
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of salt concentration on the formation of Pickering emulsions
David J.
French
*a,
Jeff
Fowler
b,
Phil
Taylor
c and
Paul S.
Clegg
 a
a
aSchool of Physics & Astronomy, The University of Edinburgh, Peter Guthrie Tait Road, Edinburgh EH9 3FD, UK. E-mail: David.French@ed.ac.uk
bSyngenta Inc., 410 Swing Rd, P.O. Box 183000, Greensboro, North Carolina 27419-8300, USA
cFormulation Technology Group, Syngenta Crop Sciences, Jealotts Hill International Research Centre, UK
First published on 8th July 2020
Abstract
Here we study emulsification in a model experimental system comprised of water, an oil and colloidal particles. The particles are charge-stabilised colloidal silica; unsurprisingly, by varying the concentration of salt the degree of flocculation of the particles can be modified. The influence of salt on the formation of particle-stabilised oil droplets goes well beyond considerations of the colloidal stability of the particles. Our results demonstrate that the influence of salt on the particle–particle interaction is less important for emulsion formation than the influence of salt on both the particle wettability and the particle–interface interaction.
1 Introduction
In previous studies we have developed a model particle-stabilised oil-in-water emulsion in which we can control whether droplets are coated by a monolayer of colloidal particles or whether the particles are shared between two droplets (to form a particle bridge).1,2 One route to avoiding bridge formation is to add salt to the system. Here we deploy our well-controlled system to investigate the process of the formation of Pickering emulsions in the presence of salt but in the absence of colloidal bridges.Initially the presence of salt has a variety of effects prior to and during emulsification. The Debye length in the aqueous phase is reduced, meaning electrostatic interactions are more strongly screened, and both the viscosity and refractive index of the water are increased.3–5 Reducing the Debye length may lead to the particles aggregating through van der Waals attractions. The addition of salt to the aqueous phase also increases the rate of coalescence between oil droplets which are not fully coated with particles, as the oil droplets carry a slight charge.6 This is important, as a slower coalescence rate allows the particles more time to adsorb to an oil–water interface, yielding smaller and more stable droplets. Finally, if the particles carry the opposite sign of charge to the oil–water interfaces, then adsorption will be hindered by the presence of salt.
Once the emulsion has formed, salt has an influence on the final position of trapped particles as described by, θw, the three-phase contact angle.7 The change in free energy, ΔG, occurring when a particle moves from its preferred phase to the oil–water interface is given by:
ΔG = −πrp2σow(1 − |cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θw|)2, θw|)2, | (1) |
Gautier et al. studied the effects of changing both pH and the sodium chloride concentration of the aqueous phase in emulsions stabilised by silica and latex particles.11 They observed several interesting phenomena, including bidisperse droplet populations at intermediate salt concentrations and droplet surface coverages which were substantially below that of hexagonal close packing at high pH and low salt concentrations. Below we consider the role of salt on emulsion formation in isolation, we study a broader range of salt concentrations and we show the contrasting behaviour of sodium chloride and sodium iodide. We also employ fluorescently labelled silica particles and confocal microscopy which allows us to extend our work to higher particle volume fractions.
2 Materials and methods
2.1 Materials
Distilled water, deionised using a Millipore Milli-Q system to a resistivity ≥18 MΩ cm, was used throughout. Dodecane (Sigma-Aldrich, ≥99%) and isopropyl myristate (Sigma-Aldrich, ≥98%) formed the oil phase. The dodecane was filtered through an alumina column twice to remove polar impurities. Isopropyl myristate was used as received, as were sodium chloride (VWR, 99.9%) and sodium iodide (Sigma-Aldrich, 99.5%).2.2 Particle phase behaviour: sample preparation
Dried silica particles and salt were dispersed in 2.33 mL of water using ultrasound (Sonics Vibracell VCX500). A typical protocol was 1 s of sonication then 5 s rest, repeated for a total time of 6 min, at an amplitude of 20% of instrument maximum. A typical sample, with ϕp = 1.7% and [NaCl] = 100 mM contained 0.09 g of silica and 0.014 g of NaCl.
