
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective colloidal bonds via polymer-mediated interactions
Joeri
Opdam†
 a,
Remco
Tuinier
*a,
Theodore
Hueckel†
b,
Thom J.
Snoeren
b and
Stefano
Sacanna
*b
a,
Remco
Tuinier
*a,
Theodore
Hueckel†
b,
Thom J.
Snoeren
b and
Stefano
Sacanna
*b
aLaboratory of Physical Chemistry, Department of Chemical Engineering and Chemistry, & Institute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, P. O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: r.tuinier@tue.nl
bMolecular Design Institute, Department of Chemistry, New York University, 29 Washington Place, New York 10003, USA. E-mail: s.sacanna@nyu.edu
First published on 7th July 2020
Abstract
Regioselectivity in colloidal self-assembly typically requires specific chemical interactions to guide particle binding. In this paper, we describe a new method to form selective colloidal bonds that relies solely on polymer adsorption. Mixtures of polymer-coated and bare particles are initially stable due to long-ranged electrostatic repulsion. When their charge is screened, the two species can approach each other close enough for polymer bridges to form, binding the particles together. By utilizing colloidal dumbbells, where each lobe is coated with polymer brushes of differing lengths, we demonstrate that the Debye screening length serves as a selective switch for the assembly of bare tracer particles onto the two lobes. We model the interaction using numerical self-consistent field lattice computations and show how regioselectivity arises from just a few nanometers difference in polymer brush length.
1 Introduction
Colloidal particles are widely studied because of their display of phase behaviour that closely mimics that of atoms and molecules.1 Like their atomic counterparts, colloids can be utilized as self-assembly building blocks that come together to form larger functional architectures and bulk materials.2,3 Most colloids, however, are limited in their assembly behaviour because of the isotropic character of their interactions. One way to achieve control over the structure and functionality of colloidal systems is to engineer particles' shapes or decorate their surface with sticky patches capable of engaging in directional and selective interparticle bonds. Several types of “colloidal glues” have been developed to serve this purpose, from simple electrostatic charges4 and hydrophobic/hydrophilic interactions,5 to more sophisticated lock-and-key entropic forces6 and DNA–mediated interactions.7In this article, we describe how to implement polymer-mediated interactions to program colloidal self-assembly. A polymer-mediated bond forms when a polymer-coated particle comes into contact with a polymer-free particle. The two species are initially stabilized by electrostatic repulsion, preventing them to make physical contact. By screening the charge, however, the particles reach a proximity where the polymer can form bridges and bind them together. We demonstrate selectivity by using dumbbell-shaped colloids with polymer brushes of varying lengths grafted onto each lobe. Bridging occurs at different screening lengths based on the brush length, leading to the programmable assembly of tracer particles at each lobe. We investigate polymer-mediated colloidal bonds by combining experiments and numerical self-consistent field (SCF) lattice computations, which are widely used to study polymeric and electrostatic interactions.8–11 With SCF, the thermodynamic free energy minimum of inhomogeneous systems containing interfaces, polymers, ions and other molecules can be determined, which in turn can be used to compute interaction potentials. An advantage of SCF over full scale computer simulations is the efficiency of the computations, which makes it possible to scan a large parameter space in a short period of time.
2 Methods
In this section we provide a summary of the experimental and theoretical methods used. A detailed description of the numerical self-consistent field method is given elsewhere;12–14 here we give a short overview of the most relevant details.2.1 Particle synthesis and assembly
The colloidal model systems we use in this study consist of polymerizable 3-(trimethoxysilyl)propyl methacrylate (TPM) emulsions and silica particles.15,16 Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO) triblock copolymers (Pluronic) of different lengths are used to create polymer brushes on TPM while silica remains bare, acting as tracers. The Pluronics used in this study are F108 (EO132PO56EO132) and F68 (EO76PO29EO76).TPM emulsions (Fig. 1a) are synthesized by introducing 100 μL TPM oil (98% MilliporeSigma) to a 100 mL solution of water and ammonia ([NH3] = 15 mM) and stirred magnetically at 300 rpm in a sealed beaker. After 2 hours, liquid TPM spheres are finished growing, at which point a solution of Pluronic F108 is added to form a polymer brush ([F108] = 30 μM) (Fig. 1b). The particles can then either be washed and utilized in their liquid state, or polymerized. Particles are washed by centrifuging them at 100 G for 4 hours, decanting the supernatant, resuspending the concentrated particles, and finally adding DI water. The washing process is repeated 3 times. To solidify the TPM, 20 mg of azobisisobutyronitrile (AIBN, 98% MilliporeSigma) is added to the suspension and stirred for five minutes, then the particles are heated for 4 hours at 80 °C. The polymerized particles are then washed in the manner described above (Fig. 1c).
 | ||
| Fig. 1 Synthesis of polymer-coated spheres and Janus dumbbells. (a) Optical micrograph of monodispersed TPM oil droplets. Scale bar 3 μm. (b) Schematic representation of the Pluronic surfactant's architecture at the TPM–water interface, with hydrophilic arms extending out into the water to form a brush and the hydrophobic core anchoring the polymer in the oil. λD represents the range of the electrostatic interactions from the negatively charged TPM surface. (c) SEM micrograph showing polymerized F108-TPM. Scale bar 3 μm. (d) Schematic of the dumbbell synthetic pathway. Liquid TPM particles are coated with F108 and polymerized. Solid F108-TPM is used as a seed to grow a new liquid TPM lobe. The lobe is functionalized with a shorter polymer (F68) and polymerized. The final product has two lobes with variable brush length. (e) SEM micrographs showing the tunability in the size of the second lobe, shown in green. Scale bar 1 μm. (f) Confocal micrograph of the dumbbells with different fluorescent dyes in each lobe. Scale bar 3 μm. (g) SEM micrograph showing the monodispersity of the dumbbell product. Scale bar 4 μm. | ||
As we show in Fig. 1d, Janus dumbbell-shaped particles with two different polymer brushes are made by nucleating a secondary lobe onto polymerized F108-coated TPM particles (F108-TPM). Briefly, 100 μL TPM oil is added to a preheated (80 °C) 20 mL F108-TPM suspension (0.1 wt%) containing 15 mM NH3. The size of the lobe directly depends on the volume of TPM oil added at this stage (Fig. 1e), while heating ensures the nucleation of a single lobe. A second Pluronic surfactant (30 μM, F68) is added to form a shorter polymer brush on the newly nucleated lobe. The dumbbells are then polymerized and purified through the processes described above (Fig. 1f and g). The silica tracers are synthesized via the Stöber process.16
Self-assembly experiments are carried out by mixing polymer-coated particles with a large excess of silica tracers (number ratio∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3000), and then adding salt to the desired final concentration. Relatively diluted NaCl stock solutions (≈25 mM) are used to avoid steep gradients during mixing. The suspension is left undisturbed for one hour to allow the particles to assemble. Unreacted silica are purified via centrifugation.
:3000), and then adding salt to the desired final concentration. Relatively diluted NaCl stock solutions (≈25 mM) are used to avoid steep gradients during mixing. The suspension is left undisturbed for one hour to allow the particles to assemble. Unreacted silica are purified via centrifugation.
2.2 Self-consistent field calculations
Numerical self-consistent field lattice computations, developed by Scheutjens and Fleer,17,18 were used to study colloidal interactions mediated by grafted polymers. Scheutjens–Fleer (SF) SCF is based upon mean-field lattice theory similar to Flory–Huggins theory,19,20 but takes concentration gradients into account and can therefore be used to find equilibrium configurations of inhomogeneous systems. In SF–SCF, every component in the system is composed of segments and interactions between the different segments are taken into account by Flory–Huggins χ-parameters.21 A potential field is assigned to the different segment types in every lattice layer based on the interactions and local volume fractions of all different segments in the lattice layer and the neighbouring layers. Based on these potential fields, the statistical probability for a certain segment to be in a certain layer is calculated. Polymer chain conformations are approximated by the freely-jointed chain model resulting from the Edwards diffusion equation. The finite segment size and volume-filling constraint are taken into account with boundary conditions.13 Based on the segment probabilities a new set of volume fractions is calculated for each layer. This problem is numerically solved in a self-consistent way within the SF–SCF machinery by starting with an initial guess for the potential fields at each layer and calculating a new volume fraction profile which in turn leads to a new set of potential fields:| {ϕj(uj)}N ↔ {uj(ϕj)}N, | (1) |
The χ-parameters used in the calculations are shown in Table 1. The parameters χEO–W, χPO–W and χEO–PO are taken from Hurter et al.23 The χTPM–EO and χTPM–PO parameters are estimated from the interaction parameters between Pluronic and benzene.23 This is a rough approximation which is based upon the fact that benzene and TPM are both apolar molecules. However, it was found that the exact values of these interaction parameters do not have a large influence on the results since the adsorption of Pluronics at the TPM/water interface is driven by the incompatibility of TPM and PPO with water. Moreover, for simplicity we chose to model the bare tracer particles in the SF–SCF computations as TPM particles. This means that the bridging attraction between the polymer-coated particle and the bare particle is also predominantly determined by the unfavourable interaction of the bare TPM surface with the water phase. In the experimental system the bare particle is a silica particle and there is a specific attraction between the PEO chains and the silica surface due to hydrogen bonding.24 However, in both cases it is the relative difference in affinity of PEO chains and water for the surface that results in a strong bridging attraction and therefore modeling the second particle as a TPM or a silica particle is not expected to lead to qualitative differences.
| χ | H2O | TPM | EO | PO |
|---|---|---|---|---|
| H2O | 0.0 | 3.7 | 0.4 | 2.0 |
| TPM | 3.7 | 0.0 | 0.14 | 0.1 |
| EO | 0.4 | 0.14 | 0.0 | 0.006 |
| PO | 2.0 | 0.1 | 0.006 | 0.0 |
The χTPM–W parameter is estimated by determining the interfacial tension of the TPM/water interface using SF–SCF in the absence of surfactants as a function of χTPM–W. The interfacial tension can be directly calculated from the grand potential of the system (Ω) which is defined as the free energy contribution of the inhomogeneities in the system, as follows from expressing the Helmholtz free energy F as:
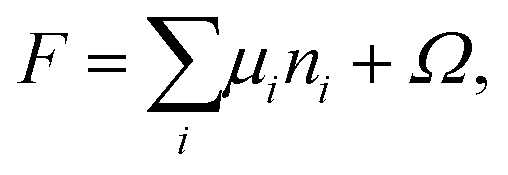 | (2) |
 | (3) |
Fig. 2 shows a comparison of polymer brushes adsorbed at liquid/liquid and solid/liquid interfaces. It shows that, although the profiles very close to the interface are different due to the dissimilar nature of the interface, the profiles of the brushes further away from the interface are quite similar for both cases. A key quantity needed to describe the selective adsorption process is the hydrodynamic layer thickness δh, which can be determined from the SF–SCF volume fraction profiles using a method described by Cohen Stuart and coworkers.29,30 The value of δh is also very close for both interfaces as can be seen in Fig. 2. The adsorbed amount of Pluronics is also directly determined from the volume fraction profiles and is expressed as the excess number of surfactants at the interface:
 | (4) |
 | ||
| Fig. 2 Self-consistent field theory results for the volume fraction profiles of an EO132PO56EO132 polymer brush layer adsorbed at (a) the liquid–TPM/water interface and (b) at the solid–TPM/water interface, in equilibrium with a polymer solution at the cmc. The volume fraction profile in (a) is positioned such that the lattice layer where ϕTPM is closest to ϕwater is taken as x = 0 nm. The adsorbed amount of polymer is equal for both interfaces. | ||
A wall is placed at the last layer of the lattice which models the second particle. The properties of the second particle, i.e. the surface charge density and the χ-parameters, are taken to be the same as for the TPM. The relative permittivity ε used for water is 80 and for all other components ε = 10. The surface potential of the particles is affected by the grafted polymers due to their different relative perimittivity with respect to water, resulting in different surface potentials for the interacting particles. The surface potentials mentioned in this article, obtained from the SF–SCF calculations, are always the values for the bare particle. When the salt concentration is varied, the surface charge density of the particles is modified accordingly, so that the surface potential of the particles is unaffected by salt concentration.
The interaction potential is determined by calculating the difference in free energy of the system at large particle separation (no interaction) and the free energy of the system at a certain interparticle distance h as a function of this distance h. Here the system is considered to be in restricted equilibrium, where the number of polymers in between the surfaces is constant and the chemical potential is allowed to change as a function of distance. In this case the dimensionless interaction potential is given by:31
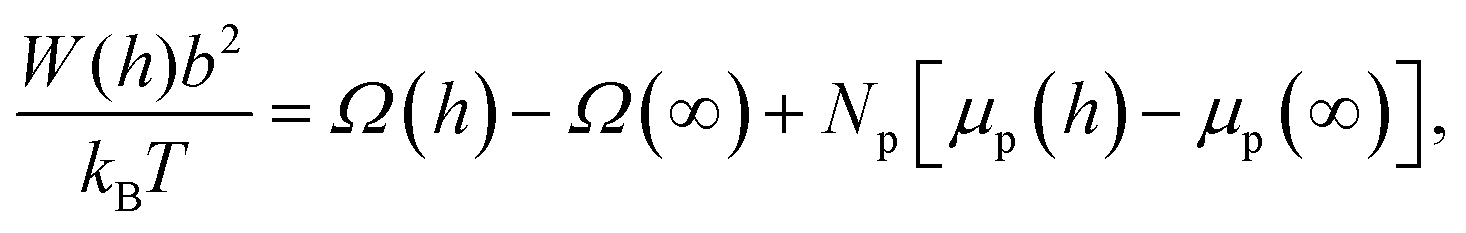 | (5) |
We emphasize here that the SF–SCF calculations in this study demonstrate the proof of principle for selective assembly of bare and polymer-coated colloidal particles obtained by changing the Debye screening length. The selective assembly is solely obtained through the competition between electrostatic repulsion and bridging attraction and does not depend on specific chemical interactions. Therefore, it is expected that the exact values for the SCF parameters used in this study do not have a qualitative influence on the results but only affect the shape of the interaction curves, the screening length needed to form bonds, and the selectivity of those bonds.
3 Results and discussion
In this section the main results of the experimental and theoretical investigations are discussed and compared. First we focus on the polymer-mediated assembly of bare particles on polymer-coated spherical particles and investigate the electrostatic and polymeric interactions that are involved. Finally we investigate the selective assembly of colloidal tracers on polymeric dumbbells and discuss how to influence the selectivity of this assembly.3.1 Polymer-mediated colloidal bonds
Polymer bridging traditionally results in random flocculation.33 We achieve specific binding in our model system comprising F108-TPM and bare silica because polymer bridging occurs exclusively between these two species. This complementarity is enabled by the strong anchoring of the hydrophobic Pluronic core to the TPM, which keeps the water phase polymer-free and allows the silica surface to remain bare.Electrostatic repulsion offers control over assembly. Negatively charged F108-TPM and silica are initially stable when mixed, however, when their charge is screened, the particles can come close to each other and bind (Fig. 3a). In this way, salt concentration is used to trigger the formation of polymer-mediated colloidal bonds. In Fig. 3b, for example, a mixture containing F108-TPM and silica particles remains stable when no salt is added to the system. A moderate 25 mM NaCl concentration, however, allows the particles to recognize each other and form clusters (Fig. 3c).
 | ||
| Fig. 3 Polymer-mediated colloidal bonds. (a) Schematic of polymer-mediated binding. Polymer-coated and bare particles are initially stable due to long-ranged electrostatic repulsion. When the charge is screened with salt, the particles can closely approach one another and the polymer can adsorb onto the bare surface to form a colloidal bond. (b and c) Optical micrographs of a mixture of F108-TPM and silica tracers at different salt concentrations. Without salt (b), the mixture is stable, and at 25 mM NaCl (c) clusters rapidly form. (b and c) Scale bars 4 μm. (d) SEM micrograph of clusters formed from a large F108-TPM core and small silica shells. (e) SEM micrograph of tetrahedral clusters formed from small F108-TPM cores and large silica shells. (d and e) Scale bars 2 μm. | ||
Different particles can be easily combined in this fashion to generate an array of colloidal products. Various cluster geometries, for instance, are easily accessible by changing the core:shell size ratio. In Fig. 3d and e, a large core and small shells lead to a colloidosome structure, while a small core and larger shells result in tetrahedrally coordinated clusters.
3.2 Selective binding
Beyond simply triggering adhesion, salt concentration can be used to make tracers select one type of polymer-coated surface over another. For example, long brushes can form bonds at a longer Debye screening length (λD) than shorter brushes. Experimentally, two different polymer brushes, F108 (long) and F68 (short), are installed onto TPM surfaces. Since the polymer is grafted to the TPM surface during polymerization, F108-TPM and F68-TPM can be mixed together without polymer exchange, retaining their distinct brush lengths. The two particles begin forming bonds with bare silica at different salt concentrations, leading to selective adhesion of tracers.Schematized in Fig. 4, a mixture of F108-TPM, F68-TPM, and silica display three assembly behaviors at varying salt concentrations. For instance, both brushes are electrostatically stabilized when λD is long (≈8 nm). At a λD of 4 nm, tracers begin adhering to F108-TPM, however the F68-TPM brush is too short to form polymer bonds. At shorter λD (≈2 nm), both brushes are activated, and tracers adhere indiscriminately. Shown also in Fig. 4 is an experimental example of selective assembly of silica tracers onto F108-TPM, while F68-TPM (fluorescent) remains stable.
 | ||
| Fig. 4 Selective bonds on polymer brushes of different lengths. An optical micrograph of a mixture of F108-TPM, F68-TPM, and tracers at λD = 4 nm reveals selective assembly, as tracers bind solely to F108-TPM while F68-TPM (fluorescent) remains stable. Scale bar 4 μm. Bottom left panel: F108-TPM particle coated in tracers. Bottom right panel: Stable F68-TPM particle. Inset: Schematic representation of the selection mechanism for tracers. For a large λD, both F108-TPM (red) and F68-TPM (green) are stable. As λD shrinks, F108-TPM can collect tracers, while F68-TPM remains stable. With a short λD both particles are able to collect tracers. | ||
3.3 Effect of block copolymer composition
The range of screening lengths where selective adsorption of the silica tracers occurs depends on the thickness of the polymer brush. The effect of the block lengths of the Pluronic surfactants on the hydrodynamic thickness of the surfactant brush at the TPM/water interface was investigated with SCF. The hydrodynamic brush thickness δh and the adsorbed amount Γ for varying number of EO units n and number of PO units m are shown in Fig. 5a and b. The brush thickness δh increases roughly linearly as a function of the hydrophilic chain length n (Fig. 5a), which is in agreement with what is expected from scaling relations for grafted polymer chains.34 There is a slight decrease in the slope of the curve due to the decrease of the adsorbed amount of polymers at the interface Γ as a function of the hydrophilic block length n (Fig. 5b). Because of this lower adsorbed amount the polymer coils can occupy more space parallel to the surface, resulting in less stretching of the polymers and therefore a slightly smaller hydrodynamic brush thickness. In contrast to the hydrophilic block length, a larger hydrophobic block length results in a slight increase of the adsorbed amount of the surfactants. As a result, the hydrodynamic brush thickness also becomes somewhat larger as a function of m due to the extra volume occupied by the PO blocks and a small increase of the stretching of the polymers. A larger hydrophobic block length of the Pluronic surfactants leads to a smaller increase in the brush thickness with respect to a larger hydrophilic block length, however it does lead to a more dense brush than a larger hydrophilic block length. Illustrative volume fraction profiles for Pluronic surfactants with different block lengths are shown in Fig. 5c–e. | ||
| Fig. 5 (a) Hydrodynamic brush thickness δh and (b) adsorbed amount Γ of an EOnPOmEOn surfactant brush layer at the TPM/water interface as a function of n (black) and m (gray) calculated using SF–SCF theory. (c–e) Illustrative volume fraction profiles for (n = 76, m = 56), (n = 160, m = 56) and (n = 76, m = 89), respectively. | ||
3.4 Colloidal interactions
To further study the interactions involved in the selective assembly, the interaction potential between polymer-coated and bare-TPM particles is investigated. The interaction potential between a negatively charged TPM particle with an F108 polymer brush and a second negatively charged bare TPM particle is shown in Fig. 6a. At large particle separation, the interaction between the particles vanishes. A repulsion develops when the particles come closer due to the overlap of the electrostatic double layers. Below a certain separation distance h the interaction turns from repulsive to strongly attractive due to a bridging attraction. It arises due to the adsorption of the polymers on the surface onto the second bare particle. Fig. 6b shows an example of the volume fraction profile of the polymer brush at h = 24 nm where there is an attraction of roughly 2kT. At this distance the surfactant covers about 10% of the surface of the second particle, leading to a bridging attraction that is strong enough to compensate the electrostatic repulsion and induce an overall effective attraction. The range of the bridging attraction is significantly larger than the hydrodynamic thickness of the polymer brush which is 13.4 nm (see Fig. 2b) for this specific Pluronic surfactant. This is because the bridging attraction is so favourable that the polymers stretch,35 reducing their configurational entropy, to adsorb at the surface of the bare particle as can be seen in the volume fraction profile of the polymer brush. It can be argued that this stretching might not occur in an experimental system since the surfactants have to cross an energy barrier to adsorb onto the surface of the second particle due to the stretching of the polymers to reach the thermodynamic energy minimum shown in Fig. 6b. However it can be expected that the range of the bridging interaction is indeed longer than the hydrodynamic brush thickness since some percentage of the surfactant tails will extend farther than the hydrodynamic thickness (as seen in Fig. 2b). Moreover, in an experimental system the polymers have a certain polydispersity and the extension of the chains is fluctuating, increasing the likelihood that the surfactants reach the second particle at distances larger then δh. | ||
| Fig. 6 (a) Interparticle separation distance h dependence of the interaction potential W between negatively charged F108-TPM and bare-TPM. The radius of the particles is 50 nm, the salt concentration is 10 mM and the surface charge density of both particles is 29.5 mC nm−2 resulting in a surface potential of −100 mV for the bare particle. (b) Volume fraction profile of the polymer brush at an interparticle distance of 24 nm corresponding to the red dot in panel (a). | ||
The height of the maximum in the interaction potential curve that represents the electrostatic repulsive barrier depends on several parameters. The effects of the salt concentration, the surface potential (ψ0) and the radius of the particles (a) on the potential curve are shown in Fig. 7. The salt concentration tunes the range of the electrostatic repulsion, however the range of the bridging attraction is hardly affected. As a result, there is an electrostatic barrier against bridging flocculation at low salt concentrations, but at higher salt concentrations, the range of the electrostatic repulsion becomes smaller and the barrier height decreases. The strength of the electrostatic repulsion can also be tuned with the surface potential and the size of the particles. When the surface potential or the size of the particles increases, the overlap of the electrostatic double layers increases leading to a stronger repulsion and therefore a higher repulsive barrier in the interaction potential curve.
 | ||
| Fig. 7 Influence of salt concentration (a), surface potential ψ0 (b) and particle radius a (c) on the interaction potential W for F108-TPM interacting with bare-TPM, determined using numerical SF–SCF lattice computations. Unless otherwise indicated, the radius of the particles is 50 nm, the salt concentration is 10 mM and the surface charge density, equal for both interacting particles, is chosen such that the surface potential of the bare particle is −100 mV. The surface charge density in the calculations for panel (b) is varied to obtain surface potentials of −50 mV, −100 mV and −200 mV for the bare particle. | ||
3.5 Regioselectivity on Janus dumbbells
With two brushes installed on a single particle, tracers can be directed to adhere to specific areas on that particle. The binary brush particles take the form of Janus colloidal dumbbells, whose synthesis is described in Section 2.1. Using SF–SCF, we show that the dumbbells have a different activation barrier for the two lobes. In Fig. 8a–c, interaction between bare-TPM, and either F108-TPM (red) or F68-TPM (green) is plotted for different salt concentrations. Because the surface charge densities and surface potentials of the particles in the experimental system are not known, the surface charge densities in the calculations were chosen such that the surface potential of the bare particles is −100 mV, which is assumed to be a reasonable value for charged colloidal particles. Due to the different brush thicknesses of δh = 13.4 nm and δh = 8.1 nm for F108 and F68, respectively, we see that the activation barrier for F108 is lower in all cases. At low salt concentrations (Fig. 8a), however, there is still a significant electrostatic repulsive barrier for both lobes, indicating that the whole dumbbell will remain tracer free. In Fig. 8b, the increased salt concentration decreases the activation barrier for F108 to roughly 1kT, leading to tracer adhesion, while the short brush still faces a large electrostatic barrier. Finally, at high salt concentrations (Fig. 8c), both barriers are negligible and tracers will adhere everywhere on the dumbbell. | ||
| Fig. 8 Regioselectivity on Janus dumbbells. (a–c) SF–SCF interaction potential curves for F108-TPM (red) and F68-TPM (green) interacting with a bare-TPM particle. The radii of all particles is 225 nm, corresponding to the average radius of the experimental tracer particles, and the salt concentrations are 10 mM (a), 16 mM (b) and 32 mM (c). The surface charge density, equal for all interacting particles, is chosen such that the surface potential of the bare particle is −100 mV. (d–f) Optical micrographs of mixtures of Janus dumbbells and tracers at various salt concentrations. At 5 mM NaCl (d), both lobes of the dumbbell are stable. At 7.5 mM (e), the smaller F108-TPM lobe is solely decorated in tracers. At 10 mM NaCl (f), the whole dumbbell is covered in tracers. Scale bars (d–f) are 3 μm. (g) Schematic representation of regioselectivity. Without salt, both lobes have sufficient electrostatic repulsion to remain stable. With salt added, tracers can adhere first to the F108-TPM lobe (red) while the F68-TPM lobe (green) is still electrostatically stabilized. With further salt, both lobes are decorated in tracers. (h) A scanning electron micrograph of a dumbbell with tracers regioselectively adhering to the F108 lobe. Scale bar 1 μm. | ||
Fig. 8d–f show optical micrographs of a dispersion containing negatively charged silica tracers and Janus dumbbells, verifying the adsorption trend that was expected from the theoretical SF–SCF calculations. At low salt concentrations, there is no tracer adhesion (Fig. 8d), and at high salt concentrations there are tracers on both lobes of the dumbbell (Fig. 8f). At an intermediate salt concentration, however, the silica particles selectively adsorb on the smaller F108-TPM lobe of the dumbbell, while the larger F68-TPM lobe remains bare. A schematic of the assembly behavior is shown in Fig. 8g, where the F108-TPM (red) surface is the first to become decorated with tracers as λD shrinks, and F68-TPM (green) is eventually decorated as well. Fig. 8h shows a SEM micrograph of a dumbbell from Fig. 8e, with tracers on the F108-TPM lobe.
The exact salt concentrations where the selective adsorption takes place in the experimental system do not match quantitatively with the theoretical predictions from Fig. 8a–c. A likely reason for this difference is that the electrostatic interactions do not correctly match with the experimental system. The surface potentials of the particles in the experimental system do not exactly match the surface potentials in the theoretical model and the size of both dumbbell lobes and the adsorbate particle in the model are taken to be equal to the size of the experimental silica particles. Also the range of the electrostatic interactions is slightly overestimated due to the value chosen for the lattice unit size as mentioned in Section 2.2.3. The bridging interactions in the calculations could also be made to resemble the experimental system more closely by modeling the adsorbate particle in the calculations specifically as a silica particle. A more realistic model of PEO adsorption on a silica surface was proposed by Postmus et al.,36 here we use a general model for the proof of principle of regioselectivity through polymer brushes with different thickness. Finally, the thickness of the polymer brushes might be underestimated in the SF–SCF model due to the mean field approximation where fluctuations are not taken into account.12 Polydispersity of the block copolymers, that can lead to an increased effective brush thickness,37 is also not taken into account in our model.
Regioselectivity is determined by the difference between the height of the repulsive barriers of the dumbbell lobes interacting with a bare particle (shown in Fig. 8a–c). This difference depends mostly on the brush thickness between the two dumbbell lobes, set by the composition of the polymers. A large difference in brush thickness leads to more selective assembly by widening the range of salt concentrations where assembly can be found. Moreover, particle properties like size and surface potential affect the strength of the electrostatic repulsion, as seen in Fig. 7, and can further enhance the contrast between the energy barriers at each lobe. This can potentially lead to a system with three distinct polymer lengths for increased complexity.
4 Conclusions
We have demonstrated the possibility of polymer-mediated selective adsorption of particles on colloidal dumbbells that contain polymer brushes with a different thickness at each lobe. The method discussed here is generally applicable for surfaces that contain polymer brushes with different thickness and does not require specific interactions such as complementary DNA strands, but is obtained simply by tuning electrostatic and polymeric interactions. Currently, polymer-mediated adsorption results in irreversible and non-reconfigurable binding, preventing ordered structures such as crystals from forming, so future systems will look to achieve reversibility between polymer-coated and bare surfaces. Further, materials such as polynipam microgels are an attractive choice for polymer-mediated interactions, since properties such as brush length can be controlled by various stimuli, allowing for dynamic tuning of the interaction range. Other applications of polymer-mediated interactions include the use of more complex polymer-coated patchy particles for directional colloidal self-assembly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF CAREER award DMR-1653465. The Zeiss Merlin FESEM was acquired through the support of the NSF under award number DMR-0923251. J. O. and R. T. are grateful for financial support from the Dutch Ministry of Economic Affairs of the Netherlands via The Top-consortium Knowledge and Innovation (TKI) roadmap Chemistry of Advanced Materials (CHEMIE.PGT.2018.006).References
- W. Poon, Science, 2004, 304, 830–831 CrossRef CAS PubMed.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- Y. A. Vlasov, X.-Z. Bo, J. C. Sturm and D. J. Norris, Nature, 2001, 414, 289–293 CrossRef CAS PubMed.
- M. E. Leunissen, C. G. Christova, A.-P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. van Roij and A. van Blaaderen, Nature, 2005, 437, 235–240 CrossRef CAS PubMed.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381–384 CrossRef CAS PubMed.
- S. Sacanna, W. T. M. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575–578 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–55 CrossRef CAS PubMed.
- J.-R. Roan and T. Kawakatsu, J. Chem. Phys., 2002, 116, 7295–7310 CrossRef CAS.
- R. Tuinier and G. J. Fleer, Macromolecules, 2004, 37, 8754–8763 CrossRef CAS.
- J. F. L. Duval, F. A. M. Leermakers and H. P. van Leeuwen, Langmuir, 2004, 20, 5052–5065 CrossRef CAS PubMed.
- M. Vis, R. Tuinier, B. W. M. Kuipers, A. Vrij and A. P. Philipse, Soft Matter, 2018, 14, 4702–4710 RSC.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Springer, Netherlands, 1998 Search PubMed.
- G. J. Fleer, Adv. Colloid Interface Sci., 2010, 159, 99–116 CrossRef CAS PubMed.
- G. H. Fredrickson, The Equilibrium Theory of Inhomogeneous Polymers, Oxford University Press, 2005 Search PubMed.
- C. van der Wel, R. K. Bhan, R. W. Verweij, H. C. Frijters, Z. Gong, A. D. Hollingsworth, S. Sacanna and D. J. Kraft, Langmuir, 2017, 33, 8174–8180 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. M. H. M. Scheutjens and G. J. Fleer, J. Phys. Chem., 1979, 83, 1619–1635 CrossRef CAS.
- J. M. H. M. Scheutjens and G. J. Fleer, J. Phys. Chem., 1980, 84, 178–190 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS.
- M. L. Huggins, J. Phys. Chem., 1942, 46, 151–158 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- B. Vincent, Colloids Surf., 1990, 50, 241–249 CrossRef CAS.
- P. N. Hurter, J. M. H. M. Scheutjens and T. A. Hatton, Macromolecules, 1993, 26, 5592–5601 CrossRef CAS.
- G. P. van der Beek, M. A. Cohen Stuart, G. J. Fleer and J. E. Hofman, Macromolecules, 1991, 24, 6600–6611 CrossRef CAS.
- S. Sacanna, W. K. Kegel and A. P. Philipse, Langmuir, 2007, 23, 10486–10492 CrossRef CAS PubMed.
- S. Sacanna, W. K. Kegel and A. P. Philipse, Phys. Rev. Lett., 2007, 98, 13–16 CrossRef PubMed.
- M. Youssef, A. Morin, A. Aubret, S. Sacanna and J. Palacci, Soft Matter, 2020, 16, 4274–4282 RSC.
- T. L. Hill, J. Chem. Phys., 1962, 36, 184111 Search PubMed.
- M. A. Cohen Stuart, F. H. W. H. Waajen, T. Cosgrove, B. Vincent and T. L. Crowley, Macromolecules, 1984, 17, 1825–1830 CrossRef.
- J. M. H. M. Scheutjens, G. J. Fleer and M. A. Cohen Stuart, Colloids Surf., 1986, 21, 285–306 CrossRef CAS.
- G. J. Fleer and J. M. H. M. Scheutjens, Colloids Surf., 1990, 51, 281–298 CrossRef CAS.
- B. Derjaguin, Kolloid-Z., 1934, 69, 155–164 CrossRef.
- R. Evans and D. H. Napper, Nature, 1973, 246, 34–35 CrossRef CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- J. G. J. L. Lebouille, F. A. M. Leermakers, M. A. Cohen Stuart and R. Tuinier, Phys. Rev. E, 2016, 94, 1–15 CrossRef PubMed.
- B. R. Postmus, F. A. M. Leermakers and M. A. Cohen Stuart, Langmuir, 2008, 24, 1930–1942 CrossRef CAS PubMed.
- W. M. de Vos and F. A. M. Leermakers, Polymer, 2009, 50, 305–316 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |