DOI:
10.1039/D0SM00537A
(Paper)
Soft Matter, 2020,
16, 6834-6840
Morphology control in metallosupramolecular assemblies through solvent-induced steric demand†
Received
27th March 2020
, Accepted 20th May 2020
First published on 7th July 2020
Abstract
Controlling the supramolecular self-assembly of π-conjugated systems into defined morphologies is a prerequisite for the preparation of functional materials. In recent years, the development of sophisticated sample preparation protocols and modulation of various experimental conditions (solvent, concentration, temperature, etc.) have enabled precise control over aggregation pathways of different types of monomer units. A common method to achieve pathway control consists in the combination of two miscible solvents in defined proportions – a “poor” and “good” solvent. However, the role of solvents of opposed polarity in the self-assembly of a given building block still remains an open question. Herein, we unravel the effect of aggregation-inducing solvent systems of opposed polarity (aqueous vs. non-polar media) on the supramolecular assembly of a new bolaamphiphilic Pt(II) complex. A number of experimental methods show a comparable molecular packing in both media driven by a synergy of solvophobic, aromatic and weak hydrogen-bonding interactions. However, morphological analysis of the respective aggregates in aqueous and non-polar media reveals a restricted aggregate growth in aqueous media into spherical nanoparticles and a non-restricted 2D-nanosheet formation in non-polar media. These findings are attributed to a considerably more efficient solvation and, in turn, increased steric demand of the hydrophilic chains in aqueous media than in nonpolar media, which can be explained by the entrapment of water molecules in the hydrophilic aggregate shell via hydrogen bonds. Our findings reveal that the different solvation of peripheral solubilizing groups in solvents of opposed polarity is an efficient method for morphology control in self-assembly.
Introduction
Supramolecular self-assembly has emerged as a powerful approach to produce functional materials for optoelectronic devices,1 biomedical applications,2 or the synthesis of adaptive membranes.3 It is widely established that the subtle interplay between different intermolecular non-covalent interactions, i.e. hydrogen bonding and aromatic interactions, plays a key role in the aggregation of small molecules in solution.4 For the particular case of aqueous solutions, additional hydrophobic and enthalpic effects must also be considered.5 Controlling these non-covalent interactions during the self-assembly of small molecules for governing the size and shape of the nanostructures is an important topic of research.6 In recent years, modulation of sample preparation protocols and experimental conditions have been identified as a promising method for controlling the size, shape and properties of supramolecular assemblies.7
Accordingly, the key influence of solvent has been recognized and exploited to drive self-assembly processes in solution.8–10 Pathway control in these systems is typically achieved by modulating the ratio between “good” and “poor” solvents.11 However, in order to achieve a fundamental understanding of the influence of solvent, a thorough investigation of the solvent-induced steric demand on the supramolecular polymerisation is required. In this context, the group of Würthner12 has reported a heat-induced desolvation of glycol chains as a driving force for entropically controlled supramolecular polymerization of amphiphilic perylene bisimides in water/THF mixtures. Very recently, Meijer and co-workers13 have reported a water-assisted self-assembly of a small biphenyl core equipped with amide moieties and long alkyl chains in apolar media (MCH). Interestingly, the potential hydrogen bonding interactions of water dissolved in oils showed a significant influence on the molecular packing due to co-assembly with the supramolecular building blocks resulting in different morphologies and intercalation motifs. Several other studies on the influence of solvent on self-assembly processes were also reported, which are based on tuneable aromatic interactions,14 equilibrium kinetics9 and morphology.15 However, a direct comparison between the effect of solvents of opposed polarity on metallosupramolecular polymerisation has been overlooked, as typically only one type of solvent system (either aqueous or non-aqueous) is selected for aggregation studies.
In this work, we have designed an amphiphilic bispyridyldichlorido Pt(II) complex C1 featuring an azobenzene-based central aromatic moiety that is connected via an amide linker to hydrophilic side groups (Scheme 1).
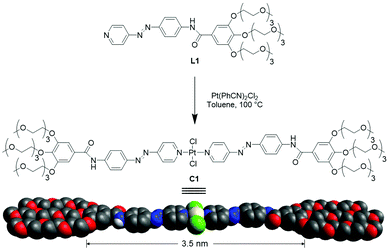 |
| | Scheme 1 Molecular structure of ligand L1 and complex C1 and cartoon representation of C1, with the length of the aromatic surface depicted below. | |
As previously shown by our group,16–18 the combination of an amide group with a central M(Pd,Pt)Cl2 moiety is a highly self-recognizing intermolecular hydrogen-bonding pattern to drive supramolecular polymerisation. In addition, our molecular design features peripheral triethylene glycol (TEG) chains, leading to a bolaamphiphilic structure. With these structural elements, we envisage a good solubility in various solvents that include aqueous as well as non-aqueous non-polar media. Additionally, this design will enable us to study the influence of solvation on the self-assembly by investigating C1 in aqueous and non-polar media separately. We hypothesize that the peripheral TEG side chains will exhibit a much greater steric demand in aqueous solution, which can be used to achieve self-assembly and morphology control.
Results and discussion
Investigations on the coordination isomerism of C1
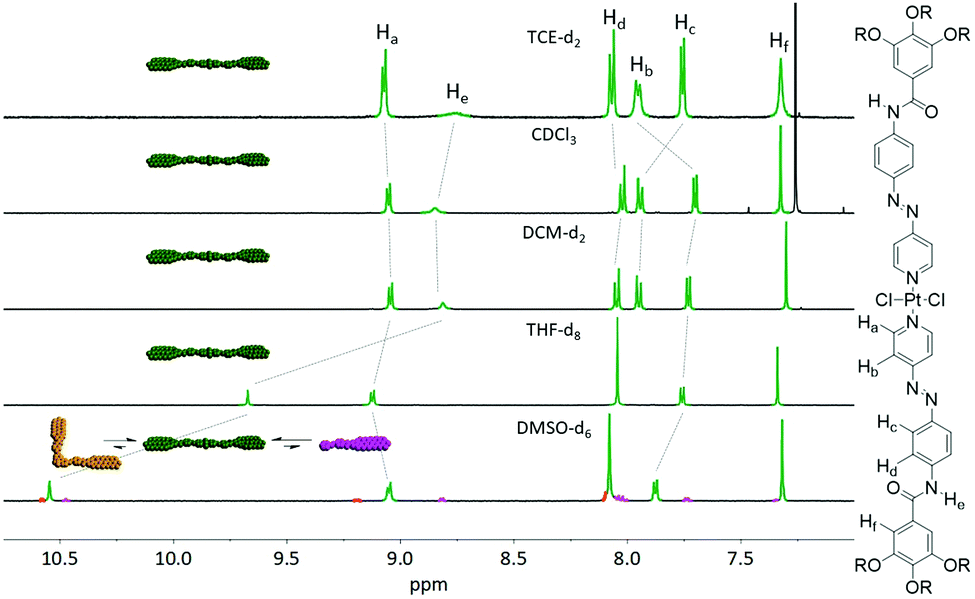
Based on our experience with an analogous hydrophobic Pt(II) complex bearing an azobenzene moiety,18 we initially probed the coordination isomerism of C1. To this end, we prepared five separate solutions of C1 in solvents of different polarity (Fig. 1) and analysed them by NMR spectroscopy. The samples were monitored at ambient conditions immediately after dissolving the compound as pure trans isomer in the respective solvent until equilibration between the two possible species (cis and trans) was reached (after one week; Fig. S5, ESI†). To our satisfaction, only the trans-isomer could be observed in all moderately polar solvents (TCE-d2, CDCl3, DCM-d2 and THF-d8), irrespective of the differences in polarity. However, due to the ability of DMSO to act as competing ligand, both the cis-isomer as well as the free pyridyl ligand could be observed after one week. This behaviour was amplified after heating the samples slightly below the boiling points of the respective solvents for one hour (Fig. S5, ESI†), which can be due to a plausible decomposition of C1 in DMSO. These results confirm that C1 is not prone to coordination isomerism under ambient conditions in a variety of solvents.
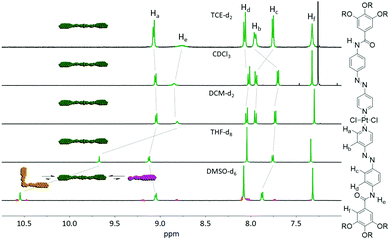 |
| | Fig. 1 Solvent-dependent 1H NMR of C1 (c = 1 × 10−3 M) after keeping all samples at ambient conditions for one week. Dashed lines indicate the changes in the chemical shift of the different signals. | |
Self-assembly behaviour of C1 in aqueous and non-polar media
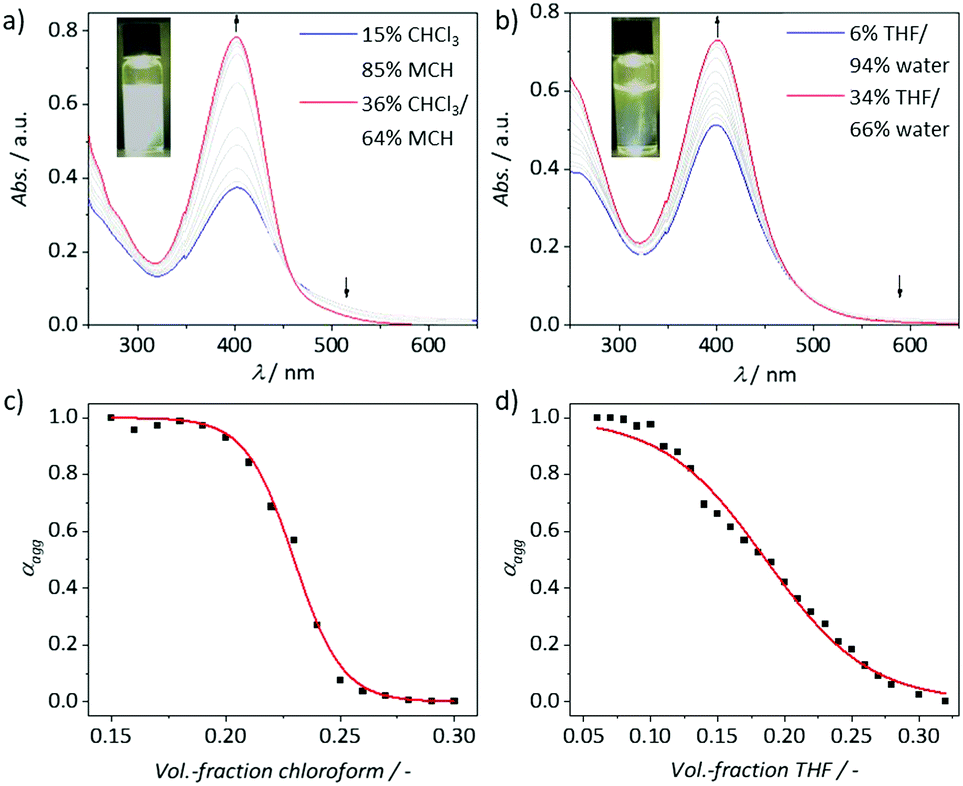
The self-assembly of C1 in solution was initially investigated by UV/vis absorption spectroscopy in different solvents (Fig. S6, ESI†). Solvents with moderate polarity, such as chloroform, DCM, THF or more polar methanol appeared as good solvents where C1 exists in a monomeric state. As expected, the absorbance in water is significantly decreased suggesting a self-assembly process, possibly due to hydrophobic and aromatic interactions of the central aromatic moiety of C1. On the other hand, C1 was only partially soluble in pure nonpolar media (such as pentane or MCH), which may be due to the presence of polar TEG chains. However, we identified that a solvent mixture such as chloroform/MCH can be a good choice to investigate the self-assembly behaviour of C1. As mentioned previously, glycol chains might have a different steric demand depending on whether a non-polar solvent or water is used. For the latter case, the hydration shell resulting from the intercalation of water molecules in the TEG region via hydrogen bonding is expected to increase the steric demand of the polar chains compared to non-polar media. Considering this point, we compared the self-assembly of C1 in THF/water and chloroform/MCH mixtures in detail. Consequently, we followed an established method for stepwise disassembly of a supramolecular polymer through addition of a monomeric solution in a good solvent.10,19 First, aggregates of C1 were prepared by dissolving in a mixture of good/poor solvent with a high content of the respective poor solvent (85% MCH/15% CHCl3; and 94% water/6% THF; c = 1 × 10−5 M, Fig. 2). Both solutions were kept at ambient conditions for 12 h to reach equilibrium. For both systems, no hints of kinetic/metastable species have been observed (Fig. S7, ESI†). The aggregate solution in chloroform/MCH was slightly opaque, indicating the formation of a poorly solvated aggregated species. In contrast, the aggregated solution obtained from THF/water was completely clear indicating sufficiently solvated aggregated species. This can be explained by the polarity mismatch between the solvent system and the peripheral TEG chains. Subsequently, the disassembly of the aggregates in 85% MCH/15% CHCl3 and 94% water/6% THF was monitored by gradual addition of the respective monomer species in CHCl3 and THF, respectively. Upon addition of the respective monomer solution, the UV/vis spectra showed a significant hyperchromism indicating the disassembly of the respective aggregates. In both cases, the increase in absorbance at the maximum (403 nm for chloroform/MCH mixture and 400 nm for THF/water mixture) is accompanied by a decrease in the broad absorption above 500 nm leading to an isosbestic point at 458 and 523 nm, respectively. The degree of aggregation (αagg) vs. solvent fraction derived from the UV/vis changes could be fitted to a sigmoidal function (Fig. 2c and d). Despite these similarities, an appreciable difference between aqueous and non-polar media was also observed, namely the range of the volume fraction in which the disassembly occurs. In chloroform/MCH, the disassembly starts at 18% of chloroform and is complete at chloroform volume fractions of 26%, whereas this process in THF/water mixtures spans from 5% to 32% of THF. These findings suggest an increased impact of solvation of the TEG chains in THF/water mixtures, which is an expected phenomenon based on the molecular design.
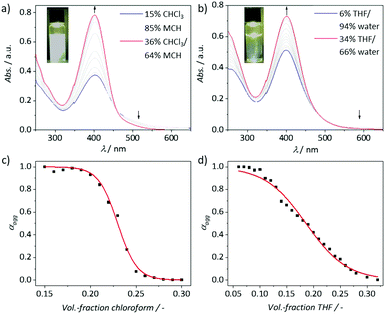 |
| | Fig. 2 Solvent-dependent UV/vis changes of C1 (c = 1 × 10−5 M; l = 1 cm) at 298 K in chloroform/MCH 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 (a) and THF/water 6:94 (b) mixtures with the corresponding denaturation curves depicted below (c and d). The red plots represent a sigmoidal fit of the experimental data points added as a visual guide. The insets of (a) and (b) show photographs of the initial solution prior to the addition of good solvent under light irradiation to amplify the opaque nature of the solution in chloroform/MCH (15:85). :85 (a) and THF/water 6:94 (b) mixtures with the corresponding denaturation curves depicted below (c and d). The red plots represent a sigmoidal fit of the experimental data points added as a visual guide. The insets of (a) and (b) show photographs of the initial solution prior to the addition of good solvent under light irradiation to amplify the opaque nature of the solution in chloroform/MCH (15:85). | |
After these preliminary self-assembly studies, we focused on the possible photoisomerization of C1 aggregates. Based on our previous report, we hypothesise a MLCT energy transfer after photoexcitation, which could in turn lead to a nonradiative decay of the excited states.20 To confirm this, we prepared an aggregated solution of C1 in THF/water (5:95) and irradiated the self-assembled structures using a LED (λ = 365 nm). Subsequent UV-vis measurement showed negligible changes (Fig. S8, ESI†), which can be attributed to a local heating associated with light irradiation. This observation suggests the dormant nature of the assemblies to light irradiation, similar to a previously reported structurally related compound.18
Unravelling the supramolecular packing of C1 in non-polar media
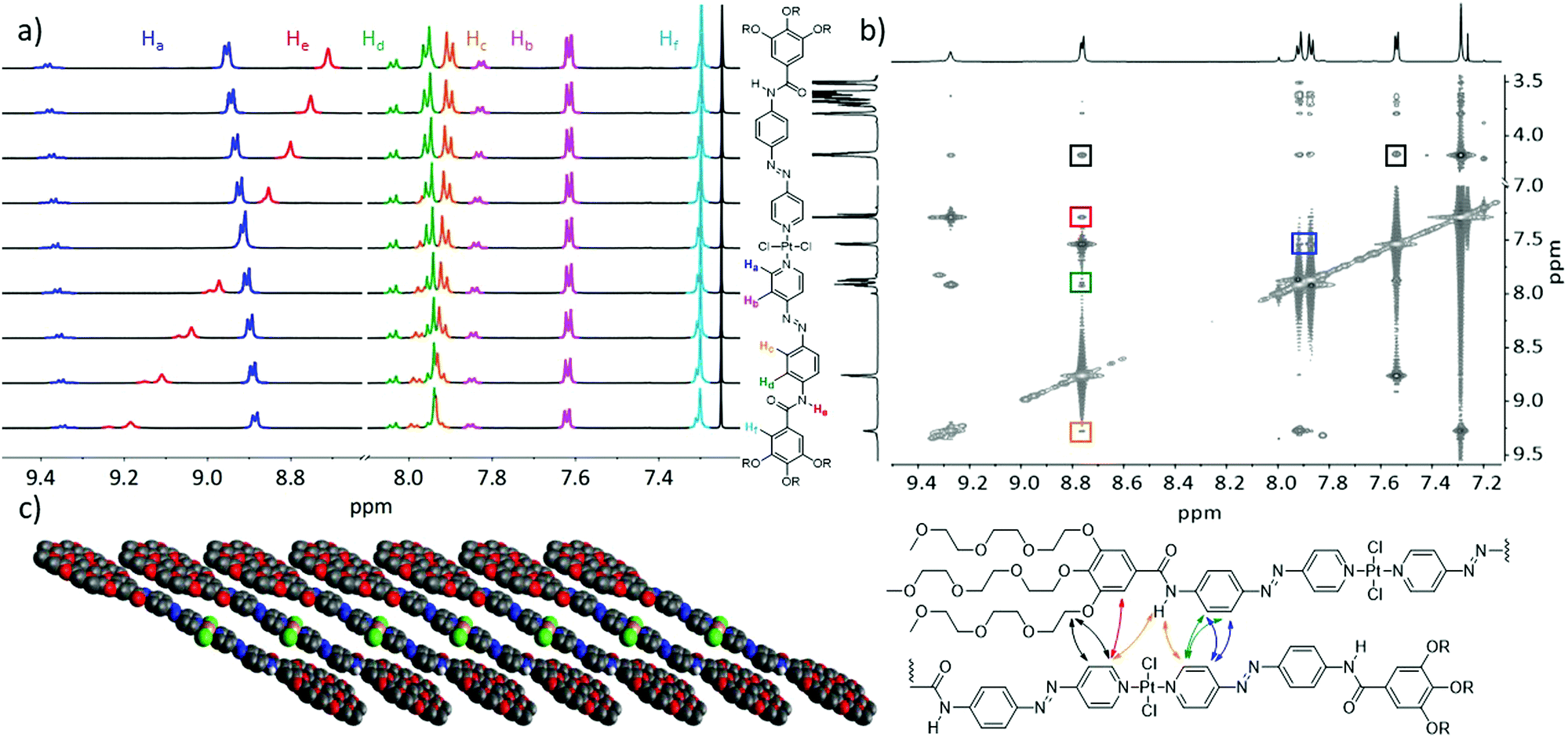
To elucidate the packing mode of the supramolecular assemblies of C1 prepared in CHCl3/MCH and THF/H2O, we employed 1D and 2D NMR techniques as well as X-ray diffraction (XRD). First, we investigated the variable temperature (VT) NMR spectral changes in chloroform at high concentration (c = 1 × 10−2 M, Fig. 3a). Upon cooling the solution from 353 K to 283 K, remarkable changes were observed. Particularly, the amide proton (He, red) shifted from 8.71 to 9.18 ppm. Such a downfield shift can be ascribed to the close proximity of N–H to an electron rich atom, i.e. Cl or O.21 Further, a notable upfield shift from 8.95 to 8.88 ppm can be observed for the signal corresponding to the alpha (α) proton of the pyridine moiety (Ha, highlighted in blue in Fig. 3a), indicating aromatic interactions.22 Without any surprise, the protons corresponding to the cis-isomer also appeared during the heating process (an identical colour code was used for clarity). Subsequently, the isomerization reached an equilibrium with a ratio of 1:4 between the cis- and trans-isomer at 353 K. The signals corresponding to the cis-isomer showed an identical behaviour when compared to the trans-isomer, indicating an identical arrangement possibly due to an end-capping of the cis-isomer to the active ends of the trans-stack.18 To avoid a possible interference of the cis-isomer in disturbing the packing mode, only ambient conditions were used for further investigations. Accordingly, we employed 2D-1H{1H} Correlated Spectroscopy (COSY) and Rotating-frame Overhauser Effect Spectroscopy (ROESY) studies of C1 at 298 K (Fig. 3b). Comparison of these two spectra allowed us to identify characteristic correlation signals corresponding to a slipped stack (Fig. 3c).16,17,23 Correlation signals between both protons of the middle phenyl ring (Hc/d) and the pyridine ring of an adjacent molecule (Ha/b) could be observed (cross signals highlighted in blue and green, Fig. 3b), which confirmed the slipped stack of C1. Further, a cross signal between the amide proton (He) and the signal corresponding to the Hα of the pyridine ring (Ha) was noticed (orange), which further supported the slipped packing. These cross signals are also in line with the VT 1H NMR results, suggesting the proximity of an electron rich group next to the amide proton. Overall, these 1D and 2D NMR experiments confirm the N–H⋯Cl interactions, which is the driving force for the self-assembly of C1 in non-polar solvents. Besides, a close contact between the Hα and the proton of the peripheral trialkoxyphenyl ring (Hf) observed in the 2D ROESY spectra (red) further supports this conclusion. Moreover, a significant number of intermolecular interactions between the peripheral glycol chains and the aromatic core can also be observed in the 2D NMR spectrum. For reasons of clarity and visibility only two of them have been highlighted (black) and will be discussed, as they are most relevant to unravel the slipped packing. Intermolecular correlation signals can be observed between the aromatic protons of the pyridine ring (Ha/b) and the first methylene unit of the glycol chains. To summarize, based on the results extracted from VT and 2D NMR, a cartoon representation of the proposed supramolecular assembly pattern is presented in Fig. 3c. This molecular packing is in perfect agreement with that exhibited by structurally related Pt(II) complexes reported by us recently. Such an intermolecular hydrogen bonding pattern throughout the stack can provide the overall stability of the self-assembled structure.17,24
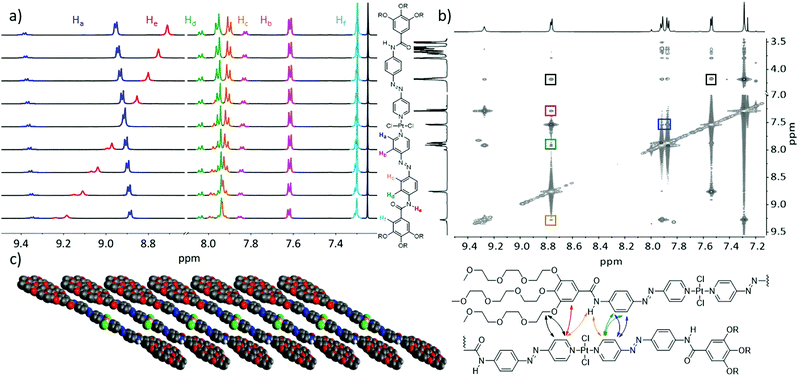 |
| | Fig. 3 (a) Variable temperature 1H NMR study of C1 in CDCl3 (c = 1 × 10−2 M) from 353 (top) to 283 K (bottom) with a 5 K interval between each measurement. The colour code of the different proton signals is depicted on the right. (b) Overlay of 2D-COSY and ROESY spectra of C1 at 298 K in CDCl3 (c = 5 × 10−2 M). Coloured boxes indicate the signals stemming from intermolecular interactions between adjacent protons in the supramolecular stack. For clarity reasons, only the most relevant cross signals between aromatic protons and the glycol chains are marked. These intermolecular interactions are presented below with an identical colour code. (c) Cartoon representation of the molecular packing stabilized by aromatic and N–H⋯Cl interactions. | |
Unravelling the supramolecular packing of C1 in aqueous media
The UV/vis disassembly studies showed minor differences in spectral changes for the assemblies in CHCl3/MCH and THF/H2O, which might be due to similarities in the molecular level interactions of C1 in solution. To address this issue, we first recorded solvent-dependent 1H NMR. For these experiments, we gradually increased the D2O content in a THF-d8 solution of C1 (c = 1 × 10−3 M, Fig. 4a) from 0 to 90%. Upon first addition of D2O (10%), the signal corresponding to the amide proton (He) disappeared, which might be due to the rapid proton exchange with the deuterated solvent.25 Additionally, a splitting of the two proton signals corresponding to the middle phenyl ring (Hc, Hd), which were completely overlapping in pure THF-d8, is observable. Upon increasing the D2O content to 50%, noticeable spectral changes were observed, for instance slight broadening and minor upfield shift of all aromatic protons. Further, a significant broadening along with a marked upfield shift were observed when the D2O content is further increased to 90%, indicating that the self-assembly is mainly driven by aromatic and hydrophobic interactions. To gain more detailed information on the packing of C1 in THF/H2O, we investigated thin films of the supramolecular aggregates by XRD at ambient conditions (Fig. 4b). As an increase in temperature can cause precipitation due to the dehydration of the solvated TEG chains, samples were prepared at ambient conditions.12 While these conditions may vary from the behaviour in solution, we envisage that the basic orientation observed in solution is transferred to the packing in thin film. A thin film suitable for XRD studies could be obtained after a stepwise drop casting and subsequent solvent evaporation. The distance derived from the diffraction at 4.09 Å is important, as it can be attributed to the d spacing between the Pt(II) moieties.26 The distance between the aromatic planes appears as 3.93 Å, common for many π-systems. Further, we tentatively attribute the reflection corresponding to a distance of 4.55 Å to the spacing between the glycol chains of neighbouring molecules in between layers, which is in good agreement with the crystal structure of previously reported analogues.18 The observation of repeating distances within the third dimension is limited to XRD investigations, as the sample preparation leads to higher concentrations than those obtained during measurements in solution. Hence, the results concerning the 3D assembly in thin film by XRD are not representative for the solution behaviour, as analysed by spectroscopy measurements. Interestingly, a reflex at 6.51 Å can also be observed, which can be ascribed to the diagonal distance between molecules within a 2D layer. The cartoon representation shown in Fig. 4c displays a plausible proposed packing motif in all three dimensions derived from the experimental data. Comparing these results with the supramolecular packing motif derived from spectroscopic studies in non-aqueous solutions leads us to conclude that in both cases C1 may be oriented in the same fashion. We assume that the overall supramolecular assembly process is driven by aromatic as well as N–H⋯Cl and C–H⋯Cl interactions. These structures can then grow in the other two dimensions, possibly by intermolecular Pt⋯Cl interactions between neighbouring stacks23,27 and in the solid state (thin film) a growth in the third dimension is possible due to the stacking of the 2D-plates.
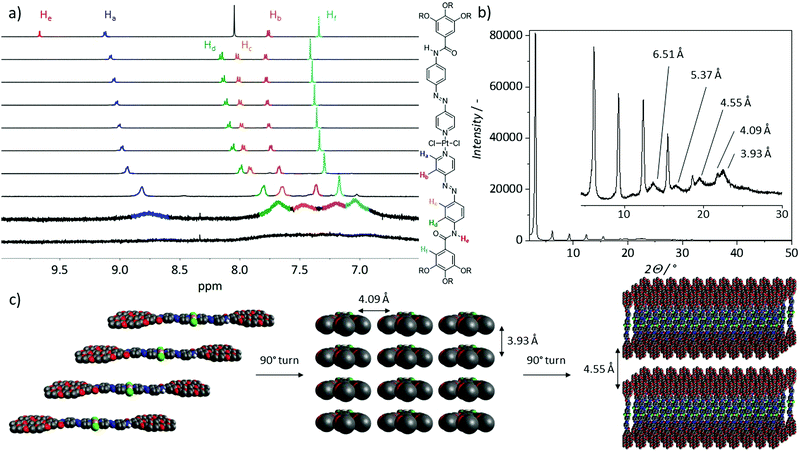 |
| | Fig. 4 (a) Solvent-dependent 1H NMR spectra of C1 at 298 K (c = 1 × 10−3 M) from 100% THF-d8 (top) to 10% THF-d8 and 90% D2O (bottom) in 10% steps with a scheme clarifying the colour code depicted on the side. (b) XRD-diffraction pattern (λ = 1.5406 Å) of a thin film of the aggregate of C1 obtained from drop casting a 1 × 10−3 M solution (THF:water 9:1) between 2 and 50° with an inset of the region between 4.5 and 30° The sharp reflexes are allocated to originate from the d-spacing between Pt atoms. (c) Cartoon representation of the 3D packing model derived from combining the results of the 1H NMR and XRD experiments with the most important intermolecular distances highlighted. The 2D layers depicted on the right are capable of stacking indefinitely during the sample preparation (drying), which enables analysis with XRD. Only two are shown for clarity. | |
Investigation of the nanoscale morphology
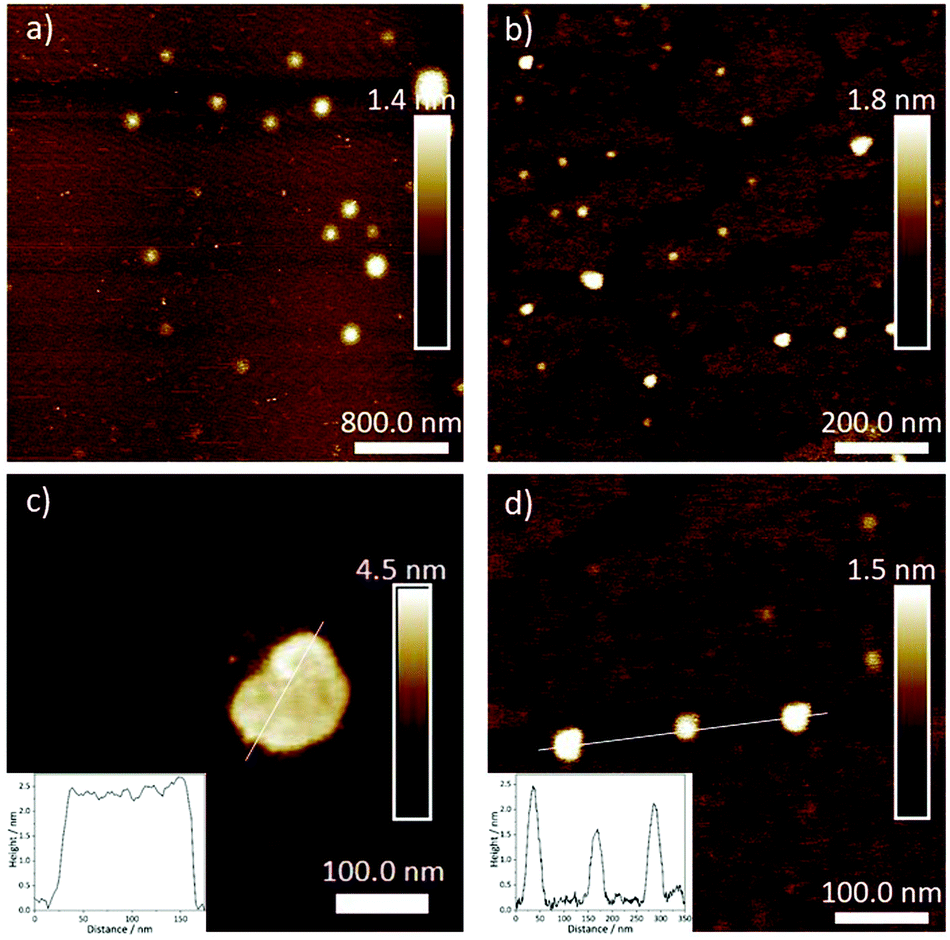
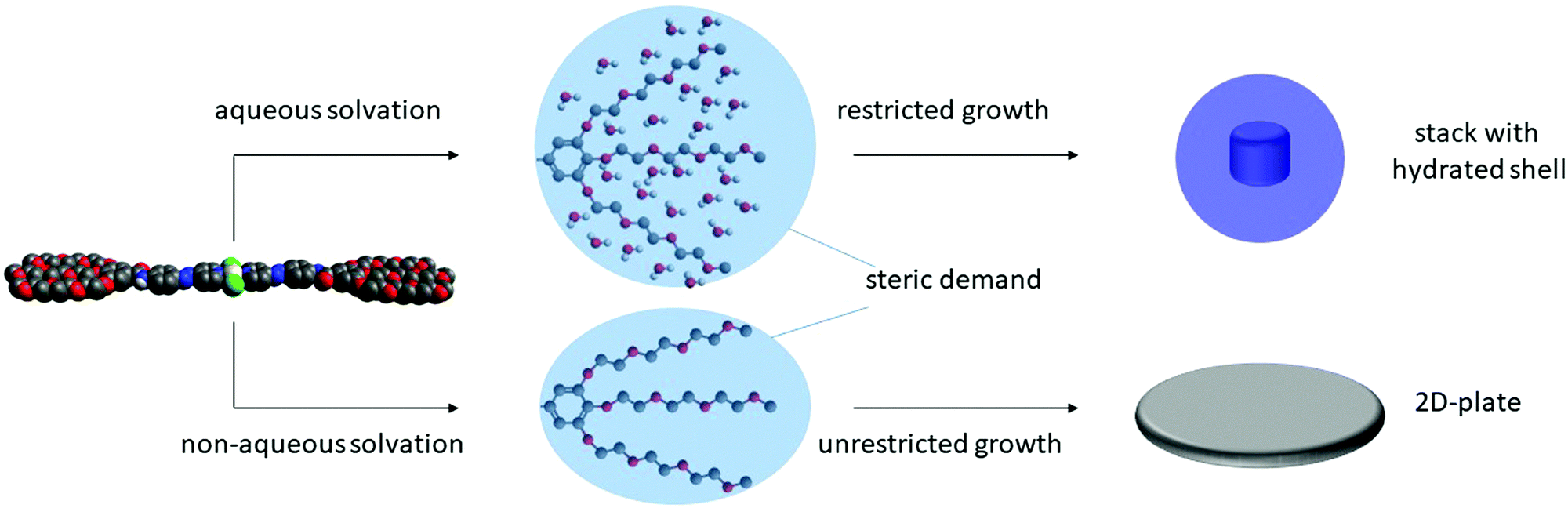
After elucidating the solvent-dependent self-assembly behaviour of C1, we questioned whether the steric demand caused by the solvation of the TEG chains in the different media could have a strong influence on the nanostructure formation. We envisage that the solvation of TEG with water can create a higher steric demand when compared to the solvation with chloroform and/or MCH. Accordingly, we speculate that the formation of extended supramolecular aggregates should be hindered in aqueous media leading to size-restricted morphologies. Conversely, the supramolecular assembly can lead to larger aggregates in non-aqueous solutions. To probe such influence of solvent on the metallosupramolecular assembly of C1, we characterised these aggregates with atomic force microscopy (AFM). All samples were prepared under identical conditions as for the previous UV/vis spectroscopic studies. After confirming the aggregate formation in solution by UV/vis, a definite volume is drop casted (c = 1 × 10−5 M, V = 20 μL) onto the respective AFM substrates (Mica) and dried at ambient conditions (see ESI† for details). As expected, the aggregate morphologies of C1 prepared with different solvent combination (THF/water (5:95) and chloroform/MCH (15:85)) differ in size and shape (Fig. 5). In non-aqueous solutions, the aggregates tend to orient in two dimensions, leading to 2D-sheet formation (d = 150 nm for the specific sheet shown in Fig. 5) with a uniform height of 2.5 nm. Considering that this experimental height is smaller than the molecular length of the aromatic scaffold of C1 (3.5 nm),18 the slipped stacks need to be necessarily tilted with respect to the mica surface, as depicted in Fig. 3c. On the other hand, the assemblies obtained in aqueous solution exhibit a spherical morphology, with diameters between 30 and 50 nm and heights between 1.5 and 2.5 nm, as shown in Fig. 5. This height again confirms the formation of a small aggregate consisting of one layer of molecules, in contrast to the multi-layered structure observed by XRD. The minor discrepancy in height can be rationalized by increased interactions between the solvated TEG chains and the polar mica surface compared to non-polar media. From the optical analysis, the spheres appear to be restricted in size as the largest one we could observe was only 50 nm in diameter. This observation can be rationalized by the steric demand of the solvated TEG chains in aqueous media (Fig. 6). Due to presence of such sterically demanding chains in solution, a potential growth of the initially formed small particles can be limited. This is in accordance with the relatively large intermolecular distances (close to 4 Å) obtained by XRD, which are associated with π–π stacking distances. Such a size restriction induced by the steric demand can detrimentally affect the polydispersity. On the other hand, an unrestricted growth can be anticipated for C1 in MCH/CHCl3, which is evident from the 2D sheet formation. Based on this experimental evidence, we conclude that the steric demand of the peripheral glycol chains in solution can be controlled by changing the solvent conditions. Such a steric demand can lead to different morphologies at the nanoscale, which can also be observed with the naked eye (opaque solution in non-aqueous solvents), while preserving the molecular level packing in the supramolecular polymer.
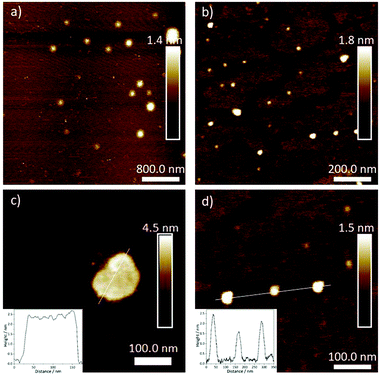 |
| | Fig. 5 AFM height images of aggregates of C1 formed in chloroform/MCH (15:85, a, c) and THF/water (5:95, b, d) with a scan size of 4 μM (a), 1 μM (b) and 500 nm (c and d) respectively. Inset: Height profile across the surfaces highlighted with a white line. | |
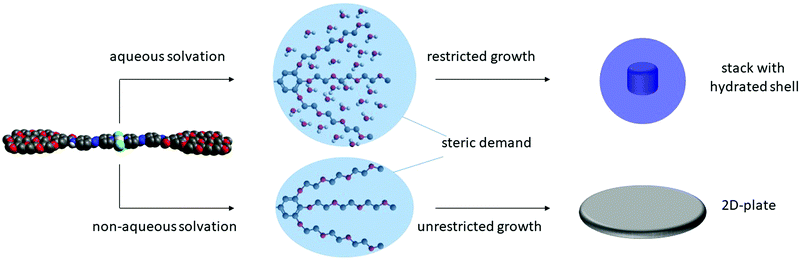 |
| | Fig. 6 Schematic representation of the self-assembly behaviour of C1 in aqueous (top) and non-aqueous (bottom) media leading to either spherical particles or 2D-plates respectively based on the steric demand of the solvated chains. | |
Conclusions
In conclusion, we have reported the impact of solvents of opposed polarity (aqueous vs. non-polar solutions) on the self-assembly of a new linear bolaamphiphilic Pt(II) complex C1. A detailed comparison of the solvent-dependent self-assembly behaviour was achieved by spectroscopic UV/vis studies, 1D and 2D 1H NMR experiments as well as XRD. According to these investigations, the self-assembled structures in both aqueous and non-polar media are stabilized, besides solvophobic interactions in the respective media, through aromatic and multiple intermolecular hydrogen bonding interactions, such as C–H⋯Cl and N–H(amide)⋯Cl. Although a similar slipped packing mode is observed in both media, the resulting nanoscale morphologies differ, as shown by AFM. Whereas in aqueous media, spheres with a narrow size distribution are visualized, discrete 2D sheets are formed in non-polar solvents. We attribute this behaviour to the different steric demand of the solubilizing chains in either solvent mixture. Water molecules can intercalate in the glycol chains through hydrogen bonding interactions leading to a higher steric demand and, in turn, to a restricted growth into spherical particles. In the absence of water, i.e. in non-polar solvents, the glycol chains exert a much lower steric demand and thus promote a growth in two dimensions. The results presented herein shed light on the complex structure–property-relationship between solubilizing chains and supramolecular morphology in metallosupramolecular polymers, which should contribute to the preparation of functional supramolecular materials.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge the European Commission (ERC-StG-2016 SUPRACOP-715923) for funding. We thank Prof. Lambert Alff for XRD measurements.
Notes and references
- J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC.
-
(a) E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141 CrossRef CAS PubMed;
(b) E. Cohen, H. Weissman, E. Shimoni, I. Kaplan-Ashiri, K. Werle, W. Wohlleben and B. Rybtchinski, Angew. Chem., Int. Ed., 2017, 56, 2203 CrossRef CAS PubMed.
-
(a) T. F. A. de Greef and E. W. Meijer, Nature, 2008, 171 CrossRef CAS PubMed;
(b) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed;
(c) C. Rest, A. Martin, V. Stepanenko, N. K. Allampally, D. Schmidt and G. Fernández, Chem. Commun., 2014, 50, 13366 RSC;
(d) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700 CrossRef CAS PubMed;
(e) A. Sikder and S. Ghosh, Mater. Chem. Front., 2019, 3, 2602 RSC.
-
(a) D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328 CrossRef PubMed;
(b) X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886 CrossRef CAS PubMed;
(c) B.-S. Kim, D.-J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333 CrossRef CAS PubMed;
(d) V. S. Talens, D. M. M. Makurat, T. Liu, W. Dai, C. Guibert, W. E. M. Noteborn, I. K. Voets and R. E. Kieltyka, Polym. Chem., 2019, 10, 3146 RSC;
(e) S. Bujosa, E. Castellanos, A. Frontera, C. Rotger, A. Costa and B. Soberats, Org. Biomol. Chem., 2020, 8, 888 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
-
(a) A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed;
(b) S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339 CrossRef CAS PubMed;
(c) T. Fukui, T. Uchihashi, N. Sasaki, H. Watanabe, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2018, 130, 15691 CrossRef;
(d) W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 129, 16224 CrossRef;
(e) D. Limón, C. Jiménez-Newman, A. C. Calpena, A. González-Campo, D. B. Amabilino and L. Pérez-García, Chem. Commun., 2017, 53, 4509 RSC;
(f) A. Sorrenti, R. Rodriguez-Trujillo, D. B. Amabilino and J. Puigmartí-Luis, J. Am. Chem. Soc., 2016, 138, 6920 CrossRef CAS PubMed;
(g) C. Oliveras-González, F. Di Meo, A. González-Campo, D. Beljonne, P. Norman, M. Simón-Sorbed, M. Linares and D. B. Amabilino, J. Am. Chem. Soc., 2015, 137, 15795 CrossRef PubMed.
- J. S. Valera, R. Sánchez-Naya, F. J. Ramírez, J. L. Zafra, R. Gómez, J. Casado and L. Sánchez, Chem. – Eur. J., 2017, 23, 11141 CrossRef CAS PubMed.
- R. P. M. Lafleur, X. Lou, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2018, 9, 6199 RSC.
- J. Buendía and L. Sánchez, Org. Lett., 2013, 15, 5746 CrossRef PubMed.
-
(a) Y. Mao, K. Liu, L. Meng, L. Chen, L. Chen and T. Yi, Soft Matter, 2014, 10, 7615 RSC;
(b) S. Yu-Lut Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228 RSC;
(c) Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761 CrossRef CAS PubMed;
(d) C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed.
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094 CrossRef PubMed.
- N. J. van Zee, B. Adelizzi, M. F. J. Mabesoone, X. Meng, A. Aloi, R. H. Zha, M. Lutz, I. A. W. Filot, A. R. A. Palmans and E. W. Meijer, Nature, 2018, 558, 100 CrossRef CAS PubMed.
- E. Y.-H. Hong, H.-L. Wong and V. W.-W. Yam, Chem. Commun., 2014, 50, 13272 RSC.
-
(a) F. Nador, K. Wnuk, C. Roscini, R. Solorzano, J. Faraudo, D. Ruiz-Molina and F. Novio, Chem. – Eur. J., 2018, 24, 14724 CrossRef CAS PubMed;
(b) Y. Tidhar, H. Weissman, S. G. Wolf, A. Gulino and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 6068 CrossRef CAS PubMed.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192 CrossRef CAS PubMed.
- K. K. Kartha, N. K. Allampally, A. T. Politi, D. D. Prabhu, H. Ouchi, R. Q. Albuquerque, S. Yagai and G. Fernández, Chem. Sci., 2019, 10, 752 RSC.
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626 CrossRef PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed.
-
(a) G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629 CrossRef PubMed;
(b) E. Borré, J.-F. Stumbé, S. Bellemin-Laponnaz and M. Mauro, Chem. Commun., 2017, 53, 8344 RSC;
(c) M. E. Moustafa, M. S. McCready, P. D. Boyle and R. J. Puddephatt, Dalton Trans., 2017, 46, 8405 RSC.
-
(a) A. Rödle, B. Ritschel, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Chem. – Eur. J., 2016, 22, 15772 CrossRef PubMed;
(b) X. Lu, Z. Guo, C. Sun, H. Tian and W. Zhu, J. Phys. Chem. B, 2011, 115, 10871 CrossRef CAS PubMed.
-
(a) S. Lahiri, J. L. Thompson and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 11315 CrossRef CAS;
(b) C. Giessner-Pettre, B. Pullman, P. N. Borer, L.-S. Kan and P. O. Tso, Biopolymers, 1976, 15, 2277 CrossRef PubMed.
- N. K. Allampally, M. J. Mayoral, S. Chansai, M. C. Lagunas, C. Hardacre, V. Stepanenko, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2016, 22, 7810 CrossRef CAS PubMed.
- A. Langenstroer, Y. Dorca, K. K. Kartha, M. J. Mayoral, V. Stepanenko, G. Fernández and L. Sánchez, Macromol. Rapid Commun., 2018, 39, 1800191 CrossRef PubMed.
-
(a) Z. Qi, C. Schlaich and C. A. Schalley, Chem. – Eur. J., 2013, 19, 14867 CrossRef CAS PubMed;
(b) Y. Bai, T. Sosnick, L. Mayne and S. W. Englander, Science, 1995, 269, 192 CrossRef CAS PubMed.
-
B. E. Warren, X-Ray Diffraction, Dover Publications, Newburyport, 2012 Search PubMed.
- J. P. Coelho, J. Matern, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2019, 25, 8960 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthethic details and additional spectroscopic analysis. See DOI: 10.1039/d0sm00537a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a