An ALV/CGS-3 goniometer and an ALV-5004 Multiple Tau Correlator were used to perform static light scattering measurements in order to measure the size of the Stöber silica particles. Calculation of rp was done by fitting the minima in the form factor to:
| qprp = tan(qprp), | (2) |
 | (3) |
 | (4) |
Particle dispersions with various aqueous phase concentrations of sodium chloride and sodium iodide were observed, with ϕp = 0.35%. The dried particles and salt were dispersed in 15 mL of deionised water using the ultrasonic probe.
2.3 Emulsion characterization
The samples were photographed regularly as the droplets creamed and any remaining particles sedimented. The droplet size distribution (measured using light scattering) was used to characterise the emulsion formation process; an increase in the droplet size indicates that more coalescence occurred during emulsification. If, for a given sample preparation route, the droplet size is small, then the emulsification process has been more efficient than if the same preparation route led to a large droplet size. To provide more insight, slides were prepared, with a drop of emulsion from each sample being diluted in some of the sample's continuous (aqueous) phase for optical and confocal microscopy.
The droplet size has been used to characterise emulsion stability; an increase in the droplet size indicates that more coalescence occurred during emulsification, either because the particle trapping energy is lower or because the particles are not able to adsorb to the interface during emulsification. If, for a given sample preparation route, the droplet size is small, then the emulsification process has been more efficient than if the same preparation route led to a large droplet size. Since optimising the emulsion system so that the emulsification process requires as little energy as possible is desirable, small droplet size can be used to indicate success.
3 Results
3.1 Characterization of particle flocculation
The sizes of particle aggregates are shown in Fig. 1(a) for sodium chloride and Fig. 1(b) for sodium iodide. When there is no added salt, the apparent particle size does not increase over the course of the experiment, which lasted 1 h, showing that the particles are indeed non-aggregating under these conditions (in fact, the particle size decreases slightly over the course of the experiment, as aggregates which were not initially fully dispersed are broken up). At non-zero salt concentrations, the apparent particle size increases relatively quickly over the first 5–10 min of the experiment, and then plateaus. The plateau value typically decreases as the salt concentration is increased, and this may be caused by the increased viscosity at higher salt concentrations leading to a greater shear stress in the stirred sample. At the highest sodium chloride concentration used here, 1000 mM, the apparent particle size starts to increase again after ≈30 min, suggesting that the particle–particle interactions at this salt concentration are strong enough to overcome the shear rate.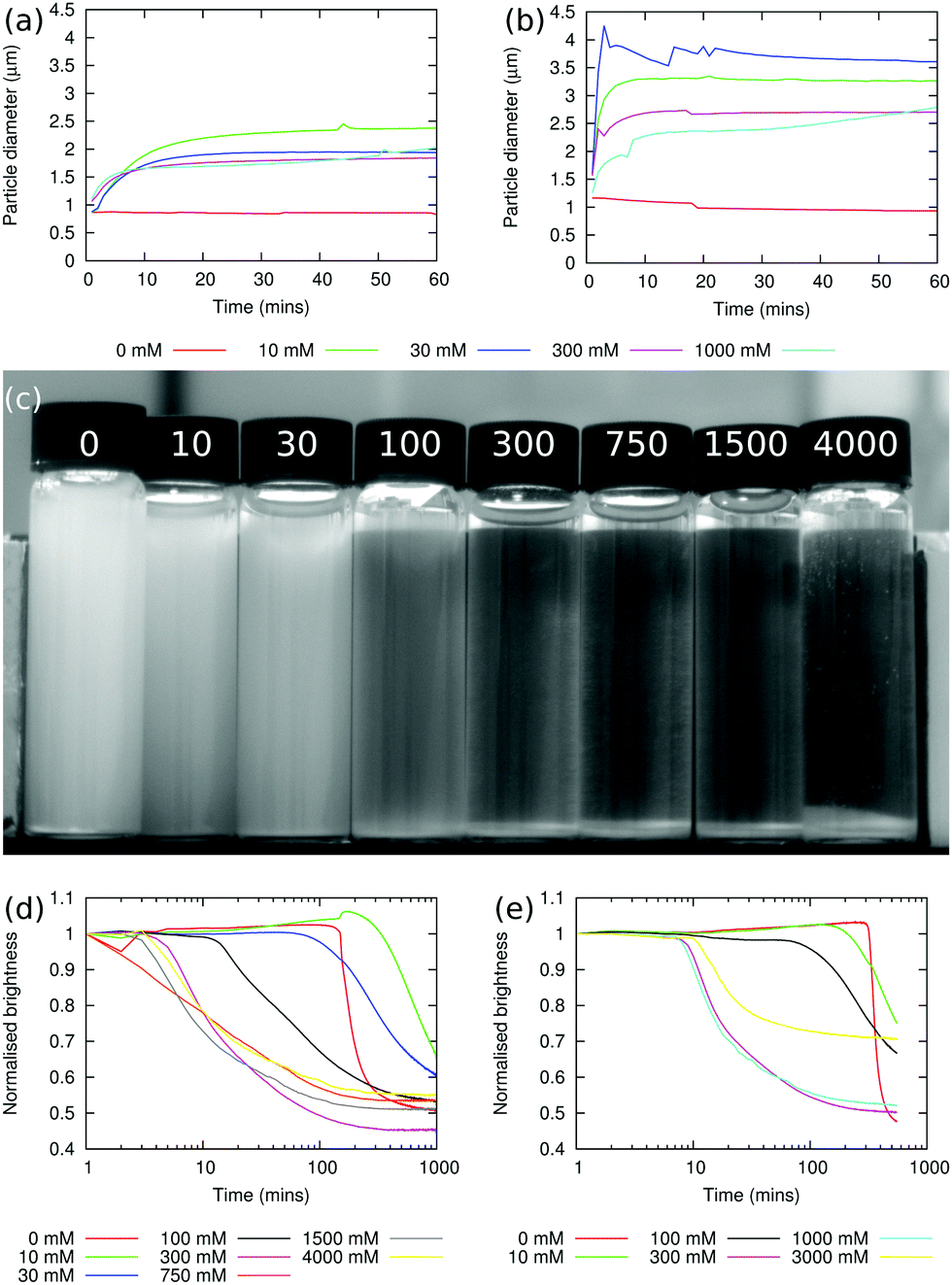 | ||
| Fig. 1 NaCl & NaI (a) and (b) Aggregation behaviour of dispersions of silica particles under shear at different sodium chloride (a) and sodium iodide (b) concentrations. (c) Photograph of Stöber silica dispersions after 120 min of sedimentation. The sodium chloride concentrations, in mM, are shown on each vial and ϕp = 0.35% in all samples. (d) and (e) Graphs showing the normalised brightness (a lower normalised brightness means that more particles have sedimented) as a function of time for the dispersions containing sodium chloride (d) and sodium iodide (e). | ||
Fig. 1(b) shows that the aggregation behaviour in the sodium iodide samples is qualitatively similar to the sodium chloride case. However, the rate of aggregation at short times is greater and the size of the aggregates in the plateau region is larger. Given the small difference between the viscosities of sodium chloride and sodium iodide solutions at concentrations below ≈500 mM,3,5 it seems more likely that the differences are due to ion specific effects. Since iodide ions are more chaotropic than chloride ions,18,19 the concentration of anions which adsorb at particle surface will be lower for iodide than for chloride, and this in turn means that the particle surface charge is likely to be higher for sodium chloride; by contrast, the degree of screening is likely to be higher for sodium iodide. The balance of these two effects will determine how strongly the particles aggregate.
When particle aggregation, due to the presence of salt, is superimposed on sedimentation, the interface between the sediment and the supernatant is no longer sharp. The average brightness, b(t), of each sample has been measured using the Plot Profile feature in ImageJ to select rectangular sections covering as much of each vial as possible. This is then repeated for multiple photographs taken at different times, with t being the time elapsed since the beginning of the experiment.21 In order to account for changing light conditions b(t) is also normalised by dividing by the average brightness of two sections of the photograph, one in the background and one in the foreground. A typical photograph from this experiment is shown in Fig. 1(c) (this photograph has been cropped such that the background and foreground sections used for normalisation aren't visible20). By plotting b(t)/b(t = 0) as a function of t, the sedimentation behaviour can be seen. Fig. 1(d) shows such a plot for dispersions containing sodium chloride. The sudden onset of sedimentation in the 0 mM sample can clearly be seen.
At higher salt concentrations, 10–30 mM, the dispersions are observed to sediment at approximately the same rate as in the sample without salt. However, in the presence of salt, the particles do not all aggregate rapidly after ≈5 h, but gradually aggregate and sediment over the course of ≈20 h. At still higher concentrations of sodium chloride, 300–4000 mM, aggregation occurs immediately and the particles sediment rapidly, with sedimentation taking less than an hour.
Fig. 1(e) shows the results of a similar experiment using sodium iodide. No qualitative differences between dispersions containing sodium chloride and sodium iodide were observable.
3.2 Characterization of droplet size
 | ||
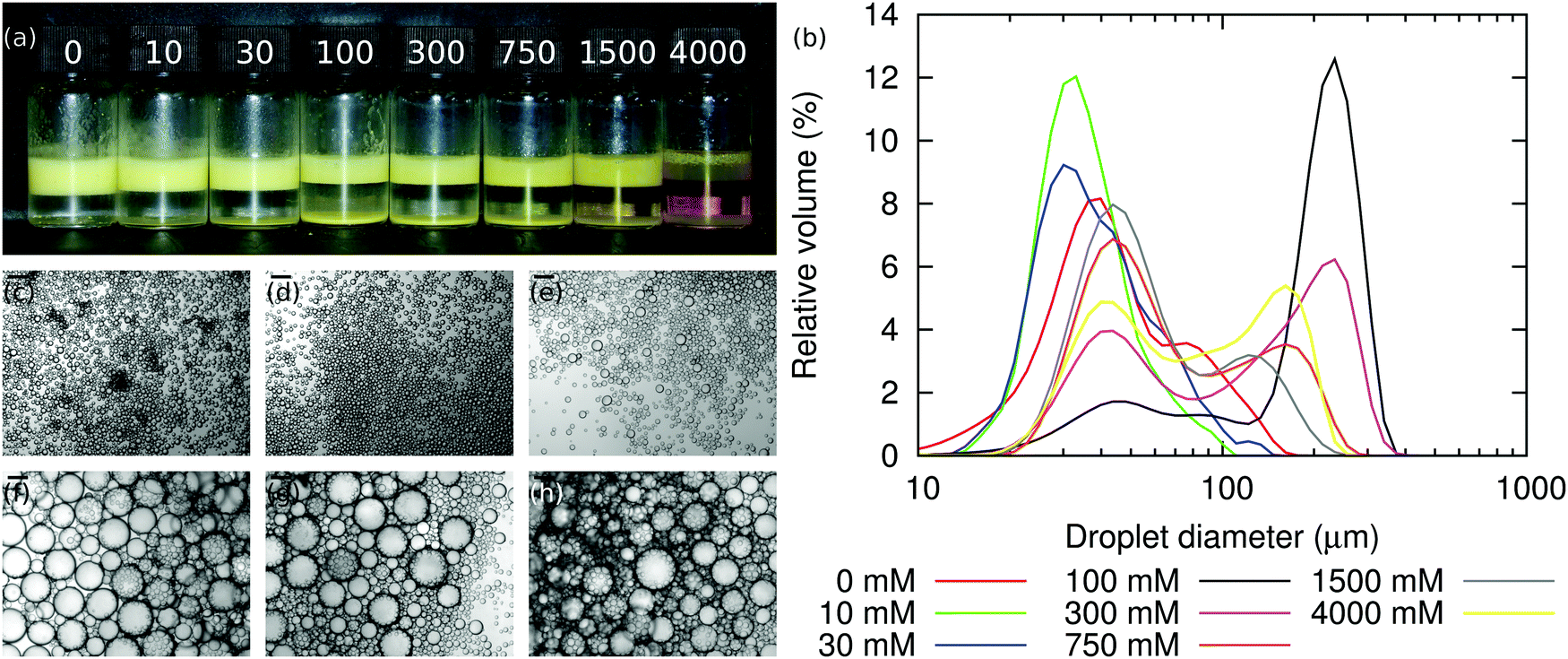
| Fig. 2 NaCl (a) Photograph showing emulsions stabilised by Stöber silica (yellow due to FITC dye) at various sodium chloride concentrations (on vial, mM). The amount of free silica at the base of the vial increases with salt concentration. (b) Size distributions of emulsions at various sodium chloride concentrations. In all samples, ϕp = 1.7%, ϕo = 20% and ΦIM = 10%. (c)–(h) Micrographs of emulsions at sodium chloride concentrations of 0 mM, 10 mM, 30 mM, 100 mM, 300 mM and 4000 mM. The scale bars are 100 μm. (i)–(k) Photographs of emulsions at salt concentrations shown on each vial, mM. In all three samples ϕp = 1.1%, ϕo = 20% and ΦIM = 0%. The times since emulsification are 0, 40 and 80 min. | ||
The size distributions of the emulsions in Fig. 2(a) are shown in Fig. 2(b), and Fig. 3 shows the average droplet size from these samples. The droplet size appears smallest when [NaCl] = 10 mM. However, this is because the droplets in the 0 mM sample are aggregating due to particle bridging.1 Hence as [NaCl] is increased from 0 mM to 1500 mM, the emulsion droplets get continuously larger, i.e., the particles are becoming increasingly ineffectual. This might be because the salt screens the particle-interface attraction during mixing, so that particles are less likely to adsorb to the interface and/or because the particle trapping energy has decreased.
 | ||
| Fig. 3 NaCl & NaI: volume-weighted mean droplet size of emulsions at different sodium chloride and sodium iodide concentrations. In all samples, ϕp = 1.7%, ϕo = 20% and ΦIM = 10%. | ||
Bright-field microscopy confirms the light-scattering data, see Fig. 2(b)–(h) and Fig. 3. The situation is slightly more complicated than Fig. 3 might initially suggest. At the lowest salt concentrations, ≤10 mM, the emulsion is relatively monodisperse. However, at a salt concentration of ≈30 mM, the emulsion becomes bidisperse, with two populations of droplets with mean diameters of ≈30 μm and 100 μm. At still-higher salt concentrations the size distribution becomes monodisperse again, with droplet diameters centered on ≈500 μm in the range 100–1500 mM, before becoming bidisperse again at the highest salt concentration studied here (4000 mM), where the droplet populations are centered around ≈80 μm and ≈500 μm.
Along with the changes in droplet size, an increase in free silica is observed when [NaCl] increases, as seen in Fig. 2(a) and (b). We attribute this to sodium chloride altering the contact angle, decreasing it initially. This means that particles are less likely to adsorb to the oil–water interface during emulsification (due to salt screening the particle-interface attraction) and that particles which do adsorb will be less strongly trapped at the oil–water interface (due to salt lowering θw). The weaker particle adsorption also means that as limited coalescence progresses, particles may be ejected from the interface either during the coalescence events themselves, or soon afterwards as jammed particles at the interface of a non-spherical droplet relax.
 | ||
| Fig. 4 NaCl (a) Confocal micrograph z-projection of 14 images, with a z-spacing of 0.44 μm (ImageJ21) showing the particle layers at two droplet interfaces. [NaCl] = 10 mM; the scale bar is 5 μm. (b) Confocal micrograph z-projection of 42 images, with a z-spacing of 0.44 μm showing the packing of particles at the interface. [NaCl] = 30 mM; the scale bar is 20 μm. (c) Confocal micrograph showing the thickened particle layer at the interface in the 100 mM sample. The scale bar is 20 μm. (d) Micrograph showing the thickening of the interfacial particle layer. The arrows indicate the thickened particle layer, which protrudes several micrometres into the continuous phase and the circled area shows that the layer can be shared by droplets. [NaCl] = 300 mM; the scale bar is 10 μm. (e) Cartoon summarizing the behaviour of droplets as the salt concentration is increased, see text. | ||
We also note that at [NaCl] = 30 mM, Fig. 2(a), not all of the particles adsorb to an interface, and a significant fraction of the particles sediment in the aqueous phase. Nonetheless, some droplets are left with large bare patches on their surfaces and are yet stable against further coalescence. Given the salt concentration in this sample, it seems unlikely that charged interfaces could be responsible for stabilizing the droplets. Alternatively, the particle attractions might be strong enough that particles on opposing droplets migrate on their respective interfaces so that they are closer together as droplets come into contact.
Overall, as [NaCl] is increased, the droplets change from being fully coated by particles to being partially coated by aggregated particles (see also ref. 22), and then to being fully coated by particles, but with particle layers which extend into the continuous phase, see cartoon in Fig. 4(e). The change from being fully coated to being partially coated appears to coincide with the change from a non-flocculating dispersion to a weakly flocculating dispersion, where the emulsification process is still capable of breaking up the particle aggregates but where screening of the particles’ charge leads to lowered particle adsorption rates and particle trapping energies. The appearance of bidisperse emulsions may be related to this; small droplets could be stabilised by dispersed particles whilst the larger droplets are stabilised by clusters of particles. The transition to a system where the particles in the continuous phase form a network linking droplets together may then occur at the point where the emulsification process is no longer capable of fully disrupting particle aggregates. The increase in particle–particle attraction on droplet interfaces as [NaCl] is increased may be responsible for an enhanced coalescence rate at higher salt concentrations leading back to fully coated droplets.
As well as the thickening of the particle layer at the interface, the amount of sedimented silica increases with [NaCl], see Fig. 2(a). Together, these contribute to the increased droplet size. The increase in the packing fraction of the emulsions as the salt concentration increases from 0 mM to 30 mM is evidence of particle bridging in the low salt concentration samples.1 The resulting dependence of droplet size on [NaCl] is non-monotonic, Fig. 5, independent of the availability of particles.
 | ||
| Fig. 5 NaCl volume-weighted median droplet size at different [NaCl] and particle volume fractions, ϕp. Measurements were made using light scattering, except for the lower two ϕp at [NaCl] = 250 mM, which were estimated from photographs. All samples had ΦIM = 0% and were emulsified using a vortex mixer for 30 s each. | ||
 | ||
| Fig. 6 NaI (a) Photograph showing emulsions stabilised by Stöber silica at [NaI] given in mM on each vial. The amount of free silica can clearly be seen to increase with salt concentration. (b) Size distributions of emulsions stabilised by Stöber silica at various [NaI]. (c)–(h) Micrographs of emulsions stabilised by Stöber silica at [NaI] of 0 mM, 10 mM and 30 mM, 100 mM, 300 mM and 1500 mM. The scale bars are 100 μm. | ||
The dramatic difference in the size of the droplets in emulsions which contain the same molarity of sodium chloride and sodium iodide is initially surprising. As was shown above, the particle dispersions containing sodium iodide aggregate more strongly than dispersions containing an equal molarity of sodium chloride, especially when sheared, an effect we attribute to iodide ions being more chaotropic than chloride ions. Particle adsorption rates have been shown to change with particle size, although the relevance of these results to high Reynolds number systems remains to be demonstrated.7,23 Since the coalescence rate also increases as salt is added, a small difference in the particle adsorption rate may make a relatively large difference to the eventual droplet size.
 | ||
| Fig. 7 NaI (a) A z-projection of 17 confocal micrographs, with a z-spacing of 0.44 μm, constructed using ImageJ with [NaI] = 30 mM.21 (b)–(e) Confocal micrographs showing surface coverage of emulsion droplets stabilised by Stöber silica, with [NaI] of 100 mM, 300 mM, 750 mM and 1500 mM, respectively. The thickening of the particle layer at the droplets' interface as the salt concentration is increased can clearly be seen. The scale bars are 20 μm. | ||
Unlike the sodium chloride case, partially coated droplets are not observed in any of the sodium iodide samples. This is surprising because the particles which remain in the continuous phase form an open and branched network. This may be because θw is high enough that the adsorbed particles repel due to electrostatic forces mediated through the oil phase.26,27 Since not all of the particles adsorb to an oil–water interface, but the droplets become fully covered by particles, the increase in droplet size observed at intermediate [NaI] can be attributed to the increased coalescence rate between droplets, which is caused by the increased screening of electrostatic charge present at the droplet interfaces. Alternatively, as discussed below, the internal structure of particle flocculations formed in NaI may be rather different than those formed in weakly flocculating, intermediate [NaCl], and this could also explain the observed differences in the extents to which droplets are covered.
4 Discussion
4.1 Particle flocculation
The observations presented are roughly in-line with expectations, but for these to illuminate emulsion formation they must be put alongside two other points. (1) The degree of aggregation in a particle dispersion will depend not only on the particle interactions, and hence the influence of salt, but also on the shear stress in the fluid. Consequently, an important factor will be whether the applied shear is capable of breaking up the particle aggregates. In this context, note the upward trend in the aggregate size at experimental times greater than 30 min and when the salt concentration is 1 M, as shown in Fig. 1(a) and (b). In contrast, at salt concentrations ≤300 mM the aggregate size is observed to plateau at a salt concentration-dependent value. Therefore, it is argued that at salt concentrations ≤300 mM the particle dispersions can be described as weakly flocculating. The aggregation state following shear emulsification and adsorption to an interface may only indirectly reflect the aggregation state observed during low shear sedimentation, but the latter observations still are informative about interparticle forces. (2) The particles initially have a positive zeta potential, but this decreases and eventually becomes negative due to water adsorption at their surfaces.14,28 It is very likely that this effect is too slow to be important here.4.2 Droplet size
Oil-in-water Pickering emulsions stabilised by fluorescent Stöber silica have been produced. The most stable emulsions are formed when the aqueous phase contains no salt and the least stable emulsions are formed at [NaCl] ≈ 1000 mM; above this concentration the emulsion stability increases. A summary of this behaviour can be seen in Fig. 2(i)–(k), which show emulsions similar to those in Fig. 2(a), but with ϕp = 1.1% and ΦIM = 0%. Samples containing sodium iodide are observed to be more stable than those containing an equal concentration of sodium chloride, despite sodium iodide causing the particle dispersions to flocculate more strongly. This is attributed to the iodide ions being more chaotropic than chloride ions, which causes the particles to be more hydrophobic and hence more likely to be trapped at a droplet interface. An alternative explanation may lie in the expected differences in internal structure between weakly- and strongly-bonded flocculations, as reported by Stradner et al.,29 that reasonably might be expected to influence how strongly they adsorb to interfaces, and their propensity to be removed by shear during emulsification. The relevance of these ideas to colloids at interfaces is not at all clear.It is valuable to focus on the measured droplet size, Fig. 5, as a function of ϕp for emulsions generated from non-flocculating, weakly flocculating and strongly flocculating particle dispersions. It can be seen that, for all particle volume fractions, the droplet size is much greater for emulsions created using weakly flocculating particle dispersions than for corresponding emulsions created from non-flocculating and strongly flocculating dispersions. i.e. when [NaCl] = 250 mM only a small proportion of the particles have become trapped at the interface. The three [NaCl] shown (0 mM, 250 mM and 4000 mM) correspond to non-flocculating, weakly flocculating and strongly flocculating particle dispersions, as evidenced by the rates at which the free silica sediments (Fig. 2(i)–(k)). Hence, this system behaves in the opposite manner from that described by, for example, Binks and Lumsdon.30 This discrepancy is likely caused by the differing surface chemistry of the silica particles used here, which have non-native amino groups and fluorescent dye at their surface. Since the fluorescent silica particles are positively charged, there is an attractive interaction between the particles and the oil–water interface. Therefore the addition of salt screens an attractive interaction, making particle adsorption less likely.
5 Conclusions
When Stöber silica is used as the emulsion stabiliser, aqueous phases containing sodium iodide are observed to lead to more stable (having a lower primary droplet size) emulsions than aqueous phases containing sodium chloride. The adsorption of ions at the particle interface is thought to play a rôle in this. For both salts, weakly flocculating particles have been shown to lower the emulsion's stability significantly. This demonstrates that the particle–particle interactions are less important than the particle–interface interactions and, whilst aggregating particles may be an indication that the particles will be attracted to the interface, there is a wealth of other aspects at play here which must be considered.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank A. B. Schofield for synthesising the particles, the EPSRC and Syngenta for a CASE award to D. J. F., and the EPSRC for grant EP/J007404/1.References
- D. J. French, P. Taylor, J. Fowler and P. S. Clegg, J. Colloid Interface Sci., 2015, 441, 30–38 CrossRef CAS PubMed.
- D. J. French, A. T. Brown, A. B. Schofield, J. Fowler, P. Taylor and P. S. Clegg, Sci. Rep., 2016, 6, 31404 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 82nd edn, 2001 Search PubMed.
- K. Bai and J. Katz, Exp. Fluids, 2014, 55, 1704 CrossRef.
- I. M. Abdulagatov, A. B. Zeinalova and N. D. Azizov, J. Chem. Eng. Data, 2006, 51, 1645–1659 CrossRef CAS.
- K. G. Marinova, R. G. Alargova, N. D. Denkov, O. D. Velev, D. N. Petsev, I. B. Ivanov and R. P. Borwankar, Langmuir, 1996, 12, 2045–2051 CrossRef CAS.
- Colloidal Particles at Liquid Interfaces, ed. B. P. Binks and T. S. Horozov, Cambridge University Press, Cambridge, 2006 Search PubMed.
- J. H. Clint and S. E. Taylor, Colloids Surf., 1992, 65, 61–67 CrossRef CAS.
- L. G. J. Fokkink and J. Ralston, Colloids Surf., 1989, 36, 69–76 CrossRef CAS.
- R. J. Hunter, Foundations of Colloid Science, Clarendon Press, Oxford, 1991, vol. 1 Search PubMed.
- F. Gautier, M. Destribats, R. Perrier-Cornet, J.-F. Dechézelles, J. Giermanska, V. H. S. Ravaine, F. Leal-Calderon and V. Schmitt, Phys. Chem. Chem. Phys., 2007, 9, 6455–6462 RSC.
- A. Imhof, M. Megens, J. J. Engelberts, D. T. N. de Lang, R. Sprik and W. L. Vos, J. Phys. Chem. B, 1999, 103, 1408–1415 CrossRef CAS.
- K. A. White, A. B. Schofield, B. P. Binks and P. S. Clegg, J. Phys.: Condens. Matter, 2008, 20, 494223 CrossRef.
- K. A. White, A. B. Schofield, P. Wormald, J. W. Tavacoli, B. P. Binks and P. S. Clegg, J. Colloid Interface Sci., 2011, 359, 126–135 CrossRef CAS PubMed.
- P. Pusey and W. van Megen, J. Chem. Phys., 1984, 80, 3513–3520 CrossRef CAS.
- H. Lamb, Hydrodynamics, Dover Publications, London, 1932 Search PubMed.
- W. J. Frith, R. Pichot, M. Kirkland and B. Wolf, Ind. Eng. Chem. Res., 2008, 47, 6434–6444 CrossRef CAS.
- K. D. Collins, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5553–5557 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- D. J. French, PhD thesis, University of Edinburgh, 2015.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- M. Derakhshandeh, B. K. Pilapil, B. Workman, M. Trifkovic and S. L. Bryant, Soft Matter, 2018, 14, 4268–4277 RSC.
- D. Reay and G. A. Ratcliff, Can. J. Chem. Eng., 1973, 51, 178–185 CrossRef CAS.
- N. Yan and J. H. Masliyah, Ind. Eng. Chem. Res., 1997, 36, 1122–1129 CrossRef CAS.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100-102, 503–546 CrossRef CAS.
- T. S. Horozov, R. Aveyard, B. P. Binks and J. H. Clint, Langmuir, 2005, 21, 7405–7412 CrossRef CAS PubMed.
- A. D. Law, M. Auriol, D. Smith, T. S. Horozov and D. M. A. Buzza, Phys. Rev. Lett., 2013, 110, 138301 CrossRef PubMed.
- L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
- A. Stradner, H. Sedgwick, F. Cardinaux, W. C. K. Poon, S. U. Egelhaaf and P. Schurtenberger, Nature, 2004, 432, 492–495 CrossRef CAS PubMed.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 2539–2547 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |