CO ester bond), 1517 cm−1 (aromatic –C6F5), and 998 cm−1 (C–F stretching bond) in the FT-IR spectra also confirmed the successful modification results. Moreover, a broad band at 3433 cm−1 attributed to –OH groups of phenylboronic acid, as well as a new ester band at 1720 cm−1 attributed to the CO stretching vibration from pyrene moieties, further demonstrated the success of the nucleophilic substitution.
 | ||
| Fig. 2 (a) 19F-NMR (377 MHz) spectra of P(DMA-co-PFPA), P1, and P(DMA-co-APB-co-PBA), P2; (b) FT-IR spectra of P(DMA-co-PFPA), P1 (blue line), and P(DMA-co-APB-co-PBA), P2 (yellow line). | ||
The interaction between pyrene moieties and SWCNTs as well as the stability of the SWCNT suspension was characterized by UV-vis analysis. As shown in Fig. 3(a), compared to the characteristic absorption peaks of P2 at 317, 331, and 347 nm originating from pyrene moieties, the corresponding absorbance peaks of P2/SWCNTs were slightly shifted to 316, 330 and 346 nm, indicating the formation of a π–π stacking hybrid between pyrene groups and SWCNTs.34 Additionally, compared to the absorbance spectrum of P2 within the range of 400–1000 nm, P2/SWCNTs displayed distinctive peaks in the range of 550–900 nm arising from interband transitions between the mirror spikes in the density of states of individualized SWCNTs,35 further demonstrating the good dispersion of SWCNTs. Besides, the stability of the standing dispersion without (sample A) and with P2 (sample B) was evaluated by monitoring their transmittance at 632 nm over time. As shown in Fig. 3(c), immediate precipitation was observed for sample A in the absence of P2 during the first day and the transmittance reached up to 90% after two days, indicating that SWCNTs without P2 modification sedimentated rapidly in aqueous solution. In contrast, the transmittance of the SWCNTs/P2 dispersion only slightly increased to 1.2% in the first day and then remained stable at around 2% during the following days, revealing its good stability. The photographs of sample A and B taken after one week of storage (see the inset of Fig. 3(c)) could further prove the effect of P2 on the stability of SWCNT dispersions. Since pyrene groups could exfoliate SWCNTs from their bundled state through a π–π stacking effect, the hydrophilic copolymer P2 was clearly capable of alleviating the aggregation of SWCNTs in aqueous media, thus forming stable dispersions even over one month, as demonstrated visually in Fig. S6 (ESI†).
 | ||
| Fig. 3 UV-vis absorbance spectra of P2 and P2/SWCNT dispersion in aqueous solution within the range of 300–400 nm (a) and 400–1000 nm (b). (c) UV-vis transmittance at 632 nm of the SWCNT aqueous suspension without and with P2, respectively sample A and B, as a function of time. The content of SWCNTs is 0.02 wt%. Inset: Photographs of the dispersed sample A and B after one week. | ||
As a next step, conductive and self-healing hydrogel composites were prepared via dynamic cross-linking of P2, PVA and SWCNTs at neutral pH 7.0, as illustrated in Fig. 4(a). For this, P2 and SWCNTs were mixed together in ultra-purified water to form a well-dispersed suspension; afterwards, PVA solution (10 wt%) was added into the suspension and the whole mixture was vigorously stirred at room temperature until gelation occurred. It should be noted that although the optimum pH for the complexation between the phenylboronic acid and diol groups is above 8.8 (the pKa of phenylboronic acid is around 8.8),36 the formation of boronate ester bonds could still happen under neutral pH due to some sufficient number of ionisable boronic acid groups as well as the rich cis-diols located along the PVA chains.37,38 Within the cross-linked system, evenly dispersed SWCNTs built conducting channels for the network, while tetrahedral boronate ester bonds were formed through reversible complexation between the phenylboronic acid groups and adjacent diol groups located along the PVA chains, therefore endowing the hydrogel with autonomous self-healing ability without any external stimuli. Indeed, SEM images (Fig. 4(c) and (d)) of the freeze-dried hydrogel samples clearly demonstrated the three dimensional interconnected network with uniformly porous microstructures, suggesting that SWNCNTs were well distributed and evenly incorporated in the hydrogels. Besides, intertwined micro-fibrils could also be observed in the cross-liked network due to the supramolecular interaction between the PVA and P2 (Fig. S7, ESI†). Further, to identify the rheological properties of the hydrogel composites, samples with different concentrations of SWCNTs were prepared and measured using an oscillatory frequency-sweep test. As shown in Fig. S8(a) (ESI†), the storage modulus (G′) exceeded the loss modulus (G′′) over the frequency region from 0.1 Hz to 10 Hz, demonstrating its solid gel-like state. Besides, the plateau of the storage modulus G′ as the stiffness response of the hydrogels increased with the higher concentration of SWCNTs, as summarized in Fig. S8(b) (ESI†).
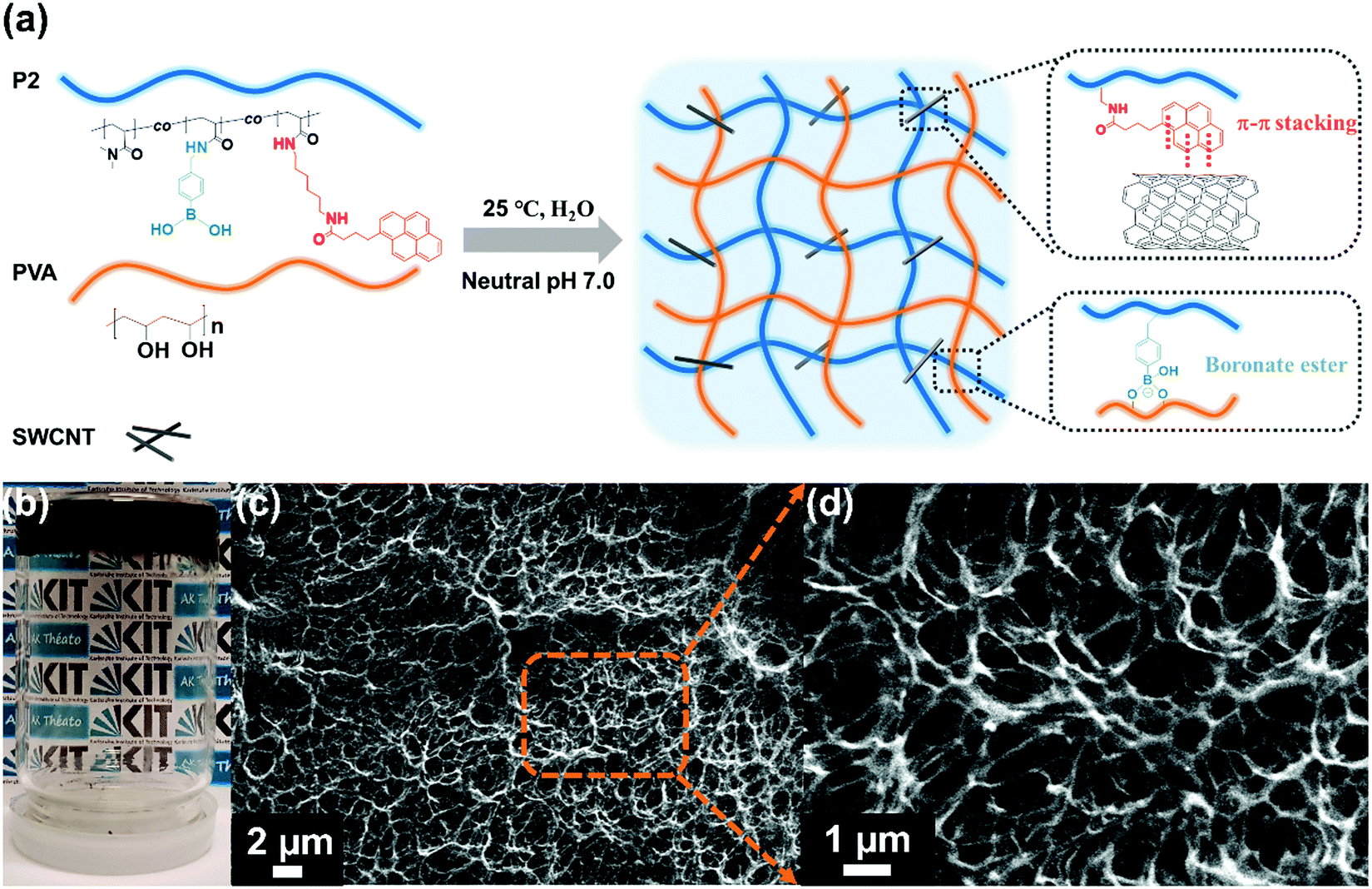 | ||
| Fig. 4 (a) Schematic illustration of the formation of conductive and self-healing hydrogels at neutral pH. (b) Photograph of the as-fabricated hydrogel. (c) and (d) SEM images of the freeze-dried hydrogel samples under different magnifications (2 μm and 1 μm, respectively). | ||
The self-healing ability of the hydrogels was tested by dynamic rheology. Oscillation strain-sweep measurements were carried out first to determine the linear viscoelastic region, as well as the critical gel–sol transition point. As shown in Fig. 5(a), the G′ and G′′ values were practically constant in the strain region from 1% to 10%. In contrast, when the strain kept increasing to 100%, G′ tended to decrease while G′′ began to increase gradually, until a crossover occurred at strain γ = 70%, therefore clearly indicating the critical point where the gel network was significantly disrupted and transformed into the sol. Furthermore, alternate strain-sweep measurement was performed to measure the self-healing ability during the cyclic change from small strain (γ = 1%) to large strain (γ = 100%). As shown in Fig. 5(b), when the strain was kept constant at 1%, G′ was larger than G′′, demonstrating the gel state; in contrast, once the sample was treated with a large strain of 100%, G′ decreased dramatically from 13500 Pa to 1550 Pa because of the disruption of the hydrogel network. Ultimately, when the strain was recovered back to 1%, G′ practically recovered to its original value. Furthermore, cyclic tests were repeated three times and exhibited a similar phenomenon, demonstrating the efficient and reversible self-healing ability of the hydrogels. In addition, cut-and-heal tests were conducted on the hydrogel samples to visually demonstrate its self-healing characteristics. As depicted in Fig. 5(c), a hydrogel disk was cut into two pieces that were then placed in contact with each other. These two hydrogel fragments were capable of supporting their weight after healing for 5 seconds at room temperature, and further integrated into one piece after 15 min, as evidenced by the disappearance of damaged sites both in the photograph of Fig. 5(c) and optical micro-images (Fig. S9, ESI†).
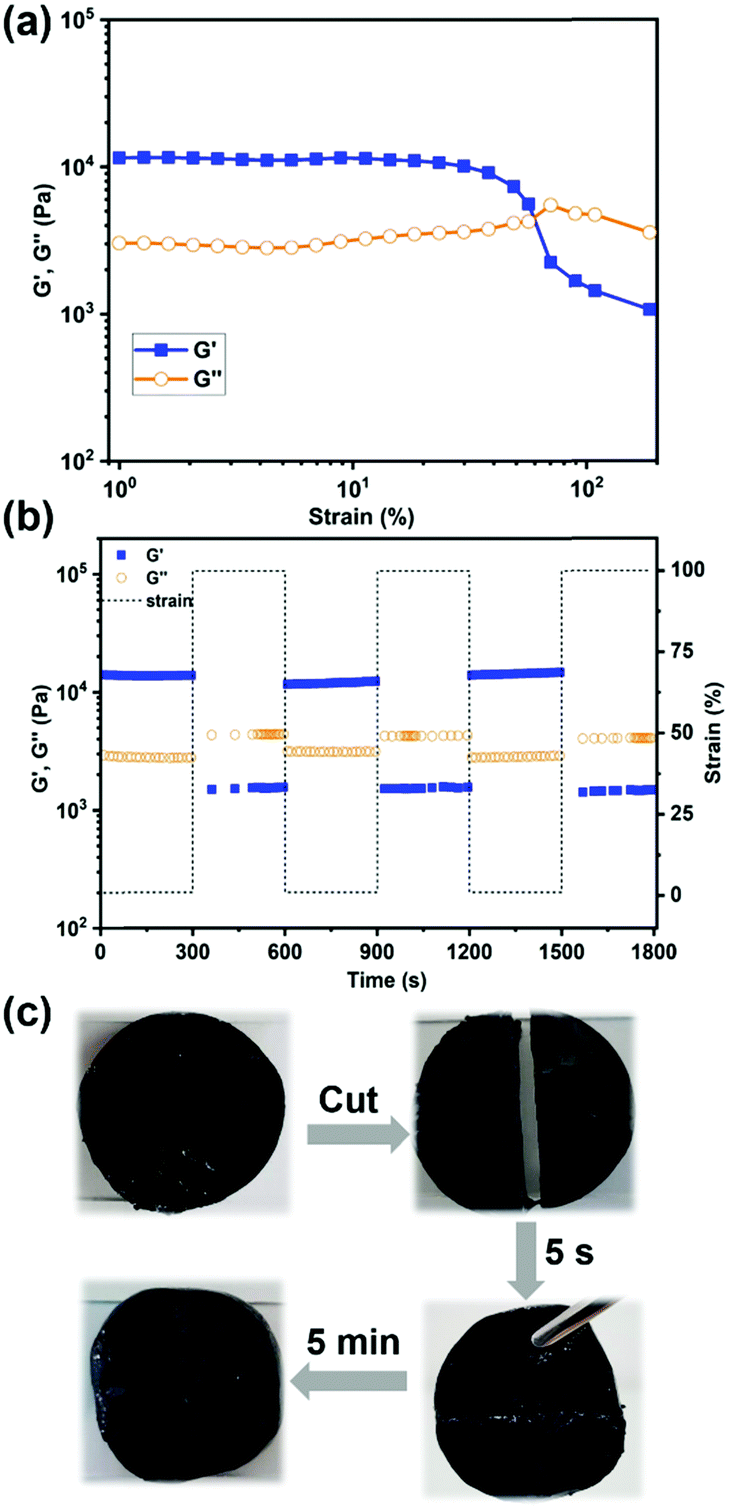 | ||
| Fig. 5 Dynamic rheology tests on hydrogel samples with 4 mg mL−1 SWCNTs. (a) Oscillation strain-sweep measurement over the strain region from 1% to 200%. (b) Cyclic strain step sweep tests from small strain (γ = 1%) to large strain (γ = 100%). (c) Photographs showing the self-recovery process of the hydrogel sample. | ||
Electrical conductivity of hydrogels with different concentrations of SWCNTs was measured, as illustrated in Fig. 6(a). Pristine hydrogels, i.e., those containing no SWCNTs, exhibited a very low conductivity of 0.04 S m−1, as a result of the conductive ions present in the system. Whereas, adding SWCNTs and gradually varying their concentration from 2 to 4, 6 and 8 mg mL−1, led to an increase in conductivity from 0.30 to 0.65, 0.83 and 1.27 S m−1, respectively, indicating the strengthening effect of the SWCNTs on the hydrogels’ conductivity. In comparison, the achieved conductivity of our hydrogel composites was relatively higher than those of the reported conductive and self-healable hydrogels, as summarized in Table S1 (ESI†). To reveal the electrical self-healing behavior of the conductive hydrogels, another additional cut-and-heal test was conducted. As displayed in Fig. 6(c), the hydrogel sample was connected into a circuit as a conductor in series with a green LED indicator. Consequently, the LED was successfully lit by an external voltage. In contrast, once the hydrogel was cut into two pieces, the LED indicator was immediately switched-off in the open-circuit state. When the split pieces were connected together, the dynamic linkages at the damaged interfaces re-associated spontaneously, thus restoring the circuit to illuminate the LED again. Additionally, real-time electrical self-healing measurement was conducted based on the resistance change during the successive cut-and-heal process at the same cut site. As shown in Fig. 6(b), electrical resistance exhibited relatively repetitive changes from stable values in a connected circuit to infinity in the open-circuit state during ten cycles. Furthermore, the electrical self-healing efficiencies defined as the ratio between the recovered conductivity and the original conductivity were calculated, and the corresponding values are listed in Fig. S10 (ESI†). The average efficiency of the ten cycle cut-and-heal process was 95% within about 10 seconds, revealing that the as-fabricated hydrogels possess repeatable and highly efficient electrical self-healing characteristics.
 | ||
| Fig. 6 (a) Electrical conductivity of different hydrogel samples with SWCNT concentrations of 0, 2, 4, 6, and 8 mg mL−1. (b) Real-time electrical self-healing measurement during ten cut-and-heal cycles. (c) Photographs showing the recovery process of electrical conductivity. | ||
Conclusions
In summary, we have demonstrated the fabrication of a conductive and self-healable hydrogel composite based on SWCNTs, PVA and a functional copolymer, P(DMA-co-APB-co-PBA). The SWCNTs were capable of evenly dispersing in aqueous solution via π–π stacking with pyrene groups of P2, and constructing conductive channels for the hydrogels at the same time. Additionally, the stiffness of the hydrogels was also strengthened by introducing the SWCNTs into the systems. The dynamic boronate ester bonds formed between the phenylboronic acid groups and PVA endowed the hydrogels with autonomous self-recovery ability without any external stimuli as determined by dynamic rheology tests. Besides, the hydrogel also exhibited a fast and repeatable electrical self-healing property (within 10 seconds) and a high recovery efficiency of 95%. Therefore, these conductive and self-healable hydrogels pave the way for potential biomedical applications in hydrogel-based areas, especially in tissue engineering, wound healing and electronic skins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. L. and X. H. gratefully acknowledge the China Scholarship Council (CSC grant: 201604910618 and 201506240019) for financial support for this work. S. M. acknowledges support from the Helmholtz Society through the program Science and Technology of Nanosystems (STN). X. L. thanks Bo Chen for his support in rheology measurements. Hongye Sun is gratefully acknowledged for her support in electrical conductivity measurements.References
- G. Huang, F. Li, X. Zhao, Y. Ma, Y. Li, M. Lin, G. Jin, T. J. Lu, G. M. Genin and F. Xu, Chem. Rev., 2017, 117, 12764–12850 CrossRef CAS
.
- J. Li, L. Mo, C.-H. Lu, T. Fu, H.-H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani and R. W. Beuerman, Nat. Mater., 2011, 10, 149–156 CrossRef CAS
- T. Distler and A. R. Boccaccini, Acta Biomater., 2020, 101, 1–13 CrossRef CAS
- L. Li, L. Pan, Z. Ma, K. Yan, W. Cheng, Y. Shi and G. Yu, Nano Lett., 2018, 18, 3322–3327 CrossRef CAS
- J. Shang, X. Le, J. Zhang, T. Chen and P. Theato, Polym. Chem., 2019, 10, 1036–1055 RSC
- C. Yang, Z. Liu, C. Chen, K. Shi, L. Zhang, X.-J. Ju, W. Wang, R. Xie and L.-Y. Chu, ACS Appl. Mater. Interfaces, 2017, 9, 15758–15767 CrossRef CAS
- Z. Lei, Q. Wang, S. Sun, W. Zhu and P. Wu, Adv. Mater., 2017, 29, 1700321 CrossRef
- S. Lee, A. Reuveny, J. Reeder, S. Lee, H. Jin, Q. Liu, T. Yokota, T. Sekitani, T. Isoyama and Y. Abe, Nat. Nanotechnol., 2016, 11, 472 CrossRef CAS
- E. Fantino, A. Chiappone, I. Roppolo, D. Manfredi, R. Bongiovanni, C. F. Pirri and F. Calignano, Adv. Mater., 2016, 28, 3712–3717 CrossRef CAS
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef
- P. Li, Z. Jin, L. Peng, F. Zhao, D. Xiao, Y. Jin and G. Yu, Adv. Mater., 2018, 30, 1800124 CrossRef
- F. H. Gojny, M. H. Wichmann, B. Fiedler and K. Schulte, Compos. Sci. Technol., 2005, 65, 2300–2313 CrossRef CAS
- Y. Huang, Y. Zheng, W. Song, Y. Ma, J. Wu and L. Fan, Composites, Part A, 2011, 42, 1398–1405 CrossRef
- Y. S. Song and J. R. Youn, Carbon, 2005, 43, 1378–1385 CrossRef CAS
- K. Balasubramanian and M. Burghard, J. Mater. Chem., 2008, 18, 3071–3083 RSC
- T. Fujigaya and N. Nakashima, Sci. Technol. Adv. Mater., 2015, 16, 024802 CrossRef
- K. C. Etika, F. D. Jochum, M. A. Cox, P. Schattling, P. Theato and J. C. Grunlan, Macromolecules, 2010, 43, 9447–9453 CrossRef CAS
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 1–6 Search PubMed
- J. Cao, C. Lu, J. Zhuang, M. Liu, X. Zhang, Y. Yu and Q. Tao, Angew. Chem., Int. Ed., 2017, 56, 8795–8800 CrossRef CAS
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12(1), 1628–1639 CrossRef CAS
- A. B. Ihsan, T. L. Sun, T. Kurokawa, S. N. Karobi, T. Nakajima, T. Nonoyama, C. K. Roy, F. Luo and J. P. Gong, Macromolecules, 2016, 49, 4245–4252 CrossRef
- H. Yu, Y. Wang, H. Yang, K. Peng and X. Zhang, J. Mater. Chem. B, 2017, 5, 4121–4127 RSC
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS
- L. Shi, P. Ding, Y. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef
- B. Marco-Dufort and M. Tibbitt, Mater. Today Chem., 2019, 12, 16–33 CrossRef CAS
- G. Vancoillie and R. Hoogenboom, Polym. Chem., 2016, 7, 5484–5495 RSC
- C. C. Deng, W. L. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef CAS
- L. He, D. Szopinski, Y. Wu, G. A. Luinstra and P. Theato, ACS Macro Lett., 2015, 4, 673–678 CrossRef CAS
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS
- W. L. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS
- W. Zhou, Y. Tang, R. Song, L. Jiang, K. S. Hui and K. Hui, Mater. Des., 2012, 37, 161–165 CrossRef CAS
- B. K. Billing, P. K. Agnihotri and N. Singh, Analyst, 2018, 143, 3343–3352 RSC
- S. Brahmachari, D. Das, A. Shome and P. K. Das, Angew. Chem., 2011, 123, 11439–11443 CrossRef
- W. L. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS
- A. J. Amaral, M. Emamzadeh and G. Pasparakis, Polym. Chem., 2018, 9, 525–537 RSC
- K. Kataoka, H. Miyazaki, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 1061–1062 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01234c |
| This journal is © The Royal Society of Chemistry 2020 |