
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the structures of organic semiconductor–quantum dot nanocomposites through ligand shell chemistry†‡
Daniel T. W.
Toolan§
 *a,
Michael P.
Weir§
*b,
Rachel C.
Kilbride
a,
Jon R.
Willmott
c,
Stephen M.
King
d,
James
Xiao
e,
Neil C.
Greenham
e,
Richard H.
Friend
e,
Akshay
Rao
e,
Richard A. L.
Jones
b and
Anthony J.
Ryan
a
*a,
Michael P.
Weir§
*b,
Rachel C.
Kilbride
a,
Jon R.
Willmott
c,
Stephen M.
King
d,
James
Xiao
e,
Neil C.
Greenham
e,
Richard H.
Friend
e,
Akshay
Rao
e,
Richard A. L.
Jones
b and
Anthony J.
Ryan
a
aDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, S3 7HF, UK. E-mail: d.toolan@sheffield.ac.uk
bDepartment of Physics and Astronomy, The University of Sheffield, Hicks Building, Hounsfield Road, Sheffield, S3 7RH, UK. E-mail: weir.mp@gmail.com
cDepartment of Electronic and Electrical Engineering, The University of Sheffield, 3 Solly Street, Sheffield, S1 4DE, UK
dISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
eCavendish Laboratory, Cambridge University, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK
First published on 31st July 2020
Abstract
Nanocrystal quantum dots (QD) functionalised with active organic ligands hold significant promise as solar energy conversion materials, capable of multiexcitonic processes that could improve the efficiencies of single-junction photovoltaic devices. Small-angle X-ray and neutron scattering (SAXS and SANS) were used to characterize the structure of lead sulphide QDs post ligand-exchange with model acene-carboxylic acid ligands (benzoic acid, hydrocinnamic acid and naphthoic acid). Results demonstrate that hydrocinnamic acid and naphthoic acid ligated QDs form monolayer ligand shells, whilst benzoic acid ligated QDs possess ligand shells thicker than a monolayer. Further, the formation of a range of nanocomposite materials through the self-assembly of such acene-ligated QDs with an organic small-molecule semiconductor [5,12-bis((triisopropylsilyl)ethynyl)tetracene (TIPS-Tc)] is investigated. These materials are representative of a wider set of functional solar energy materials; here the focus is on structural studies, and their optoelectronic function is not investigated. As TIPS-Tc concentrations are increased, approaching the solubility limit, SANS data show that QD fractal-like features form, with structures possibly consistent with a diffusion limited aggregation mechanism. These, it is likely, act as heterogeneous nucleation agents for TIPS-Tc crystallization, generating agglomerates containing both QDs and TIPS-Tc. Within the TIPS-Tc crystals there seem to be three distinct QD morphologies: (i) at the crystallite centre (fractal-like QD aggregates acting as nucleating agents), (ii) trapped within the growing crystallite (giving rise to QD features ordered as sticky hard spheres), and (iii) a population of aggregate QDs at the periphery of the crystalline interface that were expelled from the growing TIPS-Tc crystal. Exposure of the QD:TIPS-Tc crystals to DMF vapour, a solvent known to be able to strip ligands from QDs, alters the spacing between PbS–hydrocinnamic acid and PbS–naphthoic acid ligated QD aggregate features. In contrast, for PbS–benzoic acid ligated QDs, DMF vapour exposure promotes the formation of ordered QD colloidal crystal type phases. This work thus demonstrates how different QD ligand chemistries control the interactions between QDs and an organic small molecule, leading to widely differing self-assembly processes. It highlights the unique capabilities of multiscale X-ray and neutron scattering in characterising such composite materials.
Introduction
Solar energy conversion systems based upon small-molecule organic semiconductors (OSCs) with inorganic semiconducting quantum dots (QDs) have attracted significant interest due to their potential to work in tandem with existing photovoltaic technologies and boost device efficiencies.1–7 One of the most promising solar energy conversion processes, “photon multiplication”, harnesses the ability of 5,12-bis((triisopropylsilyl)ethynyl)tetracene (TIPS-Tc) and a family of similar molecules to undergo singlet exciton fission (SF), whereby one singlet exciton is converted into two triplet excitons that are subsequently harvested and emitted by appropriately matched QDs.2,4OSC:QD solar energy conversion systems have been demonstrated in the laboratory, in the solution phase,1,8,9 but practical large scale deployment will require the development of solid-state solar energy conversion materials.10 Here, a route for generating a solid-state OSC:QD system is explored, whereby high concentrations of an OSC and QD self-assemble to form a hybrid material containing both crystalline OSC and inclusions of QDs in various states of colloidal order. This work aims to provide the first steps in a structural picture of the resulting nanocomposite materials; however, the optoelectronic study of their function is left for future work.
A significant challenge for organic semiconductor–nanocrystal blends is that they typically suffer from either aggregation or phase separation of the components due to a mismatch of their size, shape and surface energies.11–14 In a number of organic:QD composite systems the QD surface ligand has been demonstrated to control not only the electronic coupling between the host material, but also lead to the formation of phase segregated QD morphologies.15,16 Here, solution phase ligand exchange is employed to control the QD surface chemistry, and hence OSC:QD self-assembly, switching the QD nature from aliphatic (native oleic acid ligands) to aromatic, employing benzoic acid (BA), hydrocinnamic acid (HCA) and naphthoic acid (NA), with the aim of investigating how subtle differences between similar short ligands (required to enable efficient OSC:QD electronic coupling) controls the self-assembly between OSC and QD. Comparative small molecule aromatic ligands have previously been employed as model systems in comparative studies providing quantitative insights into ligand exchange processes, and how such ligands tune QD band edge positions (via NMR spectroscopy and spectrophotometric absorbance, respectively).17,18
Small angle scattering (both X-ray [SAXS] and neutron [SANS]) have provided quantitative structural insight into a range of QDs systems, from the size and distribution of the inorganic core radii (via SAXS) and the coverage and conformation of the native oleic acid (OA) ligand layer of as-synthesized QDs (via SANS).19–23 Here, the ability of such scattering techniques to probe multiple length-scales is employed to quantify QD ligand exchange processes and consequent OSC:QD self-assembly. Such scattering approaches provide direct insight into the routes by which a range of QD structures are formed with an OSC, from extended and disordered fractal aggregates at the early stages in solution, to final, long-time, OSC:QD nanocomposite aggregate forms. This work shows that the different ligands employed determine how QDs are incorporated into the organic semiconductor crystals lays the foundation for the rational design and growth of targeted QD morphologies, from dispersed to superlattice via solution-phase self-assembly approaches. Further, we demonstrate how nanocomposite structures/morphologies may be tuned post-processing, via exposure to solvent vapours. These structures have the aforementioned applications in solar energy harvesting, as well as showing great potential to be incorporated into solution-processed nanocomposites, for example with a polymer phase. Such control over QD nanocomposite forms has potential for tuneable electron and phonon transport24 and thus also builds our understanding towards QD thermoelectrics and other QD devices. In addition, this work highlights the key role of multi-scale X-ray and neutron scattering techniques for understanding the highly complex morphologies of self-assembled solar energy conversion materials.
Experimental
Chemicals
Lead oxide (PbO, 99.999%) was purchased from Alfa Aesar. Ethanol and 1-butanol (Hi-Dry anhydrous) were purchased from Romil. TIPS-Tc was obtained from Ark Pharm. All other chemicals were purchased from Sigma-Aldrich. All materials were used as received without further purification.Synthesis of PbS–OA quantum dots
A master batch of oleic acid capped PbS nanocrystals was synthesised by the method of Hines & Scholes.25 Lead oxide (1.25 g, 5.6 mmol), oleic acid (OA, 90%, 4 mL, 12.6 mmol) and 1-octadecene (ODE, 90%, 25 mL, 78 mmol) was placed in a 3-necked round bottomed flask and degassed under vacuum (<10−2 mbar) at 110 °C for 2 hours with stirring, forming a colourless solution. In a nitrogen glovebox, a syringe was prepared containing ODE (13.9 mL, 43 mmol) and hexamethyldisilathiane (TMS2S, 95%, 592 μL, 2.8 mmol). The flask was put under nitrogen flow and the syringe contents rapidly injected into the flask at 110 °C and allowed to cool. Upon cooling to 60 °C, the reaction mixture was transferred to an argon glovebox. The as-formed nanocrystals were precipitated with ethanol/butanol and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 minutes. The nanocrystals were then suspended in hexane and precipitated again with ethanol. The purified nanocrystals were suspended in toluene for storage.
000g for 30 minutes. The nanocrystals were then suspended in hexane and precipitated again with ethanol. The purified nanocrystals were suspended in toluene for storage.
Ligand exchange to –BA, –NA and –HCA
A 2 molar excess of BA, HCA or NA (Sigma-Aldrich) was added to PbS–OA QDs (25 mg mL−1 in a toluene:THF, 1:1), based upon an estimated PbS:OA mass balance of 1:1.21. Then acetone (20 mL) was added to precipitate the PbS–(BA, HCA or NA) nanocrystals; the mixtures were centrifuged at 72500g for 20 min at 20 °C, the supernatant removed, and the QD residue re-dispersed in toluene (1 mL). The ligand-exchanged nanocrystals were purified using three repeated dispersion/precipitation/centrifugation cycles. For the final re-dispersion the QDs were dispersed in deuterated toluene or a blend of hydrogenous and deuterated toluene as appropriate for small-angle neutron scattering experiments. A typical particle volume fraction for these measurements is of the order of 2–3%. However a proper determination of quantum dot volume fraction is only possible when data has been fitted to a model. This is due to the contribution from the ligand shell, which may be partially swollen by solvent and, for PbS cores of this size, accounts for ∼80% of the particle volume.
Small-angle neutron scattering
SANS was carried out on the LOQ26 and SANS2D27 small-angle diffractometers at the ISIS Pulsed Neutron Source (STFC Rutherford Appleton Laboratory, Didcot, UK). TIPS-Tc/QD solutions were prepared in deuterated solvents or blends of hydrogenous and deuterated solvents, to manipulate the neutron contrast, and were contained in 1 or 2 mm path length quartz cylindrical “banjo” cells (type 120, Hellma GmbH), with the longer path length reserved for fully deuterated solvents and the shorter path length required to reduce the absorbance and incoherent background in h/d solvent blends. Sample solutions of a desired QD concentration were prepared through drying aliquots of stock solutions of PbS–OA under nitrogen, followed by the addition of the appropriate solvent (d-toluene or h/d-toluene blend).LOQ
This is a fixed-geometry time-of-flight instrument which utilizes neutrons with wavelengths between 2.2 and 10 Å. Data are simultaneously recorded on two, two-dimensional, position-sensitive, neutron detectors, to provide a simultaneous q-range of 0.007–1.0 Å−1.SANS2D
A simultaneous q-range of 0.006–1.2 Å−1 was achieved utilizing an incident wavelength range of 1.65–16.5 Å and employing an instrument set up of L1 = L2 = 4 m, where L1 and L2 are the pre- and post-sample flight paths respectively, with the rear detector offset vertically 75 mm and horizontally 100 mm.For both instruments, the beam diameter was collimated to 12 mm at the sample. On LOQ, scattering data were typically accumulated for 83 minutes per sample/background (with the actual acquisition time dependent upon the ISIS Target Station 1 beam current). For all collected data, each raw scattering data set was corrected for the detector efficiencies, sample transmission and background scattering and converted to scattering cross-section data (∂Σ/∂Ω vs. q) using the Mantid framework (version 3.13.0).28 These data were placed on an absolute scale (cm−1) using the scattering from a standard sample (a solid blend of hydrogenous and perdeuterated polystyrene) in accordance with established procedures.29 Fitting was performed using the SasView software package (version 4.2.2).30
Small-angle X-ray scattering
SAXS measurements were carried out on a Xeuss 2.0 instrument equipped with an Excillum MetalJet liquid gallium X-ray source. Scattering data were collected for 900 s using collimating slits of 0.5 × 0.6 mm (“high flux” mode). Scattering patterns were recorded on a vertically-offset Pilatus 1M detector with a sample to detector distance of 555 mm calibrated using a silver behenate standard to achieve a q-range of 0.03–1.5 Å−1 Data reduction was performed using the Foxtrot software (supplied as proprietary software from Xenocs SAS, Grenoble, France; may be requested from the developer at Soleil synchrotron31) and the IRENA32 macros for Igor Pro, before fitting was again performed using SasView.Microscopy
Fluorescence micrographs were obtained using a Zeiss Axio Scope.A1 in reflection mode, utilizing either LED-module neutral white f. Axio, EX BP 470/40, BS FT 495 and for IR fluorescence an IR a 1000 nm shortpass filter was placed between the LED illumination source and excitation bandpass filter and a 1000 nm longpass filter placed between the dichroic mirror and the detector. An IR sensitive camera (InGaAs Hamamatsu Model Number: C12741-03) coupled with a 10×/0.25 or LD Plan NeoFluer ×20/0.4 objective or Axiocam 105 colour camera were employed, to provide fluorescence micrographs in IR (matched fluorescence for QD emission) or at 525/50 nm (fluorescence of TIPS-Tc).DMF vapour exposure
PbS QD:TIPS-Tc crystals suspended via Kapton tape where placed in a 10 mL glass beaker containing 200 μL of DMF and exposed to vapours for 30 minutes.Results & discussion
The work presented herein employs a combination of SANS and SAXS to provide complementary insights into: (i) structural studies of ligand exchange from as as-synthesized PbS–OA QDs to PbS–BA, PbS–NA and PbS–HCA QDs, (ii) the early stages of self-assembly of small molecules and QDs from solution where the concentration of the small molecule is increased, approaching the solubility limit, generating aggregated structures; and (iii) characterizing and understanding the complex morphologies of QD:TIPS-Tc nanocomposite materials formed via self-assembly.In either SAXS or SANS, the contrast between two components is proportional to the square of the scattering length density (SLD) difference. In SAXS measurements, contrast between two components therefore increases in proportion to the square of the electron density difference. As such, for all QDs studied herein, the SAXS scattering signal is dominated by that of the PbS core. This unfavourable contrast for resolving QD-ligand structures is further exacerbated by the relatively poor contrast between organic ligands and organic solvents. Complementary to SAXS, SANS offers significantly greater scope for studying QD-ligand:TIPS-Tc interactions due to the favourable contrast between the PbS core, BA, organic ligands (HCA, NA), TIPS-Tc and organic solvents, with associated neutron and X-ray SLDs (respectively, nSLD and xSLD) presented in Table 1. In many situations, the COOH moiety is expected to be strongly localised at the surface of the QD, and the main functional group is highly aligned in a shell-like structure; for this reason SLDs are quoted excluding the carboxylic acid. In some cases this has a significant effect upon the measured scattering length density inside the shell.
| Substance | Empirical formula | Density (g cm−3) | nSLD (10−6 Å−2) | xSLD (Ga) (10−6 Å−2) |
|---|---|---|---|---|
| h-Toluene | C7H8 | 0.87 | 0.95 | 8.03 |
| d-Toluene (d8) | C7D8 | 0.95 | 5.68 | 8.03 |
| Lead sulphide | PbS | 7.6 | 2.34 | 50.7 |
| Oleic acid (excluding COOH) | C18H34O2 | 0.895 | 0.08 | 8.51 |
| C 17 H 33 – | 0.895 | −0.24 | 8.65 | |
| Benzoic acid [BA] (excluding COOH) | C7H6O2 | 1.27 | 1.73 | 11.3 |
| C 6 H 5 – | 1.27 | 2.10 | 11.5 | |
| Naphthoic acid [NA] (excluding COOH) | C11H8O2 | 1.40 | 2.68 | 11.4 |
| C 10 H 7 – | 1.40 | 2.67 | 12.6 | |
| Hydrocinnamic acid [HCA] (excluding COOH) | C9H10O2 | 1.07 | 1.46 | 9.7 |
| C 8 H 9 – | 1.07 | 1.20 | 9.87 | |
| TIPS-Tc | C40H52Si2 | 1.1 | 0.90 | 10.2 |
1. Characterisation of QD ligand exchange
Small-angle X-ray and neutron scattering (SAXS and SANS) measurements were used to determine the efficacy of ligand exchange procedures performed on as-synthesized PbS–OA QDs to produce PbS–BA, PbS–NA and PbS–HCA QDs. Oleic acid (OA)-ligated PbS quantum dots with an excitonic absorption peak at 1.3 eV (ESI,‡ Fig. S1) were synthesized according to previously reported methods. We have recently characterised such as-synthesised Pbs–OA QDs via combined SAXS and SANS measurements.21 Here, as synthesised PbS–OA precursor QD batch were characterised using SAXS (10 mg mL−1 in toluene) and SANS (82 mg mL−1 d-toluene), with radially integrated data shown in Fig. 1(a). It was found that the as-synthesized PbS–OA QDs could be adequately described as quasi-spherical nanoparticles with an average PbS core radius of 16.6 Å and a 10% size polydispersity (equivalent standard deviation of 1.66 Å using a lognormal distribution). However, given the concentrations involved, it was also necessary to incorporate a hard-sphere structure factor. The contribution to SAXS data from the OA envelope is minimal in this measurement, but SANS shows a packed corona of OA on the PbS QD core, namely a 16 Å thick layer with ∼20% solvent penetrations as detailed in our previous characterisations of PbS–OA via SAXS and SANS.21 | ||
| Fig. 1 (a) SANS and SAXS from the stock PbS–OA at respective concentrations of 82 mg mL−1 and 10 mg mL−1 (see also ref. 21) along with model fits from a core–shell sphere * hard sphere model and a sphere * hard sphere model respectively (b) SANS from PbS–BA, which implies the presence of smaller ligand complexes in addition to quasi-spherical QDs. (c) SANS from PbS–NA and PbS–HCA with associated core–shell-sphere model fits. | ||
SANS data (performed on LOQ) from PbS QDs post ligand exchange with benzoic acid (BA) are shown in Fig. 1(b), with panel (c) showing naphthoic acid (NA) and hydrocinnamic acid (HCA) ligand exchanges. For the PbS–NA and PbS–HCA exchanged QDs SANS data were again fitted with the simple core–shell-sphere * hard sphere model (for further details, see ESI,‡ Sections 2.2 and 2.3). In both cases, a single shell of the new ligand is visible (6.3 Å thick for NA and 11.4 Å thick for HCA) and approximately 70% ligand coverage by volume inside the shell (for further details, see ESI,‡ Section 1.2). The additionally “washed-out” appearance of the PbS–NA QD SANS data arises from a larger Gaussian polydispersity in the shell thicknesses of approximately 0.71 (ratio of standard deviation to mean thickness), compared to 0.24 for PbS–HCA. For HCA, this is another piece of evidence for the presence of both chemisorbed and physisorbed ligands as is also hypothesised to occur for native oleic acid ligands on PbS QDs.21
In the case of BA, elucidation of the final structure of the exchanged QDs was not as straightforward as for PbS–HCA and PbS–NA, as data could not be adequately fitted using the same model as described above. Assuming PbS–BA QDs can be accounted for using such a model, and keeping the core size and polydispersity fixed, then the data appear to also show additional scattering objects, with a radius of gyration of around 4 Å. In the fitting these are accounted for by combining the core–shell sphere model with a simple Guinier model. This length-scale is of a commensurate size with BA and so is attributed to excess free BA ligands that are not attached to QD surfaces. This fitting approach indicates that PbS–BA QDs possess a BA shell with a thickness of 19.1 Å. The BA ligand shell thickness is significantly greater than that of a single extended BA molecule and thus indicates that QD ligand corona must consist of both chemisorbed and physisorbed BA ligands. A significant advantage of the SAXS/SANS approach demonstrated here is the detection of two populations of ligands, one bound to the QD surface and the other in free solution.
2. Self-assembly of small molecules and QDs from solution
To quantify the self-assembly of QD with the TIPS-Tc small organic molecule, first, the behaviour of increasing concentrations of TIPS-Tc was investigated, with SANS measured for TIPS-Tc in deuterated toluene at concentrations of 25, 50, 100, 200 and 300 mg mL−1 as presented in Fig. 2(a). To ensure the scattering from the molecular shape of TIPS-Tc was properly understood, especially since it was later required for a background subtraction, the 25 mg mL−1 SANS data were fitted to a SAXS profile for TIPS-Tc in H2O generated by the FOXS server34,35 (for further details, see ESI,‡ Section 2). A reasonable match was obtained between the shape of TIPS-Tc measured from SANS with that calculated from the SAXS model, and was enough to conclude that the TIPS-Tc SANS signal originates from isolated TIPS-Tc molecules in solution, at least at the lowest concentration (25 mg mL−1). In order to make an approximate measurement of how the SANS from TIPS-Tc scaled with nominal concentration, a Guinier model was used up to q = 0.34 Å−1, in order to capture the overall size of the scattering objects but to avoid the smaller scale feature appearing at q = 0.4–0.5 Å−1. The scale factor of this Guinier fit is, to a reasonable approximation, proportional to the TIPS-Tc solution concentration. The scale factor increases with nominal TIPS-Tc concentration (i.e. the amount added) in a non-linear fashion at concentrations above 100 mg mL−1, indicating that the solution is approaching saturation around these values. However, in addition to this effect, the reduced separation between TIPS-Tc molecules leads to a repulsive structure factor which starts to suppress the scattering intensity. Importantly, we find that saturated TIPS-Tc solutions in toluene at concentrations between 100–300 mg mL−1 are highly stable and do not readily form crystallites in solution at these high concentrations.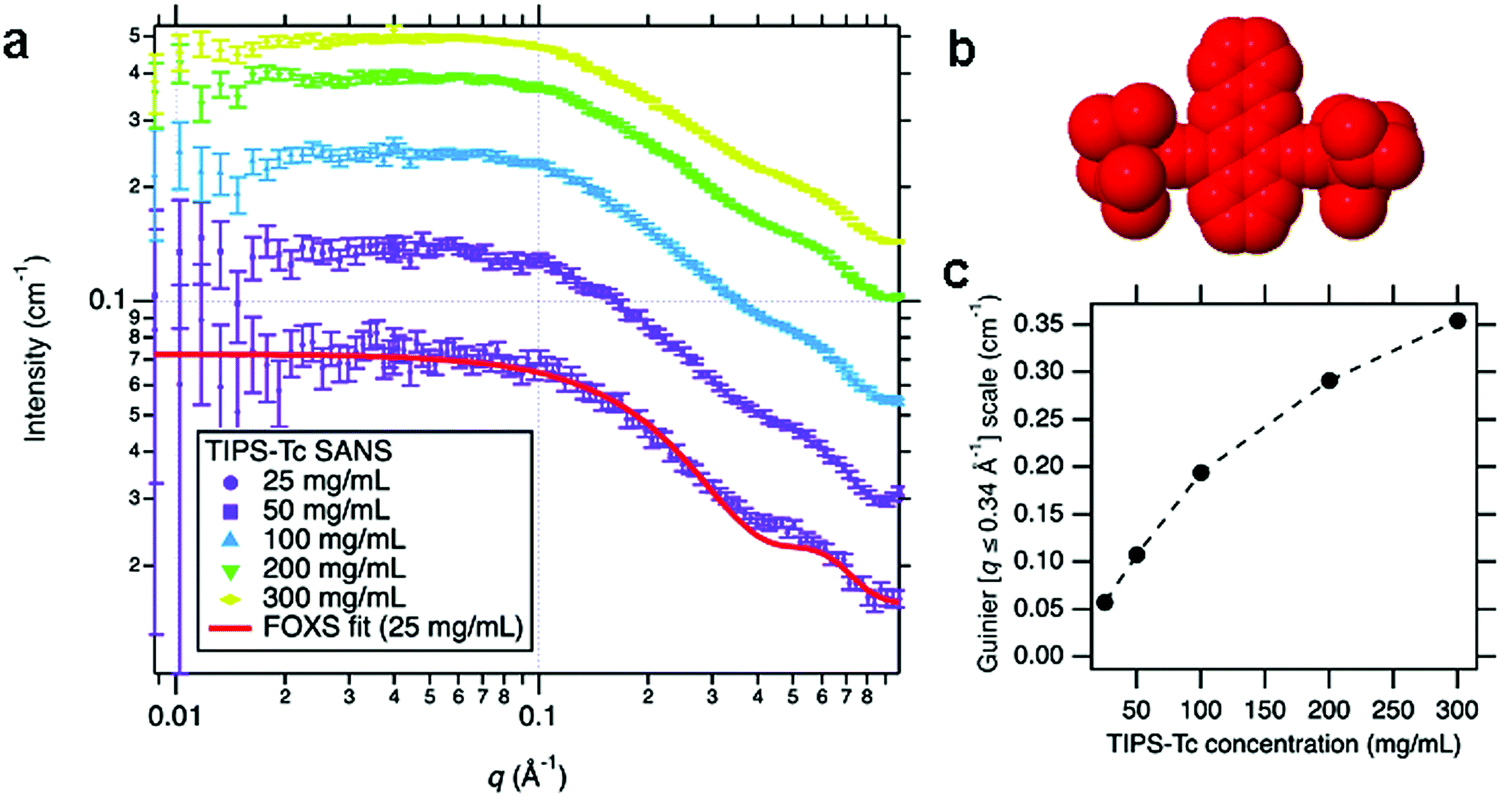 | ||
| Fig. 2 (a) Small-angle neutron scattering (SANS) of TIPS-Tc in deuterated toluene at concentrations ranging from 25 to 300 mg mL−1, with the associated fit to an X-ray scattering model of TIPS-Tc in H2O (using the FOXS34,35 server), to confirm the molecule's overall shape only (b) representation of TIPS-Tc shape used by FOXS to calculate the molecular scattering, rendered using an implementation of Jmol/JSmol.36 (c) The scale factor of the Guinier fit to the SANS data in the range q = 0.008–0.34 Å−1 indicative of the relative volume fractions of TIPS-Tc, as a function of nominal TIPS-Tc concentration. | ||
SANS experiments were conducted to gain insights into the self-assembly behaviour of QDs possessing different ligand chemistries (PbS: –BA, –HCA & –NA) with TIPS-Tc. In these experiments QD concentrations remained constant (100, 50 & 78 mg mL−1, for PbS–BA, PbS–HCA & PbS–NA, respectively) and TIPS-Tc was sequentially added to reach cumulative concentrations of 25, 50, 100, 200, and 300 mg mL−1. The SANS data obtained represents snapshots of structural evolution after ∼4.5 hours at each concentration. Ideally QD concentrations would have remained constant for the different PbS–BA, PbS–HCA & PbS–NA ligand exchanged species, however, due to losses from the ligand exchange processes, coupled with the need to utilise all available QDs to maximise the neutron scattering signal, this was not possible.
Due to the relatively complex nature of background subtraction involved when measuring QDs in TIPS-Tc solutions (compared to the direct removal of simple solvent backgrounds), the SANS curves were obtained by subtracting the appropriate empty quartz cell background followed by a subtraction of the appropriate TIPS-Tc/d-toluene background in accordance with expected volume fractions. In all cases, a very high proportion (up to 100%) of the TIPS-Tc remained in solution with the QD, indicating that TIPS-Tc only participated sparingly in the formation of aggregates. This is illustrated in Fig. 3a for PbS–BA with the highest cumulative TIPS-Tc concentration of 300 mg mL−1. The scattering from TIPS-Tc in solution is always a component of the PbS–BA + TIPS-Tc curve. The scattering attributed to the aggregation of the quantum dots, in the presence of TIPS-Tc but separated from TIPS-Tc solution scattering, is shown in Fig. 3b as a function of the cumulative TIPS-Tc concentration. Two features are clearly visible, namely (i) a decrease in the scale factor of the PbS–BA (pure, zero TIPS-Tc) quantum dot scattering as isolated PbS–BA is removed either into aggregates or precipitates, and (ii) an low-q power-law-like (being insufficient in q-range to fully describe as such) upturn in scattering whose scale factor increases as TIPS-Tc concentration increases. Both factors (i) and (ii) point to the formation of PbS–BA aggregates driven by small amounts of TIPS-Tc. Similar data is shown for PbS–NA and PbS–HCA in the ESI‡ (Fig. S3). The proportion of isolated, well-dispersed quantum dots remaining in solution may be estimated at any TIPS-Tc concentration by estimating the scale factor of the PbS–R (R = BA, NA, or HCA with zero TIPS-Tc) scattering that is required to recover the Fig. 3b scattering at high q away from the low-q upturn. This scale factor relative to the zero TIPS-Tc PbS–R gives the relative concentration [PbS–R]/[PbS–R]0. This is most useful plotted against the TIPS-Tc concentration normalised by the initial quantum dot concentration, [TIPS-Tc]/[PbS–R]0, to adjust for the initial concentrations for PbS–BA, PbS–NA and PbS–HCA detailed above. This comparison is shown in Fig. 3c and clearly shows the greater propensity of PbS–BA to aggregate or precipitate by interacting with TIPS-Tc, through the moderate PbS–NA to the least-interacting PbS–HCA. Simple exponential fits to the relative concentration data are shown as dotted or dashed lines, with the form y = exp(−ax) where a takes values of 0.69 ± 0.05, 0.38 ± 0.02 and 0.24 ± 0.02 for PbS–BA, PbS–NA and PbS–HCA respectively.
 | ||
| Fig. 3 (a) Comparison of SANS from PbS–BA + TIPS-Tc 300 mg mL−1 (squares) with SANS from TIPS-Tc 300 mg mL−1 (circles), along with the TIPS-Tc-subtracted curve (triangles). (b) TIPS-Tc-subtracted data for PbS–BA as TIPS-Tc is sequentially added at 0, 25, 50, 100, 200 and 300 mg mL−1. (c) Estimate of the relative concentration of PbS–R (R = BA, NA, HCA) remaining in solution [PbS–R]/[PbS–R]0 as a function of the concentration of TIPS-Tc added, normalised to the initial QD concentration, [TIPS-Tc]/[PbS–R]0. Lines are exponential fits of the form y = exp(−ax). (d) The low-q scattering (q = 0.01–0.1 Å−1) observed in PbS–R QD (R = BA, NA, HCA) isolated by subtraction of the appropriately scaled zero TIPS-Tc curve, PbS–RTIPS-Tc-0, with straight lines representing negative power law exponents of 2.0 and 2.5 for illustration. | ||
The low-q upturn is analysed in more detail in Fig. 3b for each of the samples at the maximum TIPS-Tc concentration of 300 mg mL−1. Here the low-q scattering was isolated by subtracting an appropriately-scaled version of the pure (zero TIPS-Tc) QD scattering as an approximation of the form factor. This was achieved by using the high-q intensity, furthest away from the influence of the low-q upturn, to estimate the maximum possible volume fraction of isolated quantum dots remaining in solution. The PbS–BA:TIPS-Tc sample exhibits power-law type scattering at low q with an exponent of approximately −2.5 possibly indicative of mass fractals formed by a diffusion-limited aggregation process. It is, however, important to stress that at least two or three decades in q would be required to make a definitive identification of the fractal character of these aggregates.37,38 Some limited insight may be gained from using a mass fractal model to estimate the number of scatterers in the aggregates causing the low-q scattering. At 300 mg mL−1 added TIPS-Tc, the PbS–HCA and PbS–NA samples respectively produced aggregates with 0.07 and 0.09 times the number of scatterers than for PbS–BA (see ESI,‡ Section 3.2 and Fig. S4).
It is highly likely that the greater propensity for aggregation observed for PbS–BA:TIPS-Tc is a consequence of a combination of factors including: (i) PbS–BA QD possessing the thickest ligand corona (of the BA, HCA, NA ligand series), (ii) the higher concentration of QDs in solution (relative to PbS–NA and PbS–HCA QDs), and (iii) the clear excess of BA also in solution (identified as 4 Å scattering objects in Fig. 1b), contributing to the overall amount of material solubilised by the toluene solvent. During the course of the experiment mm-sized circular precipitates formed on the surface of the neutron scattering sample cell for the PbS–BA:TIPS-Tc system, clearly evidence of much larger scale features forming through the identified aggregation process. No such visible, large-scale precipitates were observed for the PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc solutions over the duration of the SANS experiments.
3. Morphology of small molecule: QD nanocomposites
The preceding section identified that the early stages of solution based self-assembly for all of the PbS(–BA, –HCA & –NA):TIPS-Tc solutions involved the formation of QD mass fractal objects formed via diffusion-limited aggregation processes. Over much longer timescales >72 hours, large mm sized agglomerated precipitates were present in all PbS(–BA, –HCA & –NA):TIPS-Tc solutions. Both the supernatant and agglomerates were separated via filtration and analysed further.SAXS was performed on the supernatants and compared to the original SANS data in order to identify any potential large scale changes in QD structure that may have occurred during precipitation process (data and analysis shown, ESI,‡ Fig. S5), with the conclusion that the QDs remaining dispersed in all three supernatants [from PbS(–BA, –HCA & –NA):TIPS-Tc] were isolated, possessing similar structures to their initial QDs solutions.
Fig. 4 shows radially integrated SAXS data (a) and fluorescence micrographs (b–d), detected at λemission > 1000 nm (QD fluorescence emission) and λemission = 525/50 nm (TIPS-Tc fluorescence emission) for agglomerates formed from PbS(–BA, –HCA & –NA):TIPS-Tc solutions, respectively. All of the QD:TIPS-Tc agglomerates show clear QD scattering features between q = 0.03–0.3 Å−1, along with sharp scattering features q > 0.6 Å−1 commensurate with TIPS-Tc crystallites. The fluorescence microscopy, conducted at emission wavelengths sensitive to either QDs (IR) or TIPS-Tc (green) provides further confirmation of the presence of both QDs and TIPS-Tc. The following analysis and discussion of SAXS data aims to gain insight into the morphology of the PbS(–BA, –HCA & –NA):TIPS-Tc agglomerates.
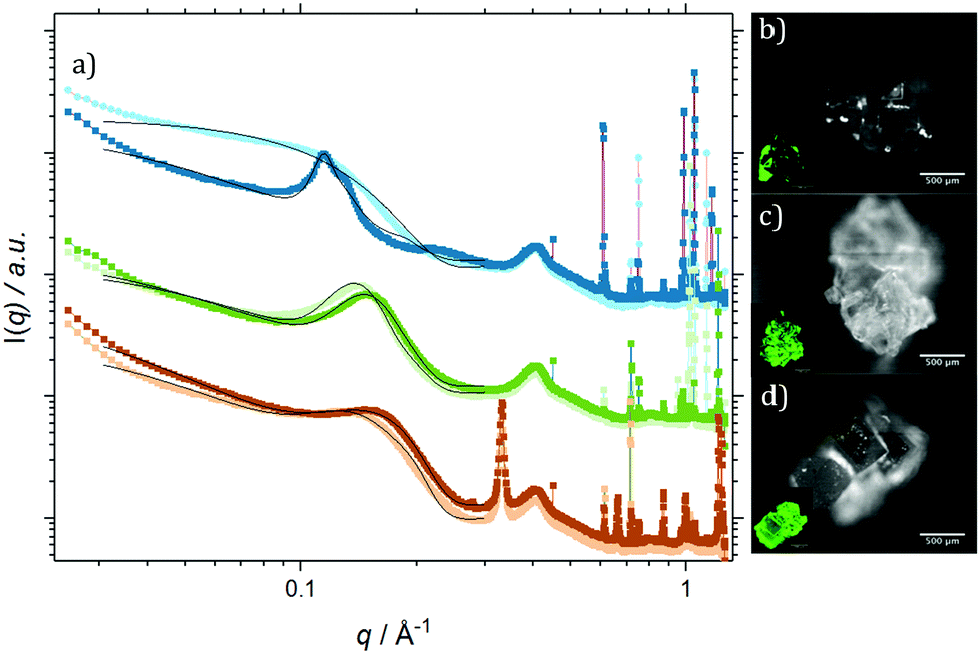 | ||
| Fig. 4 (a) Radially integrated SAXS data for PbS:BA:TIPS-Tc (light blue, dark blue), PbS:HCA:TIPS-Tc (light green, dark green) & PbS:NA:TIPS-Tc (light orange, dark orange) co-crystals, as formed and post DMF solvent annealing, respectively, with corresponding fits (black) and IR fluorescence micrographs (showing QD fluorescence λex 470 nm, λem > 1000 nm), PbS:BA:TIPS-Tc (b), PbS:HCA:TIPS-Tc (c) & PbS:NA:TIPS-Tc (d), with insert micrographs (showing TIPS-Tc fluorescence λex 470 nm, λem 525 nm). | ||
Focusing first on the high q region of scattering data (q > 0.5 Å−1), all agglomerates formed from PbS(–BA, –HCA or –NA):TIPS-Tc solutions exhibit sharp scattering peaks at q = 0.61 & 0.72 Å−1 consistent with the 110 and 111 reflections for crystalline TIPS-Tc (as previously identified, CCDC database, dx.doi.org/10.5517/cc119qsv). To estimate the size of the TIPS-Tc crystalline domains within the agglomerates, the apparent crystal grain size was calculated from the full width half maximum (FWHM) of the 111 diffraction peak using the Scherrer equation.39,40
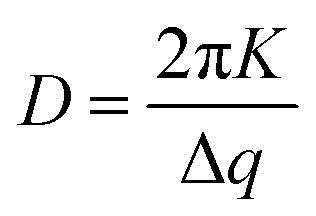 :TIPS-Tc (1:1) is shown in ESI,‡ Fig. S6).
:TIPS-Tc (1:1) is shown in ESI,‡ Fig. S6).
Data in the low q region (0.03–0.3 Å−1, commensurate with QD features) for the PbS–BA:TIPS-Tc agglomerate was fitted to a spherical form factor with a sticky hard sphere structure factor. In contrast, the QD scattering features for the PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates could only be effectively fitted by also adding in a Gaussian peak. For PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates, an effective volume fraction of QDs attributed to the scattering arising from sticky hard spheres (ϕSHS) or the Gaussian feature (ϕGaussian) were obtained through integration of the two separate components of the model fits (with data shown in Table 2 and the constituent fits in ESI,‡ Fig. S7). ϕSHS describes QD order that is more correlated than if the QDs were completely uncorrelated (compared to non-interacting hard-spheres), whilst ϕGaussian describes randomly distributed QD aggregate type features. For both PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc ϕSHS > ϕGaussian, with ϕSHS(PbS–NA) > ϕSHS(PbS–HCA). The presence of the Gaussian/aggregate scattering feature provides insight into the relative QD solubilities for PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc solutions at 300 mg mL−1 TIPS-Tc, with HCA-ligated QDs exhibiting the greatest propensity to form agglomerated features (despite being the lowest concentration of QDs studied) and hence a lower relative solubility compared to that of PbS–NA QDs, which still formed a large portion of agglomerated QD species. The interparticle separation for the HCA:TIPS-Tc and PbS–NA:TIPS-Tc aggregates was calculated, first obtaining the QD centre-to-centre distance from the q value at the peak centre and applying d = 2π/q, and then subtracting the QD-PbS-core diameter from the obtained centre-to-centre distance, with values shown in Table 2. The interparticle spacing of 12.35 and 14.86 Å, respectively for the PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates are slightly smaller than the extended lengths of two HCA or NA ligands and thus indicate either tilted or partially interdigitated ligands, between neighbouring QDs.
| ϕ SHS | ϕ FCC | |
|---|---|---|
| PbS–BA:TIPS-Tc | 1.0 | — |
| PbS–BA:TIPS-Tc (DMF anneal) | 0.57 | 0.43 |
| ϕ SHS | ϕ Gaussian | Gaussian scattering feature | ||||
|---|---|---|---|---|---|---|
| Peak position [q/Å−1] | Peak FWHM [q/Å−1] | Centre to centre distance [Å] | Inter-particle separation [Å] | |||
| PbS–HCA:TIPS-Tc | 0.59 | 0.41 | 0.1379 | 0.0191 | 46 | 12 |
| PbS–HCA:TIPS-Tc (DMF anneal) | 0.59 | 0.41 | 0.1463 | 0.0214 | 43 | 10 |
| PbS–NAA:TIPS-Tc | 0.7 | 0.3 | 0.1307 | 0.0287 | 48 | 15 |
| PbS–NA:TIPS-Tc (DMF anneal) | 0.7 | 0.3 | 0.1476 | 0.0378 | 43 | 9 |
Whilst the SAXS models described provided adequate fits to the QD scattering data between q = 0.03 and 0.3 Å−1 they are insufficient for q < 0.03 Å−1, where a significant turn-up at low q is observed, consistent with the fractal morphologies identified in the earlier SANS data, which showed an early snapshot of the QD self-assembly. However, the X-ray camera-length employed here was insufficient to probe this small-angle scattering feature of the agglomerates in more detail.
In order to gain further insights into the role of ligand in controlling both the QD sticky hard sphere and aggregated phases, the PbS(–BA, –HCA, –NA):TIPS-Tc agglomerates were exposed to DMF solvent vapours with the aim of stripping the QD ligands, employing a similar approach to that adopted by Baumgardner et al. where QD thin-films were immersed in DMF to form connected QD solids.42
For PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates exposed to DMF vapours, the same model employed to fit the SAXS data from unexposed agglomerates was sufficient to fit these data too, though with a shift of the Gaussian peak to higher q, indicating a shrinking in the dimensions of the QD aggregates. [n.b. adequate fits for the as formed PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc crystals could also be obtained using a sphere * sticky hard sphere + fractal sphere model, however, this approach was discounted as it was unable to adequately fit the PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc crystals post-exposure to DMF vapours.] Upon exposure of PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc crystals to DMF vapours, no change in ϕSHS or ϕGaussian is observed, indicating that no bulk migration of QDs occurs and that the QDs arranged as sticky hard spheres are sufficiently well dispersed/spaced so that no further aggregate features are formed. Analysis of the Gaussian peak position for the aggregate type features reveals that the interparticle spacing decreases for both PbS–HCA and PbS–NA to a value on the order of ∼9.5 Å.
For the as-formed PbS–BA:TIPS-Tc agglomerates, the QDs were initially distributed entirely with sticky hard sphere order, in stark contrast to the behaviour of PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates. However, upon exposure of the PbS–BA:TIPS-Tc agglomerate to DMF vapours, a pronounced broad scattering feature emerged with distinct peaks at q = 0.114 and 0.128 Å−1. Scattering data for the PbS–BA:TIPS-Tc agglomerates post DMF vapour annealing were adequately fitted to a sphere form factor with a sticky hard sphere structure factor plus an FCC colloidal crystal, with the integrated effective volume fractions of both components being ϕSHS = 0.57 and ϕFCC = 0.43, representing a substantial rearrangement of QD packing morphology upon DMF vapour annealing. The QD FCC colloidal crystal has a unit cell dimension of 96 Å, giving a centre-to-centre distance of 68 Å and an interparticle separation of 34.8 Å, which is commensurate with the ∼19 Å BA ligand layers identified in the earlier SANS analysis, providing further validation of the core–shell-sphere model with a Guinier approximation employed to describe the PbS–BA QDs, with the presence of excess BA in solution. The ability of the PbS–BA QDs possessing such large ligand shells to form an FCC phase further indicates that the overall QD dimensions (PbS core + ligand shell) must be relatively monodisperse.
Unlike PbS–HCA and PbS–NA, where DMF solvent annealing resulted in no change to the fractions of the QD present as either ϕSHS or ϕGaussian, but a change in interparticle spacing between QDs, DMF solvent annealing of the PbS–BA:TIPS-Tc agglomerate resulted in the conversion of a fraction of QDs dispersed as ϕSHS to ϕFCC. Critically the interparticle spacing derived from the obtained FCC unit cell indicates that DMF exposure did not change the thickness of the BA ligand envelope. Thus, the conversion of QDs from SHS to FCC must be mediated by the ability of DMF to solvent anneal the TIPS-Tc crystal phase, facilitating the phase segregation of QDs to form ordered colloidal crystal morphologies. This result is somewhat surprising, in that rather than stripping the small organic ligands as observed for PbS–HCA and PbS–NA, in the case of PbS–BA, we observe that the extended ligand layer is not only retained, but sufficiently unperturbed and well-defined to facilitate the formation of a colloidal crystal QD morphology.
Detailed analysis of SAXS data for the QD:TIPS-Tc nanocomposite agglomerates formed from solution reveals a highly complex morphology spanning a wide range of length scales. A potential scheme of how the different TIPS-Tc and QD morphological features are organised is shown in Fig. 5. The mass fractal QD structures formed early on (and identified via SANS) act as heterogeneous nucleation sites for TIPS-Tc crystallization. The presence of QDs does not impact on the unit cell of TIPS-Tc crystallites, which grow to ∼150 nm and form the larger mm-sized agglomerate materials studied. Two distinct QD morphologies have been identified as described by either a sticky hard sphere or QD aggregates. For the PbS–HCA:TIPS-Tc and PbS–NA:TIPS-Tc agglomerates, exposure to DMF vapours stripped the ligands from the aggregate QD domains, resulting in a reduction of the length scale of the aggregate features, commensurate with the PbS QD cores coming together within the aggregates. As such, we hypothesise that such QD agglomerates are likely to be present at the TIPS-Tc crystal grain boundaries. For the PbS–BA:TIPS-Tc, agglomerates DMF exposure instead lead to the formation of an FCC QD colloidal crystal, with a lattice spacing commensurate with the thick ligand shell identified in earlier SANS experiments. We propose that for the PbS–BA:TIPS-Tc nanocomposite, exposure to DMF vapours preferentially allows sufficient “plasticity” of the TIPS-Tc small organic molecules to sufficient mobility of the PbS–BA QDs, facilitating the phase segregation of QDs into the observed FCC colloidal crystal phase and that this process outcompetes the ligand displacement ability of DMF for such thick BA ligand shells.
 | ||
| Fig. 5 Representations of different QD distributions identified in PbS(–BA, –HCA & –NA):TIPS-Tc co-crystals, showing; (i) fractal like QDs at the centre of TIPS-Tc crystallites, (ii) QDs trapped within the growing crystallite (giving rise to sticky hard sphere QD features), aggregate QDs at the periphery of the crystalline interface and how these QD morphologies change upon exposure to DMF vapours. | ||
Fig. 5 presents a proposed interpretation of how the structural insights obtained from complementary SANS and SAXS measurements could best describe the ensemble structure and morphology of the QD:TIPS-Tc nanocomposites under investigation in this study. SANS reveals the formation of fractal QD features that form via diffusion limited aggregation. These fractal QD structures likely act as heterogeneous nucleation agents for TIPS-Tc crystallization and the crystal motif is unaffected by the nanoscale QD nucleating agents. The TIPS-Tc crystals most likely contain QDs at the crystallite centre (fractal-like QD nucleating agents), trapped within the growing crystallite (giving rise to sticky hard sphere QD features) and additionally a portion of QDs are likely to be expelled from the growing TIPS-Tc crystal front, giving rise to a further population of aggregate QDs at the periphery of the crystalline interface. Exposure of the QD:TIPS-Tc crystals to DMF vapour, a solvent known to be able to strip ligands from QDs alters the spacing between PbS–HCA and PbS–NA QD aggregate features but for PbS–BA QDs promotes the formation of ordered QD colloidal phases.
To conclude, this work demonstrates the power of utilizing a combination of SANS and SAXS techniques to study how functionalised QD self-assemble with TIPS-Tc to form hybrid QD/TIPS-Tc nanocomposite materials, where the QD distribution is controlled by the QD ligand, but may be further manipulated via solvent annealing approaches (as summarized in Fig. 5). In recent studies,19,21–23 small-angle scattering has been able to unambiguously elucidate the structures of both QD core and ligand structures when QDs are isolated in solution or in the regularly arranged in the solid state as thin-film colloidal crystals. In contrast, this work demonstrates that when QDs self-assemble together in solution with a small organic molecule the assembly process generates much less well-defined, fractal or disordered agglomerate features. Through controlling QD ligand chemistry it is possible to direct self-assembly at length scales from nanometres to microns, generating materials with potential applications as solar energy conversion materials where well dispersed QD nanostructures are desired, but also in thermoelectric and QD semiconducting devices where interconnected QD nanomorphologies are advantageous. The approach demonstrated here enables greater control and understanding of QD nanostructured assemblies and lays important groundwork, taking inspiration from colloid science, for rationally designing self-assembly approaches to obtain targeted QD nanomorphologies. However, this work also highlights the wide range of length scales over which structures manifest in such materials and the high degree of complexity they present, meaning that a great deal of careful study remains outstanding in order to master the rational design of energy harvesting nanocomposites using such approaches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding through the Winton Programme for the Physics of Sustainability and the Engineering and Physical Sciences Research Council (UK) via grants EP/P027814/1 and EP/P027741/1, The authors would like to acknowledge beamtime awarded at the ISIS Pulsed Neutron and Muon Source through experiment numbers RB1810513 on SANS2D (DOI: 10.5286/ISIS.E.RB1810513) and RB1820367 on LOQ (DOI: 10.5286/ISIS.E.RB1820367). This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 Research and Innovation Programme under the SINE2020 project, grant agreement no. 654000.References
- N. J. L. K. Davis, et al., Singlet Fission and Triplet Transfer to PbS Quantum Dots in TIPS-Tetracene Carboxylic Acid Ligands, J. Phys. Chem. Lett., 2018, 9(6), 1454–1460 CrossRef PubMed.
- M. H. Futscher, A. Rao and B. Ehrler, The Potential of Singlet Fission Photon Multipliers as an Alternative to Silicon-Based Tandem Solar Cells, ACS Energy Lett., 2018, 3(10), 2587–2592 CrossRef PubMed.
- Z. Huang and M. Lee Tang, Semiconductor Nanocrystal Light Absorbers for Photon Upconversion, J. Phys. Chem. Lett., 2018, 9(21), 6198–6206 CrossRef PubMed.
- A. Rao and R. H. Friend, Harnessing singlet exciton fission to break the Shockley–Queisser limit. Nature Reviews, Materials, 2017, 2(11), 17063 CAS.
- H. Sakai, et al., High-Yield Generation of Triplet Excited States by an Efficient Sequential Photoinduced Process from Energy Transfer to Singlet Fission in Pentacene-Modified CdSe/ZnS Quantum Dots, Chem. – Eur. J., 2018, 24(64), 17062–17071 CrossRef CAS PubMed.
- N. Nishimura, et al., Photon upconversion utilizing energy beyond the band gap of crystalline silicon with a hybrid TES-ADT/PbS quantum dots system, Chem. Sci., 2019, 10(18), 4750–4760 RSC.
- N. Yanai and N. Kimizuka, New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption, Acc. Chem. Res., 2017, 50(10), 2487–2495 CrossRef CAS PubMed.
- J. R. Allardice, et al., Engineering Molecular Ligand Shells on Quantum Dots for Quantitative Harvesting of Triplet Excitons Generated by Singlet Fission, J. Am. Chem. Soc., 2019, 141(32), 12907–12915 CrossRef CAS PubMed.
- V. Gray, et al., Direct vs Delayed Triplet Energy Transfer from Organic Semiconductors to Quantum Dots and Implications for Luminescent Harvesting of Triplet Excitons, ACS Nano, 2020, 14(4), 4224–4234 CrossRef CAS PubMed.
- V. Gray, et al., Towards efficient solid-state triplet–triplet annihilation based photon upconversion: Supramolecular, macromolecular and self-assembled systems, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- N. Greenham, X. Peng and A. Alivisatos, Charge separation and transport in conjugated polymer/cadmium selenide nanocrystal composites studied by photoluminescence quenching and photoconductivity, Synth. Met., 1997, 84(1–3), 545–546 CrossRef CAS.
- D. Ginger and N. Greenham, Photoinduced electron transfer from conjugated polymers to CdSe nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(16), 10622 CrossRef CAS.
- J. Kwak, et al., Characterization of quantum dot/conducting polymer hybrid films and their application to light-emitting diodes, Adv. Mater., 2009, 21(48), 5022–5026 CrossRef CAS PubMed.
- E. Martínez-Ferrero, J. Albero and E. Palomares, Materials, nanomorphology, and interfacial charge transfer reactions in quantum dot/polymer solar cell devices, J. Phys. Chem. Lett., 2010, 1(20), 3039–3045 CrossRef.
- J. D. Olson, G. Gray and S. A. Carter, Optimizing hybrid photovoltaics through annealing and ligand choice, Sol. Energy Mater. Sol. Cells, 2009, 93(4), 519–523 CrossRef CAS.
- Y. Zhou, et al., Improved efficiency of hybrid solar cells based on non-ligand-exchanged CdSe quantum dots and poly(3-hexylthiophene), Appl. Phys. Lett., 2010, 96(1), 013304 CrossRef.
- D. M. Kroupa, et al., In situ spectroscopic characterization of a solution-phase X-type ligand exchange at colloidal lead sulphide quantum dot surfaces, Chem. Commun., 2016, 52(96), 13893–13896 RSC.
- D. M. Kroupa, et al., Tuning colloidal quantum dot band edge positions through solution-phase surface chemistry modification, Nat. Commun., 2017, 8, 15257 CrossRef CAS PubMed.
- J. Maes, et al., Size and concentration determination of colloidal nanocrystals by small-angle X-ray scattering, Chem. Mater., 2018, 30(12), 3952–3962 CrossRef CAS.
- D. Moscheni, et al., Size-Dependent Fault-Driven Relaxation and Faceting in Zincblende CdSe Colloidal Quantum Dots, ACS Nano, 2018, 12(12), 12558–12570 CrossRef CAS.
- M. P. Weir, et al., Ligand Shell Structure in Lead Sulfide–Oleic Acid Colloidal Quantum Dots Revealed by Small-Angle Scattering, J. Phys. Chem. Lett., 2019, 10(16), 4713–4719 CrossRef CAS PubMed.
- S. W. Winslow, et al., Quantification of a PbClx Shell on the Surface of PbS Nanocrystals, ACS Mater. Lett., 2019, 1, 209–216 CrossRef CAS.
- S. W. Winslow, et al., Characterization of colloidal nanocrystal surface structure using small angle neutron scattering and efficient Bayesian parameter estimation, J. Chem. Phys., 2019, 150, 244702 CrossRef.
- E. Alonso-Redondo, et al., A new class of tunable hypersonic phononic crystals based on polymer-tethered colloids, Nat. Commun., 2015, 6, 8309 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Colloidal PbS nanocrystals with size-tunable near-infrared emission: observation of post-synthesis self-narrowing of the particle size distribution, Adv. Mater., 2003, 15(21), 1844–1849 CrossRef CAS.
- R. Heenan, J. Penfold and S. King, SANS at pulsed neutron sources: present and future prospects, J. Appl. Crystallogr., 1997, 30(6), 1140–1147 CrossRef CAS.
- R. Heenan, et al., Small angle neutron scattering using Sans2d, Neutron News, 2011, 22(2), 19–21 CrossRef.
- T. Ogawa, et al., Aggregation-free sensitizer dispersion in rigid ionic crystals for efficient solid-state photon upconversion and demonstration of defect effects, J. Mater. Chem. C, 2018, 6(21), 5609–5615 RSC.
- G. T. Wignall and F. Bates, Absolute calibration of small-angle neutron scattering data, J. Appl. Crystallogr., 1987, 20(1), 28–40 CrossRef CAS.
- A. Thampi, et al., Elucidation of excitation energy dependent correlated triplet pair formation pathways in an endothermic singlet fission system, J. Am. Chem. Soc., 2017, 140(13), 4613–4622 CrossRef.
- FOXTROT may be requested from: Soleil synchrotron SWING beamline (“Beamline Software”) https://www.synchrotron-soleil.fr/en/beamlines/swing, accessed 3rd June 2020.
- J. Ilavsky and P. R. Jemian, Irena: tool suite for modeling and analysis of small-angle scattering, J. Appl. Crystallogr., 2009, 42(2), 347–353 CrossRef CAS.
- L. Fan, et al., The absolute calibration of a small-angle scattering instrument with a laboratory X-ray source, Journal of Physics: Conference Series, IOP Publishing, 2010 Search PubMed.
- D. Schneidman-Duhovny, et al., FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles, Nucleic Acids Res., 2019, 44(W1), W424–W429 CrossRef.
- D. Schneidman-Duhovny, et al., Accurate SAXS Profile Computation and its Assessment by Contrast Variation Experiments | Elsevier Enhanced Reader, Biophys. J., 2019, 105, 962–974 CrossRef.
- M. Nishio, The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates, Phys. Chem. Chem. Phys., 2011, 13(31), 13873–13900 RSC.
- G. Beaucage, Small-Angle Scattering from Polymeric Mass Fractals of Arbitrary Mass-Fractal Dimension, J. Appl. Crystallogr., 1996, 29(2), 134–146 CrossRef CAS.
- D. F. R. Mildner and P. L. Hall, Small-angle scattering from porous solids with fractal geometry, J. Phys. D: Appl. Phys., 1986, 19, 1535–1545 CrossRef.
- R.-J. Roe, Methods of X-ray and neutron scattering in polymer science, Oxford University Press on Demand, 2000, vol. 739 Search PubMed.
- J. Rivnay, et al., Quantitative analysis of lattice disorder and crystallite size in organic semiconductor thin films, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045203 CrossRef.
- J. I. Langford and A. J. C. Wilson, Scherrer after sixty years: A survey and some new results in the determination of crystallite size, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- W. J. Baumgardner, K. Whitham and T. Hanrath, Confined-but-Connected Quantum Solids via Controlled Ligand Displacement, Nano Lett., 2013, 13(7), 3225–3231 CrossRef CAS PubMed.
Footnotes |
| † The underlying data from this article are available at https://doi.org/10.15131/shef.data.12752630. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01109f |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |