
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic drug–peptide–polymer conjugates based on poly(ethylene glycol) and hyperbranched polyglycerol for epidermal growth factor receptor targeting: the effect of conjugate aggregation on in vitro activity†
Lilla
Pethő‡
a,
György
Kasza‡
 *b,
Eszter
Lajkó
c,
Orsolya
Láng
c,
László
Kőhidai
c,
Béla
Iván
*b and
Gábor
Mező
*ad
*b,
Eszter
Lajkó
c,
Orsolya
Láng
c,
László
Kőhidai
c,
Béla
Iván
*b and
Gábor
Mező
*ad
aMTA-ELTE Research Group of Peptide Chemistry, Eötvös Loránd University, H-1117 Budapest, Pázmány Péter sétány 1/A, Hungary. E-mail: gmezo@elte.hu
bPolymer Chemistry Research Group, Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, H-1117 Budapest, Magyar tudósok körútja 2, Hungary. E-mail: kasza.gyorgy@ttk.hu; ivan.bela@ttk.hu
cDepartment of Genetics, Cell- and Immunobiology, Semmelweis University, H-1089 Budapest, Nagyvárad tér 4, Hungary
dEötvös Loránd University, Faculty of Science, Institute of Chemistry, H-1117 Budapest, Pázmány Péter sétány 1/A, Hungary
First published on 19th May 2020
Abstract
Numerous peptide–drug conjugates have been developed over the years to enhance the specificity and selectivity of chemotherapeutic agents for tumour cells. In our present work, epidermal growth factor receptor targeting drug–peptide conjugates were prepared using GE11 and D4 peptides. To ensure the drug release, the cathepsin B labile GFLG spacer was incorporated between the targeting peptide and the drug molecule (daunomycin), which significantly increased the hydrophobicity and thereby decreased the water solubility of the conjugates. To overcome the solubility problem, drug–peptide–polymer conjugates with systematic structural variations were prepared, by linking poly(ethylene glycol) (PEG) or a well-defined amino-monofunctional hyperbranched polyglycerol (HbPG) directly or via a pentaglycine spacer to the targeting peptides. All the drug–peptide–polymer conjugates were water-soluble as confirmed by turbidimetric measurements. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells proved that the HbPG and the PEG highly influenced the biological activity. The conjugation of the hydrophilic polymer resulted in the amphiphilic character of the conjugates, which led to self-aggregation and nanoparticle formation that decreased the cellular uptake above a specific aggregation concentration. On the other hand, the hydrodynamic volume and the different polymer chain topology of the linear PEG and the compact hyperbranched HbPG also played an important role in the biological activity. Therefore, in similar systems, the investigation of the colloidal properties is inevitable for the better understanding of the biological activity, which can reveal the structure–activity relationship of amphiphilic drug–peptide–polymer conjugates for efficient tumour targeting.
Introduction
Cancer is a leading cause of death worldwide, hence intensive research focuses on finding effective and patient-friendly ways (e.g., new potent cytotoxic molecules,1–3 the encapsulation of chemotherapeutically used drugs,4 or photodynamic therapy5) to treat the different types of cancers. Generally, drug delivery systems are used to enhance the specificity and selectivity of these therapeutic approaches for tumour cells, where small organic compounds (e.g. biotin6 or folic acid7), peptides or antibodies are usually used as homing devices, which can specifically bind to a receptor overexpressed on tumour cells. Peptide–drug conjugates usually have a high drug loading capacity and can be easily prepared in a homogenous form with straightforward and well-defined conjugation chemistry. These conjugates enter the tumour cells via receptor-mediated endocytosis, and their degradation occurs in the lysosomes, where the drug molecule or its active metabolite gets released, which leads to an antitumour effect.8Numerous peptide–drug conjugates have been developed over the years, where the drug molecules were attached through an enzyme labile or pH-sensitive spacer to the targeting moiety.9 Peptide spacers sensitive to cathepsin B, a lysosomal enzyme overexpressed in different cancer cells (e.g., breast,10 pancreatic,11 lung12 and colon cancers13), enable a selective drug release in the targeted cells.14 The GFLG spacer15 is a well-known and widely used cathepsin B labile short peptide sequence, which enables the lysosomal metabolism of the bioconjugates.
However, the application of such bioconjugates is often limited by their lack of solubility. There are some strategies to enhance the hydrophilicity and water solubility of the drug delivery systems like the incorporation of ionic and/or hydrophilic amino acids (β-sulphoalanine16) or short peptide sequences (repeating Gly and Ser residues,17 polycationic or polyanionic tags18) or rather the conjugation of water-soluble polymers. In the latter case, the most applied approach is PEGylation, the conjugation of poly(ethylene glycol) (PEG).19 Several mono- and homo- or heterobifunctional PEG derivatives with various functionalities, such as amine, carboxyl, maleimide, azido, alkyne etc., have already been produced, and most of these are commercially available with different molecular weights. Based on the known disadvantages of the PEG (non-biodegradability and causinghypersensitivity),20 nowadays, nonlinear PEG-like analogues, e.g. hyperbranched polyglycerol (HbPG), are intensively investigated as biocompatible nanocarriers.21–28 HbPG has outstanding water solubility and biocompatibility; furthermore its synthesis and functionalization can be performed easily.29 Moreover, procedures to produce HbPGs by conjugation of relevant functionalities, such as amine, carboxylic, chloroacetamide and maleimide in well-defined numbers and positions, have already been developed.30–32 Such functionalized HbPGs can be utilized to synthesize novel biomaterials, drug delivery systems and/or enhance the water solubility and biocompatibility of the linked residues. Peptide–HbPG conjugates have already been synthesized by the substitution of the multiple hydroxyl groups through thiol–ene33 or azide–alkyne reactions34 or ester linkages.35 In the literature, biotinylated PEG–HbPG was conjugated in a noncovalent manner with avidin and streptavidin,36 but to the best of our knowledge, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 covalent peptide–polymer conjugates with well-defined monofunctional HbPG have not been reported so far.
:1 covalent peptide–polymer conjugates with well-defined monofunctional HbPG have not been reported so far.
During the design of the peptide-based bioconjugates, the identification of a target receptor that is overexpressed on tumour cells is essential. The epidermal growth factor receptor (EGFR) is upregulated within a high percentage (>60%) of solid tumours (e.g., lung, liver, breast, and colon cancer cells), and hence it is an attractive target for targeted tumour therapies. The EGFR belongs to the ErbB transmembrane receptor family with intrinsic tyrosine kinase activity. The ligand-receptor binding activates the kinase moiety and leads to dimerization, autophosphorylation and downstream signalling, which may result in proliferation, differentiation, enhanced cell migration and adhesion or inhibition of apoptosis.37–39
Various EGFR targeting peptides were developed and applied as drug targeting agents for cancer therapy.40–44 Phage display studies identified a dodecapeptide, GE11 (YHWYGYTPQNVI) that specifically binds to the EGFR (Kd ≈ 22 nM) as an antagonist with much lower mitogenic activity than the EGF. GE11 was shown to be able to mediate the target-specific delivery of reporter genes to EGFR overexpressing tumour cells in vitro and in vivo.45 It was also shown that GE11 was internalized efficiently without EGFR signalling activation using an alternative actin-dependent pathway.46 GE11 has already been used successfully for the delivery of nanoparticles,47,48 micelles,49 genes,50 adenoviral vectors51 and antitumour microRNAs52 as well as in photodynamic therapy.53–55
D4 (LARLLT) was identified by computer-aided design as a potential targeting peptide.56 This hexapeptide has a specific type of binding to the EGFR, to a binding pocket different from the EGF binding site. Liposomes modified with D4 were tested efficiently in in vitro and in vivo models. The D4 mediated delivery of liposomal doxorubicin was effective against EGFR overexpressing cell lines (H1299 human non-small cell lung carcinoma cells and SPCA-1 human lung adenocarcinoma) in vitro, and the conjugated liposomes were shown to gradually concentrate at the tumour site and be preferentially retained there for more than 80 hours after injection in vivo.56 D4 has also already been successfully used as a targeting moiety of a near-IR fluorescent imaging agent57 and as a photosensitizer.53,55
Since the EGFR is a potent target receptor in tumour therapy, our aim was to prepare different EGFR targeting drug–peptide conjugates to study the structure–activity relationship. These bioconjugates were synthesized using the above-mentioned peptides (GE11 and D4) as targeting moieties, and daunomycin (Dau) as a drug molecule. Daunomycin is commonly used in cancer therapy. It belongs to the family of anthracycline antibiotics that can intercalate into the DNA and interact with the minor groove while poisoning the topoisomerase IIα enzyme.58 This cytotoxic agent was attached to the N-terminal of the peptides via oxime linkage using an aminooxyacetyl linker. Based on our previous research, the oxime bond between the Dau and the aminooxyacetyl linker is stable under physiological conditions. Therefore, a proper enzyme labile spacer should be inserted into the structure between the targeting peptide and the Dau that ensures the release of an active metabolite. The GFLG spacer is proved to be cleaved by an enzyme (cathepsin B) overexpressed in the lysosomes of the tumour cells between the glycine and the phenylalanine resulting in a metabolite containing one amino acid (Dau![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Aoa–Gly–OH). This formed metabolite was confirmed to bind to the DNA similarly to the free drug and it has only slightly lower cytotoxic activity than Dau.59 However, the GFLG spacer highly increased the hydrophobicity and thereby decreased the water solubility of the conjugates. Therefore, amino-PEG and a well-defined amino-monofunctional HbPG were used to prepare drug–peptide–polymer conjugates to overcome the solubility problems. The placement of a pentaglycine (G5) spacer between the peptide and the polymers was also performed to investigate the hindrance of the polymer in receptor binding. These novel drug–peptide–polymer conjugates were tested in vitro in cytotoxicity and internalization tests on HT-29 human colon adenocarcinoma cells.
Aoa–Gly–OH). This formed metabolite was confirmed to bind to the DNA similarly to the free drug and it has only slightly lower cytotoxic activity than Dau.59 However, the GFLG spacer highly increased the hydrophobicity and thereby decreased the water solubility of the conjugates. Therefore, amino-PEG and a well-defined amino-monofunctional HbPG were used to prepare drug–peptide–polymer conjugates to overcome the solubility problems. The placement of a pentaglycine (G5) spacer between the peptide and the polymers was also performed to investigate the hindrance of the polymer in receptor binding. These novel drug–peptide–polymer conjugates were tested in vitro in cytotoxicity and internalization tests on HT-29 human colon adenocarcinoma cells.
Results and discussion
The epidermal growth factor receptor (EGFR) is a well-known target receptor for cancer therapy. Two targeting strategies are known in chemotherapy: the use of ligands that block the extracellular domain (monoclonal antibodies, e.g. cetuximab, or peptides) and the use of ATP-competitive small molecules (e.g. gefitinib) that inhibit the kinase activity.60 In terms of targeted tumour therapy, the first strategy is easier to achieve. Therefore, numerous different EGFR-targeting peptides were identified, and several of them have already been successfully used in biological tests.40–45,56 In this work, Dau-containing EGFR targeting conjugates with systematic structural variations were prepared by using GE11 and D4 peptides as targeting moieties.First, the drug–peptide conjugates were synthesized using GE11 (YHWYGYTPQNVI) and D4 (LARLLT) as peptide backbones. Derivatives containing the GFLG enzyme labile spacer were also prepared. This spacer was coupled to the N-termini of the sequences, and aminooxyacetic acid was applied in all cases as a functional linker for the drug conjugation. Daunomycin was conjugated to all 4 peptides (GE11 and D4 with and without GFLG) via oxime linkage, in solution under slightly acidic conditions (pH 5.1). The obtained conjugates were purified by RP-HPLC and analysed via ESI-MS (Table 1; analytical chromatograms and mass spectra are presented in the ESI,† Fig. S1–S4). As the data in the table and figures indicate, the drug–peptide conjugates were obtained in high purity and with the expected molecular weights.
| Conjugate | R t (min)a | MWcalc./MWmeas. | Turbidity (100-T%)b |
|---|---|---|---|
|
a Gradient elution (0 min 0% B, 5 min 0% B, and 50 min 90% B) in 0.1% TFA in water and 0.1% TFA in acetonitrile–water 80:20% (v/v).
b 100 μM, measured in 10% (v/v) DMSO containing complete cell culture medium (RPMI-1640).
|
|||
| DauAoa–GE11 |
30.2 | 2121.9/2122.1 | 18.2 |
| DauAoa–GFLG–GE11 |
33.6 | 2496.3/2496.5 | 36.8 |
| DauAoa–D4 |
33.9 | 1267.1/1267.1 | 1.9 |
| DauAoa–GFLG–D4 |
39.0 | 1641.5/1641.6 | 18.7 |
Since the targeting peptides contain several hydrophobic amino acids and the enzyme labile spacer further increases the hydrophobicity of the conjugates, solubility tests were performed before the in vitro assays. We found that all daunomycin–peptide conjugates were insoluble in water, therefore, stock solutions in DMSO were prepared and diluted with 9 volume excess of serum-containing cell culture medium (RPMI-1640). The turbidimetric measurements were performed directly after the preparation of the solutions by using a UV-Vis spectrophotometer and the transmittance of light of the solutions was measured at 630 nm, since the drug–peptide conjugates have no absorbance at this wavelength (Fig. S5, ESI†). Turbidity, expressed in 100-T%, is the reduction of transparency of a liquid caused by the presence of undissolved matter, so it is the opposite of clarity. The obtained turbidity results are listed in Table 1. As shown by the observed data, with the exception of the DauAoa–D4, all conjugates partially precipitated in spite of the large amount of DMSO. In addition, the turbidity steadily decreased with time, clearly indicating the slow sedimentation of the precipitated conjugates, which was visible to the naked eye as well. Because of the low solubility and the observed sedimentation of these conjugates beside the 10% (v/v) DMSO concentration, which is far above the amount that is tolerable (non-toxic) for the cells, these conjugates are not suitable for cancer treatment, hence in vitro assays were not performed. Our aim was to overcome this solubility problem, therefore, we designed water-soluble polymer containing drug–peptide–polymer conjugates. PEGylation is a well-known and straightforward method to increase the water solubility of drugs, peptides and conjugates, but its disadvantages are also already known.20 Hence, not only the commercially available amine-terminated PEG, but also a mono-amino functional hyperbranched polyglycerol (HbPG), which can be considered as a non-linear PEG-like analogue known to have outstanding water solubility and biocompatibility,30 were used to investigate the effect of the hydrophilic polymer on the solubility and the in vitro bioactivity of the conjugates. Since such polymers may cause steric hindrance in receptor–ligand binding, analogues containing a pentaglycine (G5) spacer between the polymer and the peptide were also designed. As shown in Fig. 1, for the synthesis of these drug–peptide–polymer conjugates, the GFLG spacer containing derivatives were used, because this spacer ensures an effective release of the drug,15 on the one hand. However, due to its hydrophobic character, it significantly decreases the solubility, on the other hand.
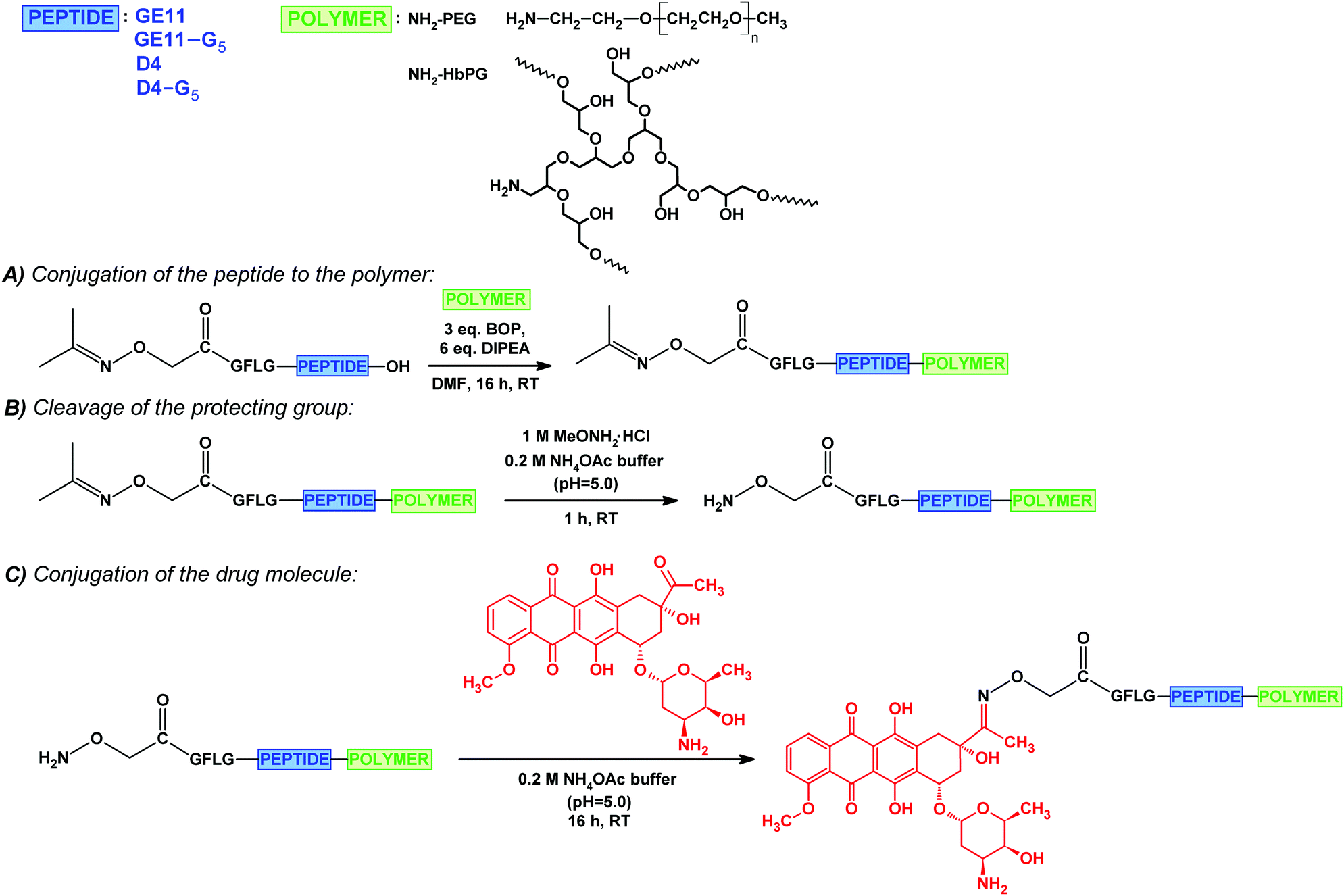 | ||
| Fig. 1 Synthesis of the drug–peptide–polymer conjugates: the conjugation of the isopropylidene protected aminooxyacetylated peptide to the amino-monofunctional polymer (PEG or HbPG) (A); cleavage of the isopropylidene protecting group (B); conjugation of daunomycin via oxime linkage (C). | ||
First, the GFLG spacer containing GE11 and D4 derivatives were synthesized with a carboxyl end, and these peptides were functionalized with isopropylidene protected aminooxyacetic acid61 on the N-terminal. Derivatives bearing a G5 spacer on the C-terminal were also prepared. The purified peptides were conjugated to the amine-functionalized polymers (amino-PEG or amino-HbPG) in solution through the C-terminal carboxylic groups by using a BOP coupling agent in the presence of DIPEA (Fig. 1A). The isopropylidene protecting group was cleaved with methoxyamine in 0.2 M ammonium acetate buffer (pH 5.0, 1 M solution to MeONH2) (Fig. 1B), and subsequently the free aminooxyacetylated peptide–polymer bioconjugates were reacted with daunomycin by the formation of an oxime linkage (Fig. 1C). The drug–peptide–polymer conjugates were purified by RP-HPLC and analysed using ESI-MS (Table 2; analytical chromatograms and mass spectra in the ESI,† Fig. S6–S13). As the data indicate, the drug–peptide–polymer conjugates were obtained in high purity.
| Conjugate | R t (min)a | MWavg.b | Turbidity (100-T%)c |
|---|---|---|---|
|
a Gradient elution (0 min 0% B, 5 min 0% B, 50 min 90% B) in 0.1% TFA in water and 0.1% TFA in acetonitrile–water 80:20% (v/v).
b Average MW, determined from the most intensive peak in the mass spectrum.
c 100 μM, measured in 10% (v/v) water containing complete cell culture medium (RPMI-1640).
|
|||
| DauAoa–GFLG–GE11–PEG |
35.6 | 3563.4 | 4.2 |
| DauAoa–GFLG–GE11–G5–PEG |
34.6 | 3632.4 | 3.3 |
| Dau Aoa–GFLG–GE11–HbPG |
32.4 | 3163.3 | 2.7 |
| DauAoa–GFLG–GE11–G5–HbPG |
32.2 | 3447.3 | 2.1 |
| DauAoa–GFLG–D4–PEG |
38.9 | 2757.2 | 3.7 |
| DauAoa–GFLG–D4–G5–PEG |
39.1 | 2954.0 | 3.5 |
| DauAoa–GFLG–D4–HbPG |
35.0 | 2382.2 | 2.2 |
| DauAoa–GFLG–D4–G5–HbPG |
35.4 | 2666.9 | 2.9 |
All synthesized drug–peptide–polymer conjugates were water-soluble. Nevertheless, turbidimetric measurements under the conditions used for the in vitro tests were performed to justify the positive effect of the hydrophilic polymers on the solubility of the conjugates. The conjugates were dissolved in double distilled water and diluted with serum-containing medium (RPMI-1640), then directly after the preparation of the solutions, the turbidity was measured at 630 nm. The polymer containing conjugates showed low turbidity (see Table 2), which was constant over time. This proves that the solubility problems of peptide–drug conjugates could be solved not only by the conjugation of the well-known PEG but also by HbPG. However, it is worth mentioning that the turbidity of the PEG containing conjugates was slightly higher than the HbPG-based analogues.
We presumed that HbPG can cause lower receptor binding hindrance due to the branched structure, since the hydrodynamic volume (Vh) of HbPG is lower than the Vh of PEG with similar molecular weight. To investigate and clarify the relationship between the polymer structure and the in vitro internalization ability and biological efficacy of the conjugates, different assays were performed.
The in vitro internalization of the conjugates was studied on HT-29 human colon adenocarcinoma cells by flow cytometry. In the case of an attached cell, the cell membrane can be divided into two parts: the free membrane, which is available for the conjugates, and a part of the cellular junction that might have limited accessibility to a polymer-containing conjugate.
The cells in suspension are roundish, and although the total membrane surface can be considered accessible for the conjugates, the membrane area could be smaller than that of a cell spread on a solid surface. Supposing that the internalization ability of the cells might depend on the free membrane surface, the treatment was performed on attached cells and on cells in suspension as well. The compounds were added to the cells with a final concentration of 10 μM and co-incubated for 30 min, and then the cells were analysed via flow cytometry. In all cases, one part of the cells was treated with trypsin–EDTA to investigate exclusively the amount of the internalized conjugates, since trypsin can remove the non-specifically membrane-bound compounds. As demonstrated in Fig. 2, most of the conjugates were found intracellularly. This means that with the exception of DauAoa–GFLG–GE11–G5–PEG, all drug–peptide–polymer conjugates could enter the cells. Moreover, no significant difference was observed between the uptake of the cells in suspension (Fig. 2A and B) and the attached cells (Fig. 2C and D). The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake DauAoa–GFLG–GE11–PEG and DauAoa–GFLG–D4–G5–HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11–PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding.
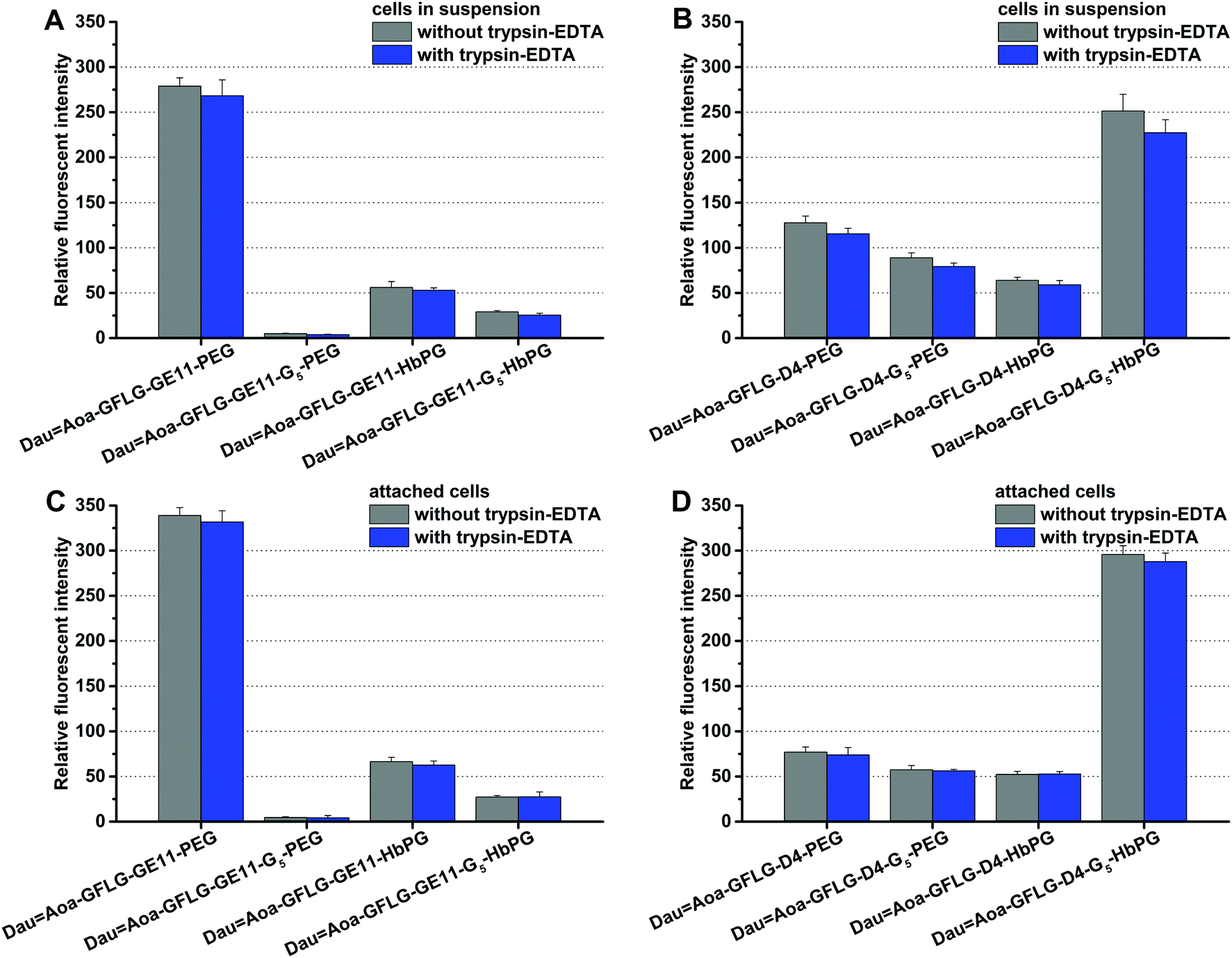 | ||
| Fig. 2 In vitro cellular uptake of the different daunomycin–peptide–polymer conjugates on HT-29 cells after 30 minutes of incubation time. The uptake was studied on cells in suspension with GE11 (A) and D4 (B) containing conjugates and on attached cells with GE11 (C) and D4 (D) containing conjugates. | ||
One of the best conjugates, the DauAoa–GFLG–D4–G5–HbPG was chosen for time-dependent cellular uptake studies. Cells in suspension, as well as attached cells, were treated for 15 min, 30 min or 60 min with this conjugate. Then after washing, the cells were treated with trypsin–EDTA and analysed by flow cytometry. As presented in Fig. 3, the fluorescence intensity increased under both conditions over time, though the initial uptake of the conjugate was lower in the case of the attached cells than in suspension. The effect of the prepared daunomycin–peptide–polymer conjugates on the in vitro cell viability was determined on HT-29 human colon cancer cells using the real-time cell analyser xCELLigence SP device. The cells were treated with the conjugates having different concentrations (0.1–50 μM) and monitored for 72 hours. The cell viability (%) values as the function of the conjugate concentration are presented in Fig. 4. The IC50 values were determined after 48 hours of treatment (Table 3). The parent cytotoxic agent (Dau) has a lower IC50 value (0.342 ± 0.067 μM) than the drug–peptide–polymer conjugates on the used cell line, but the application of the produced conjugates may be more beneficial based on the increased selectivity, which leads to fewer side effects. It must be noted that the used peptides, as well as the used polymers, had no toxic effect in the utilized concentration range (IC50 ≫ 50 μM). There was one outstanding conjugate from each group (DauAoa–GFLG–GE11–PEG and DauAoa–GFLG–D4–G5–HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide.
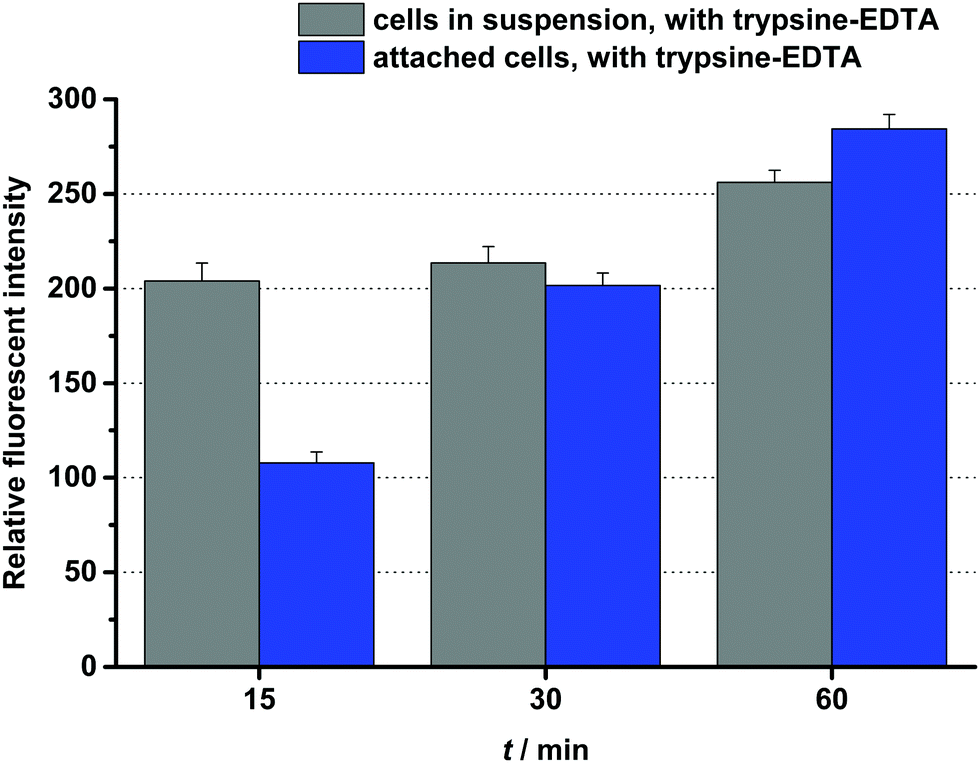 | ||
| Fig. 3 Time-dependent in vitro cellular uptake of DauAoa–GFLG–D4–G5–HbPG on HT-29 cells. | ||
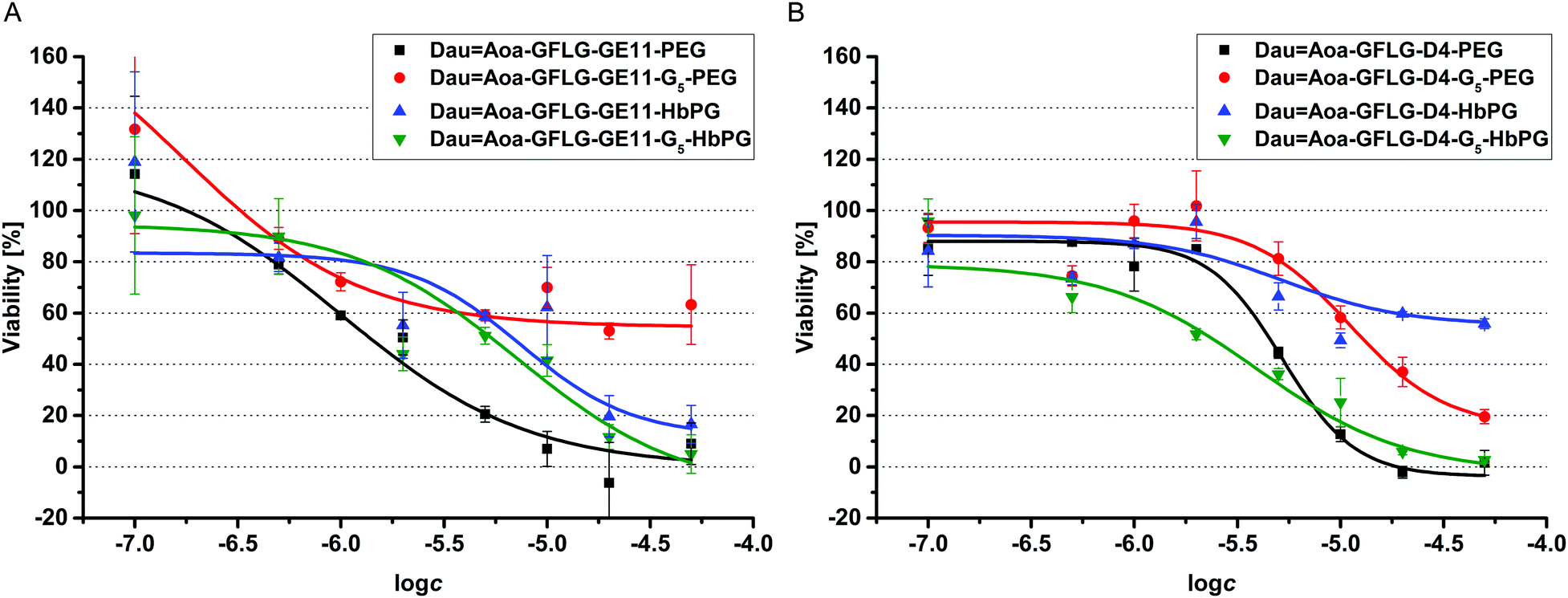 | ||
| Fig. 4 Results of the cell viability assay performed on HT29 cells with GE11 (A) and D4 (B) containing drug–peptide–polymer conjugates. | ||
| Conjugate | IC50 (μM) | d (nm) | PDI | cac (μM) |
|---|---|---|---|---|
| DauAoa–GFLG–GE11–PEG |
1.2 | 370.0 | 0.082 | 28.6 ± 4.4 |
| DauAoa–GFLG–GE11–G5–PEG |
9.1 | 171.8 | 0.137 | 9.3 ± 0.9 |
| DauAoa–GFLG–GE11–HbPG |
9.3 | 83.8 | 0.144 | 16.6 ± 1.6 |
| DauAoa–GFLG–GE11–G5–HbPG |
6.0 | 52.3 | 0.092 | 20.2 ± 2.9 |
| DauAoa–GFLG–D4–PEG |
5.1 | 189.3 | 0.130 | 16.1 ± 1.3 |
| DauAoa–GFLG–D4–G5–PEG |
13.5 | 157.4 | 0.076 | 56.9 ± 3.2 |
| DauAoa–GFLG–D4–HbPG |
>50.0 | 76.5 | 0.057 | 6.4 ± 1.2 |
| DauAoa–GFLG–D4–G5–HbPG |
3.8 | 71.3 | 0.042 | 13.4 ± 0.5 |
We observed that some of the conjugates (DauAoa–GFLG–D4–HbPG and DauAoa–GFLG–GE11–G5–PEG) could not cause complete cell death, i.e., ∼0% viability value – characteristic for cell-free culturing medium – was not achieved even at the highest concentration, since their dose–response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates. Therefore, to prove this, the aggregation dynamic light scattering (DLS) measurements were carried out. The size (d) and dispersity index (PDI) of the conjugates were compared with conjugates having 100 μM concentration and the obtained results are presented in Table 3. The size distribution curve of the DauAoa–GFLG–D4–G5–HbPG conjugate as one representative sample is presented in Fig. 5 (for the DLS size distribution curves of the other conjugates, see Fig. S14–S20 in the ESI†). It is visible from these data that the formation of nanoscale aggregates was observed for each conjugate. It can be stated that the HbPG conjugates resulted in the formation of smaller aggregates compared to the PEG-containing conjugates. In addition, the size of the GE11-PEG conjugates was bigger than the D4-PEG analogues (e.g. DauAoa–GFLG–GE11–PEG: d = 370 nm, and DauAoa–GFLG–D4–PEG: d = 189 nm). Moreover, the size of all HbPG-containing conjugates was in the range of 50–80 nm. These can be explained by the compact highly branched structure and the lower molar hydrodynamic volume of the HbPG than that of the PEG at the same molar mass. As observed, the incorporation of the G5 spacer into the conjugates also decreases the size of the formed nanoparticles in all cases. The self-aggregation of the amphiphilic molecules takes place above a certain concentration, called the critical aggregation concentration (cac). For the determination of the cac, the intensity of scattered light was measured by DLS in a broad concentration range (0.5–500 μM) of the conjugates. The recorded scattered intensity (kcps) plotted as a function of the concentration of the DauAoa–GFLG–D4–G5–HbPG conjugate, as a representative sample, is presented in Fig. 5 (right). As shown in this figure, the scattered intensity is nearly constant at lower concentrations and increases linearly at higher concentrations. The cac was defined as the intersection of the straight lines fitted to the constant and the increasing sections. The determined cac values of the conjugates are listed in Table 3 (for the scattered intensity vs. concentration curves of the other conjugates, see Fig. S14–S20 in the ESI†). As observed, the cac of all conjugates was in the range of 6–60 μM but there is a significant difference between the PEG and HbPG conjugates, which may be explained by the structural differences of the polymers. In the case of the HbPG-based conjugates, it can be stated that the G5 spacer between the targeting peptide and the polymer slightly increases the cac. Furthermore, the GE11–HbPG conjugates have higher cac values compared to the D4 analogues. This can be explained by the higher hydrophobicity of the D4 sequence and the compact structure of the HbPG that provides lower steric hindrance on the peptide part, which is primarily responsible for the self-aggregation behaviour. However, at first sight, there is no direct correlation between the structure and the determined cac in the case of the PEG-conjugates. In the case of the GE11–PEG conjugates, the incorporation of the G5 spacer between the peptide and the polymer increases the accessibility of the hydrophobic part, which leads to a lower cac value, on the one hand. On the other hand, the D4 analogue consists of only six amino acids, and in this case, the pentaglycine spacer can drastically decrease the hydrophobicity of the peptide part, which then highly increases the cac value.
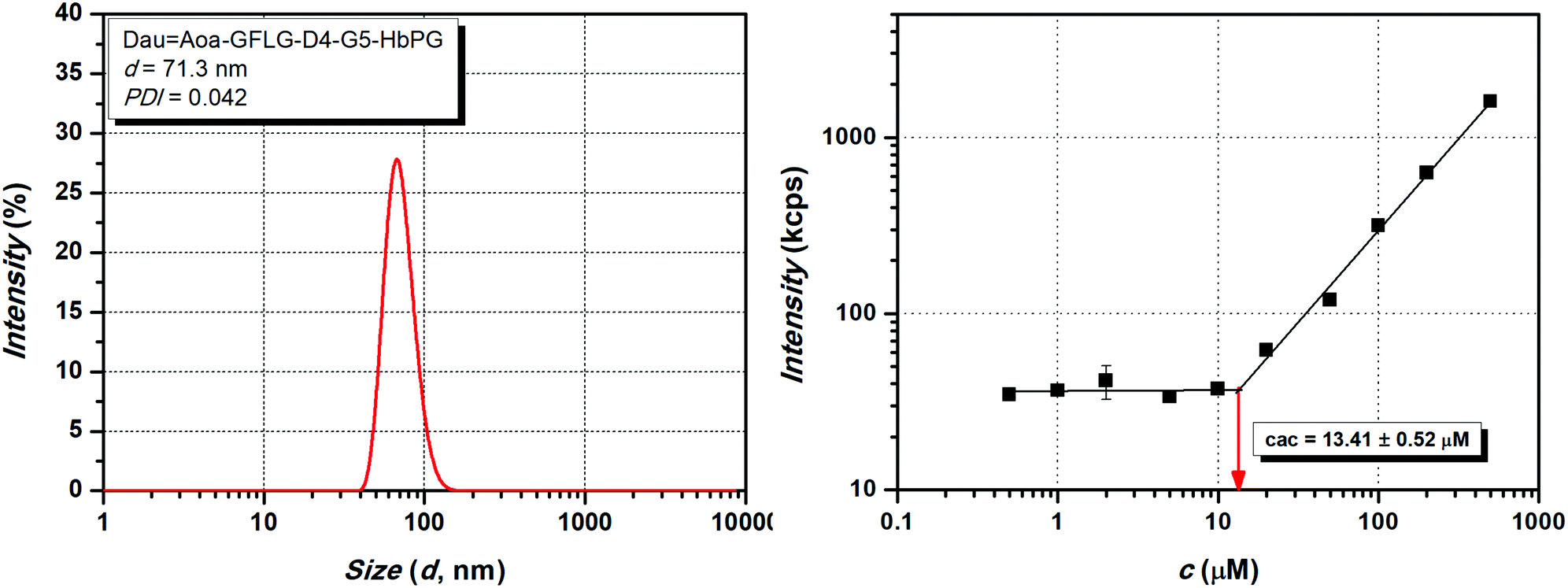 | ||
| Fig. 5 Dynamic light scattering size distribution at 100 μM concentration (left) and the scattered intensity (kcps) as a function of concentration (μM) (right) of the DauAoa–GFLG–D4–G5–HbPG conjugate. | ||
The obtained DLS data contribute greatly to the understanding of the in vitro cell viability and cellular uptake results. By comparing the GE11–PEG and PEG–G5–GE11 conjugates, there is a significant difference between the cellular uptake and the IC50 values of these conjugates. Although the largest nanoparticle was formed by the GE11–PEG conjugate (370 nm, at 100 μM), its cac value is around 28 μM, which is far above the utilized concentration for the cellular uptake measurements (10 μM). In contrast, the cellular uptake of the GE11–G5–PEG conjugate was the lowest in the investigated series, and the cell viability does not follow a classical sigmoid curve (see Fig. 4A). 50% cell viability is reached around 5–10 μM but does not further decrease by increasing the concentration, i.e., the mortality is saturated. In this case, the cac is around 9 μM, therefore, above this concentration the resulting aggregates probably cover the targeting peptide part, which inhibits the receptor-mediated internalization, and thus this results in decreased toxicity. Nevertheless, the cell viability curves of the HbPG-based GE11 conjugates follow a similar pattern, and no significant differences can be observed in the calculated IC50 values and the cell internalization abilities, respectively.
In the series of D4 conjugates, the HbPG-based D4 conjugate has the worst biological efficacy. In addition, the cell viability curve of this conjugate follows a sigmoid pattern but does not reach 50% mortality even at the highest concentration. The reason for this may be that the D4–HbPG conjugate has the lowest cac (around 6 μM), which also coincides with the saturation part of the cell viability curve. This means that small-sized (∼76 nm) aggregates are already formed at low concentrations, which may enter the cells through a receptor-mediated mechanism only in the low concentration range. In contrast, D4–G5–HbPG has the lowest IC50 value (3.8 μM) and it also shows outstanding cellular uptake in the series of the D4 conjugates. Here, the G5 spacer decreases the hydrophobicity and increases the accessibility of the peptide part. Therefore, it can be concluded that in the case of a shorter targeting peptide, the HbPG is more advantageous with a short G5 spacer. Nevertheless, in the case of using PEG, the incorporation of the G5 spacer decreases the biological efficacy irrespective of the type of targeting peptide used.
Experimental
Materials and methods
All amino acid derivatives, the Fmoc-Rink Amide MBHA resin, the Wang resin, N,N′-diisopropylcarbodiimide (DIC) and trifluoroacetic acid (TFA) were purchased from Iris Biotech GmBH (Marktredwitz, Germany). Boc-aminooxyacetic acid (Boc-Aoa-OH), 1-hydroxybenzotriazole hydrate (HOBt), 4-(dimethylamino)pyridine (DMAP), triisopropylsilane (TIS), and N,N-diisopropylethylamine (DIPEA) were obtained from Sigma Aldrich Kft (Budapest, Hungary). Aminooxyacetic acid (Aoa·1/2HCl), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and methoxyamine hydrochloride were obtained from TCI Europe N.V. (Zwijndrecht, Belgium), while piperidine was purchased from Molar Chemicals Kft (Budapest, Hungary). Daunomycin hydrochloride was a kind gift from IVAX (Budapest, Hungary). All solvents used for the synthesis and purification and ammonium acetate were purchased from VWR International Kft. (Debrecen, Hungary). Isopropylidene protected aminooxyacetic acid was prepared in our laboratory, as described earlier, by the reaction of aminooxyacetic acid and acetone.61Methoxypoly(ethylene glycol) amine (amino-PEG; MW: 1000) was obtained from Alfa Aesar, Thermo Fisher Scientific (Ward Hill, MA, USA). Amino-monofunctional HbPG was produced and characterized in our laboratory, as described earlier.32 Briefly, phthalimide monofunctional HbPG was synthesized by the ring-opening multibranching polymerization of freshly distilled glycidol with a phthalimide/potassium phthalimide initiating system (the monomer/initiator ratio was 15) and by using a slow monomer addition technique (feed rate: 2.5 mL h−1) at 95 °C under an inert atmosphere. The phthalimide moiety was transformed to an amine functionality through a reaction with hydrazine monohydrate in ethanol at room temperature. The number average molar mass of the used NH2-HbPG was 1230 g mol−1 (by NMR) with narrow polydispersity (Đ = 1.3) (for further characterization data see our previous article32).
For the in vitro assays, RPMI-1640 medium, trypsin–EDTA and PBS were obtained from Sigma-Aldrich Ltd (St. Louis, MO, USA), FBS was purchased from Lonza Group Ltd. (Basel, Switzerland), while penicillin/streptomycin and L-glutamine were obtained from Gibco®/Invitrogen Corporation (New York, NY, USA). TrypLe was obtained from Thermo Fisher Scientific (Waltham, MA, USA) and the E-plates were purchased from ACEA Biosciences (San Diego, CA, USA).
Synthesis of EGFR targeting peptides
The peptides were synthesized manually using solid-phase peptide synthesis according to the Fmoc/tBu strategy. The Fmoc protecting group was removed with 2% piperidine + 2% DBU in DMF (2 + 2 + 5 + 10 min), and the α-Fmoc-protected amino acid derivatives were coupled with DIC and HOBt (3 eq. each to the resin capacity).Peptides for the drug–peptide conjugates were synthesized on Fmoc-Rink Amide MBHA resin, and a Boc-aminooxyacetic acid (Boc-Aoa-OH) was coupled to the N-terminal. The peptides were cleaved from the resin with the 95% TFA/2.5% TIS/2.5% H2O (v/v/v) mixture and 10 eq. Aoa·1/2HCl at room temperature for 2 h. The crude products were purified by RP-HPLC and the solvent was evaporated. The pure peptides were used directly, without lyophilization for the daunomycin conjugation.
The peptides for the drug–peptide–polymer conjugates containing the GFLG spacer on the N-terminal were synthesized on Wang resin. The first amino acid derivative was always coupled with DIC and DMAP (2:2:0.2 eq. to the resin capacity) for 3 hours. Isopropylidene protected aminooxyacetic acid was coupled to the N-termini of the peptides. The peptides were cleaved from the resin in 2 h with the 95% TFA/2.5% TIS/2.5% H2O (v/v/v) mixture, purified by RP-HPLC and the pure compounds were lyophilized.
Synthesis of peptide–polymer conjugates
The polymer (1 eq. amino-PEG or amino-HbPG), the peptide (1.1 eq.), BOP (3 eq.) and HOBt (3 eq.) were, respectively, transferred into a 4 mL vial and dissolved in 500 μL DMF. DIPEA (6 eq.) was added after complete dissolution and the reaction mixtures were stirred for 16 h at room temperature. The crude products were purified by RP-HPLC and lyophilized. The acetone protecting group was removed by using 1 M MeONH2·HCl in DMF/0.2 M NH4OAc buffer (pH 5.0) (50:50%; v/v) for 1 h at room temperature. The reactions were purified by RP-HPLC, the solvent was evaporated, and the pure peptide–polymer conjugates were used directly for the next reaction (daunomycin conjugation) without lyophilization.
Conjugation of daunomycin
Daunomycin (Dau) was conjugated via oxime linkage to the peptides or peptide–polymer conjugates. The peptides/peptide–polymer conjugates (1 eq.) and the Dau (1.3 eq.) were transferred into a 4 mL vial and were dissolved in 500 μL DMF and 500 μL 0.2 M NH4OAc buffer (pH 5.0). The reaction was stirred for 16 h at room temperature, then the conjugates were purified by RP-HPLC and the pure compounds were lyophilized.Characterization of the drug–peptide and the drug–peptide–polymer conjugates
The purity of the conjugates was determined by analytical RP-HPLC using a Phenomenex Aeris PEPTIDE XB-C18 column (5 μm, 100 Å; 250 × 4.6 mm). The chromatograms were acquired on a Knauer RP-HPLC system (Bad Homburg, Germany) using gradient elution (0 min 0% B, 5 min 0% B, 50 min 90% B) with eluents 0.1% TFA in water (A) and 0.1% TFA in acetonitrile–water 80:20%, v/v (B) by a flow rate of 1 mL min−1, and the peaks were detected at λ = 220 nm.
The pure compounds were analysed on a Bruker Daltonics Esquire 3000+ electrospray ionization ion trap mass spectrometer (Bruker Daltonics GmbH, Bremen, Germany), operating with continuous sample injection at a flow rate of 10 μL min−1. Samples were dissolved in 0.1% acetic acid containing the acetonitrile–water (50:50%, v/v) eluent and mass spectra were recorded in positive ion mode in the m/z 50–2000 range.
Solubility test of the drug–peptide and drug–peptide–polymer conjugates
Solubility tests were performed through turbidity measurements. The drug–peptide conjugates (dissolved in DMSO) and the drug–peptide–polymer conjugates (dissolved in water) were diluted by 9 volume excess of serum-containing RPMI medium, homogenized and kept for 15 min at room temperature. The transmittance (T%) of the samples (100 μM) was recorded using a Jasco V-650 spectrophotometer at 630 nm with a 0.2 × 1 cm quartz cuvette tempered at 25 °C with the DMSO–RPMI medium (1:9) or water–RPMI medium (1:9) mixture as a reference. The turbidity of the samples is expressed as 100-T%.
Determination of size, dispersity and critical aggregation concentration through dynamic light scattering measurements
The produced drug–peptide–polymer conjugates (100 μM aqueous solutions) were characterized by their intensity mean diameter (d) and dispersity index (PDI) measured by using the dynamic light scattering (DLS) method. The critical aggregation concentration (cac) of the produced polymer–peptide–drug conjugates was determined according to the literature62via the recorded scattered intensity by DLS as a function of the conjugate concentration in the range of 0.5–500 μM. All DLS measurements were carried out at 25 °C using a Malvern Zetasizer, NANO ZS (Malvern Instruments Limited, UK) equipped with a 4 mW He–Ne laser operating at a wavelength of 633 nm. The measurements were repeated three times.Cell culture
HT-29 human colon adenocarcinoma cells, purchased from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK), were cultured in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine and 100 μg mL−1 penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere.In vitro cellular uptake studies
The in vitro cellular uptake of the daunomycin–peptide–polymer conjugates was measured by the FACSCalibur flow cytometer (Becton–Dickinson, San Jose, CA, USA) using the fluorescence property of daunomycin. The treatments were performed in two ways: on attached HT29 cells as well as on HT29 cells in suspension. In the former case, the cells were seeded on 12-well plates with a density of 105 cells per well and cultured for 24 h. The compounds were added to the attached cells with a final concentration of 10 μM and co-incubated for 30 min. The cells were washed with PBS (phosphate-buffered saline pH = 7.2) and treated with either trypsin–EDTA or TrypLE, composed of recombinant cell-dissociation promoting enzymes (Thermo Fisher Scientific, Waltham, MA, USA), for 5 min at 37 °C. Then, the effect of the dissociation reagents was inhibited with fresh serum-containing cell culture medium. The cells were transferred to FACS-tubes and after centrifugation and a washing step with PBS, they were analysed by a flow cytometer.For the experiments on cells in suspension, the TrypLE reagent was added to the monolayer cultures of HT29 cells. The effect of the TrypLE was stopped after 5 min by adding fresh medium. The cells were transferred to FACS-tubes (105 cells/FACS-tube), centrifuged and washed with fresh cell culture medium. The treatment was carried out as described above. To remove the conjugates the cellular samples were centrifuged and washed with PBS. One part of the cells was treated with trypsin–EDTA for 5 min at 37 °C and its digestion was stopped with fresh cell culture medium. After centrifugation and a washing step with PBS, the cells were analysed by a flow cytometer. The other part of the cells, after a washing step with PBS, was used directly for the measurements.
For each measurement, 10000 cells were used. The geometric mean of relative fluorescence intensity was used to quantify the membrane-bound or intracellular daunomycin-containing conjugates, and this value was adjusted with the autofluorescence of the non-treated control. For the measurement and analysis of data, CellQuest Pro software was used. There were two independent experiments with the use of two parallels and a representative result of these experiments was plotted.
In vitro cell viability assay
The in vitro cytotoxic effect of the daunomycin–peptide–polymer conjugates was determined by using the xCELLigence SP System (ACEA Biosciences, San Diego, CA, USA). This allows monitoring of the cellular events by measuring electrical impedance in a real-time manner. The detected impedance depends on the number and/or the spreading of the cells attached to the surface of the electrodes found on the bottom of a special tissue culture plate (E-plate). The change in impedance is represented by the Cell Index (CI). The CI is calculated by the following formula:| CI = (Zi − Z0)/Fi |
The background measurement for 1 h time period with 1 min interval was done by using only cell culture medium. Then, the cells were seeded into the E-plate with a density of 104 cells per well and cultured for 24 h. The cells were treated with the compounds in the concentration range of 0.1–50 μM and monitored for 72 h every 20 s.
The CI change as a result of the treatment with the daunomycin–peptide–polymer conjugates was expressed as the so called normalized Cell Index, calculated by dividing the CI at a given time point by the CI at the last time point before the treatment. Viability (%), plotted on the Y-axis of Fig. 4, was calculated as the ratio of the normalized CI recorded for conjugate-treated cells and the normalized CI of the control group. To compare the cytotoxic effects of the conjugates, the IC50 values were calculated from the viability percentages obtained at 48 h for each concentration by the integrated software (RTCA 2.0) of the xCELLigence System and OriginPro 2016 (OriginLab Corporation, Northampton, MA, USA). IC50 values were determined as the concentration that resulted in 50% reduction in the normalized CI (in cell viability). Each datum represents the mathematical average of three parallels. In the case of the control wells, an adequate volume of pure cell culture medium was loaded.
Conclusions
The epidermal growth factor receptor (EGFR) is a widely used target receptor for cancer therapy. In this work, GE11 and D4 EGFR targeting peptides containing drug conjugates with systematic structural variations were prepared and investigated. As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide–polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug–peptide–polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely DauAoa–GFLG–GE11–PEG and DauAoa–GFLG–D4–G5–HbPG. Based on the performed DLS measurements, the conjugates have amphiphilic character and all conjugates aggregate above a specific concentration (cac) which leads to nanoparticle formation with 60–370 nm size, which depends on the composition of the conjugates. In general, the size of the HbPG containing conjugate nanoparticles is lower than that of the PEG analogues. This is related to the lower hydrodynamic volume of the HbPG, which has a compact hyperbranched structure in contrast to the linear PEG. The incorporation of the pentaglycine (G5) spacer between the targeting peptide and the polymer influences the philicity and also increases the accessibility of the peptide segment. It was found that the G5 spacer influences not only the biological activity (internalization and toxicity) but also the aggregation. This latter property of the drug–peptide–polymer conjugates is also affected by the steric hindrance of the hydrophilic polymer. Hence, it can be generally concluded that the investigation of the colloidal properties, such as self-aggregation behaviour and nanoparticle formation, is necessary for the better understanding of the biological activity of bioconjugates with amphiphilic character, which can reveal the structure–activity relationship of amphiphilic drug–peptide–polymer conjugates for tumour targeting. Our findings strongly indicate that the solubilization or apparent solubilization of drugs or bioconjugates is not a sufficient criterion for increased bioactivity. The formation of nanoparticle aggregates may hinder the targeting moiety for the high extent of receptor binding, on the one hand. On the other hand, the enzyme labile spacer may also be blocked in such nanoparticles. In order to overcome these difficulties, the structure of the receptor binding moiety and the solubilizing polymer must be well aligned for highly efficient drug targeting.
According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Research, Development and Innovation Office, Hungary (NKFIH K119552, NVKP_16-1-2016-0036), and the MedInProt Protein Science Research Synergy Program (MedInProt). This work was also supported by the BIONANO_GINOP-2.3.2-15-2016-00017 project.References
- S. M. Kupchan, Y. Komoda, A. R. Branfman, A. T. Sneden, W. A. Court, G. J. Thomas, H. P. Hintz, R. M. Smith, A. Karim, G. A. Howie, A. K. Verma, Y. Nagao, R. G. Dailey, V. A. Zimmerly and W. C. Sumner, J. Org. Chem., 1977, 42, 2349 CrossRef CAS PubMed.
- R. E. Schwartz, C. F. Hirsch, D. F. Sesin, J. E. Flor, M. Chartrain, R. E. Fromtling, G. H. Harris, M. J. Salvatore, J. M. Liesch and K. Yudin, J. Ind. Microbiol., 1990, 5, 113 CrossRef CAS.
- G. R. Pettit, Y. Kamano, C. L. Herald, A. A. Tuinman, F. E. Boettner, H. Kizu, J. M. Schmidt, L. Baczynskyj, K. B. Tomer and R. J. Bontems, J. Am. Chem. Soc., 1987, 109, 6883 CrossRef CAS.
- A. Kumari, R. Singla, A. Guliani and S. K. Yadav, EXCLI J., 2014, 13, 265 Search PubMed.
- T. Nomoto and N. Nishiyama, Photodynamic Therapy. Photochemistry for Biomedical Applications, ed. Y. Ito, 2018, Springer, Singapore, pp. 301–313 Search PubMed.
- S. Chen, X. Zhao, J. Chen, J. Chen, L. Kuznetsova, S. S. Wong and I. Ojima, Bioconjugate Chem., 2010, 21, 979 CrossRef CAS PubMed.
- Y. Zhang, J. Zhou, C. Yang, W. Wang, L. Chu, F. Huang, Q. Liu, L. Deng, D. Kong, J. Liu and J. Liu, Int. J. Nanomed., 2016, 11, 1119 CAS.
- G. Mező and M. Manea, Exp. Opin. Drug Delivery, 2010, 7, 79 CrossRef PubMed.
- D. Böhme and A. G. Beck-Sickinger, J. Pept. Sci., 2015, 21, 186 CrossRef PubMed.
- M. Sameni, K. Moin and B. F. Sloane, Neoplasia, 2000, 2, 496 CrossRef CAS PubMed.
- W. Halangk, M. M. Lerch, B. Brandt-Nedelev, W. Roth, M. Ruthenbuerger, T. Reinheckel, W. Domschke, H. Lippert, C. Peters and J. Deussing, J. Clin. Invest., 2000, 106, 773 CrossRef CAS PubMed.
- H. H. Heidtmann, U. Salge, M. Abrahamson, M. Bencina, L. Kastelic, N. Kopitar-Jerala, V. Turk and T. T. Lah, Clin. Exp. Metastasis, 1997, 15, 368 CrossRef CAS PubMed.
- A. T. Chan, Y. Baba, K. Shima, K. Nosho, D. C. Chung, K. E. Hung, U. Mahmood, K. Madden, K. Poss, A. Ranieri, D. Shue, R. Kucherlapati, C. S. Fuchs and S. Ogino, Cancer Epidemiol., Biomarkers Prev., 2010, 19, 2777 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855 CrossRef CAS PubMed.
- P. Rejmanová, J. Kopeček, J. Pohl, M. Baudyš and V. Kostka, Makromol. Chem., 1983, 184, 2009 CrossRef.
- A. Romieu, T. Bruckdorfer, G. Clavé, V. Grandclaude, C. Massif and P.-Y. Renard, Org. Biomol. Chem., 2011, 9, 5337 RSC.
- T. H. Evers, E. M. W. M. van Dongen, A. C. Faesen, E. W. Meijer and M. Merkx, Biochemistry, 2006, 45, 13183 CrossRef CAS PubMed.
- V. Paraskevopoulou and F. H. Falcone, Microorganisms, 2018, 6, 47 CrossRef PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- N. Rades, K. Achazi, M. Qiu, C. Deng, R. Haag, Z. Zhong and K. Licha, J. Controlled Release, 2019, 300, 13 CrossRef CAS PubMed.
- P. Ray, M. Ferraro, R. Haag and M. Quadir, Macromol. Biosci., 2019, 19, 1900073 CrossRef PubMed.
- K. Pant, C. Neuber, K. Zarschler, J. Wodtke, S. Meister, R. Haag, J. Pietzsch and H. Stephan, Small, 2019, 1905013 Search PubMed.
- A. Kadhim, L. K. McKenzie, H. E. Bryant and L. J. Twyman, Mol. Pharm., 2019, 16, 1132 CrossRef CAS PubMed.
- X. Yang, C. Zhang, A. Li, J. Wang and X. Cai, Mater. Sci. Eng., C, 2019, 95, 104 CrossRef CAS PubMed.
- X. Zhou, L. Xu, J. Xu, J. Wu, T. B. Kirk, D. Ma and W. Xue, ACS Appl. Mater. Interfaces, 2018, 10, 35812 CrossRef CAS PubMed.
- J. N. Lockhart, D. B. Beezer, D. M. Stevens, B. R. Spears and E. Harth, J. Controlled Release, 2016, 244, 366 CrossRef CAS PubMed.
- Y. Zhong, M. Dimde, D. Stöbener, F. Meng, C. Deng, Z. Zhong and R. Haag, ACS Appl. Mater. Interfaces, 2016, 8, 27530 CrossRef CAS PubMed.
- S. Abbina, S. Vappala, P. Kumar, E. M. J. Siren, C. C. La, U. Abbasi, D. E. Brooks and J. N. Kizhakkedathu, J. Mater. Chem. B, 2017, 5, 9249 RSC.
- E. Moore, A. T. Zill, C. A. Anderson, A. R. Jochem, S. C. Zimmerman, C. S. Bonder, T. Kraus, H. Thissen and N. H. Voelcker, Macromol. Chem. Phys., 2016, 217, 2252 CrossRef CAS.
- D. B. Beezer and E. Harth, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2820 CrossRef CAS.
- G. Kasza, G. Kali, A. Domján, L. Petho, G. Szarka and B. Iván, Macromolecules, 2017, 50, 3078 CrossRef CAS.
- J. G. Zhang, O. B. Krajden, R. K. Kainthan, J. N. Kizhakkedathu, I. Constantinescu, D. E. Brooks and M. I. C. Gyongyossy-Issa, Bioconjugate Chem., 2008, 19, 1241 CrossRef CAS PubMed.
- B. L. Wilkinson, S. Day, L. R. Malins, V. Apostolopoulos and R. J. Payne, Angew. Chem., Int. Ed., 2011, 50, 1635 CrossRef CAS PubMed.
- H. Bao, X. Jin, L. Li, F. Lv and T. Liu, J. Mater. Sci.: Mater. Med., 2012, 23, 1891 CrossRef CAS PubMed.
- F. Wurm, J. Klos, H. J. Räder and H. Frey, J. Am. Chem. Soc., 2009, 131, 7954 CrossRef CAS PubMed.
- E. K. Rowinsky, Annu. Rev. Med., 2004, 55, 433 CrossRef CAS PubMed.
- R. Roskoski, Biochem. Biophys. Res. Commun., 2004, 319, 1 CrossRef CAS PubMed.
- R. Bianco, T. Gelardi, V. Damianoa, F. Ciardiello and G. Tortora, Int. J. Biochem. Cell Biol., 2007, 39, 1416 CrossRef CAS PubMed.
- M. Hamzeh-Mivehroud, A. Mahmoudpour and S. Dastmalchi, Chem. Biol. Drug Des., 2012, 79, 246 CrossRef CAS PubMed.
- C. Y. Han, L. L. Yue, L. Y. Tai, L. Zhou, X. Y. Li, G. H. Xing, X. G. Yang, M. S. Sun and W. S. Pan, Int. J. Nanomed., 2013, 8, 1541 Search PubMed.
- C. Han, Y. Li, M. Sun, C. Liu, X. Ma, X. Yang, Y. Yuan and W. Pan, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 161 CrossRef CAS PubMed.
- S. Sachdeva, H. Joo, J. Tsai, B. Jasti and X. Li, Sci. Rep., 2019, 9, 997 CrossRef PubMed.
- Y. Yin, N. Ochi, T. W. Craven, D. Baker, N. Takigawa and H. Suga, J. Am. Chem. Soc., 2019, 141, 19193 CrossRef CAS PubMed.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978 CrossRef CAS PubMed.
- F. M. Mickler, L. Möckl, N. Ruthardt, M. Ogris, E. Wagner and C. Bräuchle, Nano Lett., 2012, 12, 3417 CrossRef CAS PubMed.
- L. Milane, Z. Duan and M. Amiji, Nanomedicine, 2011, 7, 435 CrossRef CAS PubMed.
- E. Kopansky, Y. Shamay and A. David, J. Drug Targeting, 2011, 19, 933 CrossRef CAS PubMed.
- H. Ren, C. Gao, L. Zhou, M. Liu, C. Xie and W. Lu, Drug Delivery, 2015, 22, 785 CrossRef CAS PubMed.
- H. Ren, L. Zhou, M. Liu, W. Lu and C. Gao, Med. Oncol., 2015, 32, 185 CrossRef PubMed.
- A. Vetter, K. S. Virdi, S. Espenlaub, W. Rödl, E. Wagner, P. S. Holm, C. Scheu, F. Kreppel, C. Spitzweg and M. Ogris, Mol. Pharm., 2013, 10, 606 CrossRef CAS PubMed.
- S. Ohno, M. Takanashi, K. Sudo, S. Ueda, A. Ishikawa, N. Matsuyama, K. Fujita, T. Mizutani, T. Ohgi, T. Ochiya, N. Gotoh and M. Kuroda, Mol. Ther., 2013, 21, 185 CrossRef CAS PubMed.
- B. G. Ongarora, K. R. Fontenot, X. Hu, I. Sehgal, S. D. Satyanarayana-Jois and M. G. H. Vicente, J. Med. Chem., 2012, 55, 3725 CrossRef CAS PubMed.
- S. Dixit, K. Miller, Y. Zhu, E. McKinnon, T. Novak, M. E. Kenney and A. M. Broome, Mol. Pharm., 2015, 12, 3250 CrossRef CAS PubMed.
- K. R. Fontenot, B. G. Ongarora, L. E. LeBlanc, Z. Zhou, S. D. Jois and M. G. H. Vicente, J. Porphyrins Phthalocyanines, 2016, 20, 352 CrossRef CAS PubMed.
- S. Song, D. Liu, J. Peng, H. Deng, Y. Guo, L. X. Xu, A. D. Miller and Y. Xu, FASEB J., 2009, 23, 1396 CrossRef CAS PubMed.
- N. E. M. Kaufman, Q. Meng, K. E. Griffin, S. S. Singh, A. Dahal, Z. Zhou, F. R. Fronczek, J. M. Mathis, S. D. Jois and M. G. H. Vicente, J. Med. Chem., 2019, 62, 3323 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727 CrossRef CAS PubMed.
- E. Orbán, G. Mező, P. Schlage, G. Csík, Ž. Kulić, P. Ansorge, E. Fellinger, H. M. Möller and M. Manea, Amino Acids, 2011, 41, 469 CrossRef PubMed.
- A. Jimeno and M. Hidalgo, Crit. Rev. Oncol. Hematol., 2005, 53, 179 CrossRef PubMed.
- K. N. Enyedi, Sz. Tóth, G. Szakács and G. Mező, PLoS One, 2017, 12, e0178632 CrossRef PubMed.
- Ö. Topel, B. A. Çakır, L. Budama and N. Hoda, J. Mol. Liq., 2013, 177, 40 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Analytical chromatograms, mass spectra, DLS and critical aggregation concentration results of the conjugates; UV-Vis spectra of daunomycin. See DOI: 10.1039/d0sm00428f |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |