
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the effect of particle surface chemistry in film stratification and morphology regulation†
Archana
Samanta
 and
Romain
Bordes
*
and
Romain
Bordes
*
Department of Chemistry and Chemical Engineering, Applied Chemistry, Chalmers University of Technology, Sweden. E-mail: bordes@chalmers.se
First published on 17th June 2020
Abstract
Combinations of colloids and binders are often used to formulate functional coatings. In these mixtures, competition between particle migration, polymer chain diffusion, evaporation and sedimentation affects their respective spatial location and therefore can govern the surface features. In addition to this, the surface chemistry of the nanoparticles (NPs) and the resulting interparticle interactions can play a significant role in dictating the morphology and the properties of resultant films. Hence it would be possible to tune the surface and bulk topology of the films by controlling these parameters. A combination of various acrylic binders with two types of silica sols, bare (BSiO2) and modified silica (MSiO2), differing in their ability to gel, were formulated and dried under controlled conditions. Factors influencing the mobility and migration of binder and silica particles were evaluated with respect to particle concentration and drying rate. MSiO2 films showed prominent pores with gradual increase in Si% across the cross-section of the films, whereas, BSiO2 films had no pores and showed a uniform Si content across the cross-section of the films. This difference is explained by the variation in gelation between BSiO2 compared to MSiO2, that hindered the NPs migration and affects the infiltration and stratification process. This study paves a path forward to achieve desired surface and bulk porosity from colloidal silica coatings by effective control of chemistry of particles along with process parameters.
Introduction
Complex colloidal coatings are widely used for fabrication of functionalized surfaces and interfaces, as a thin layer of these deposited particles has the advantage of significantly altering the surface properties of the material while keeping the bulk properties intact. Properties such as surface hardness, corrosion resistance, self-cleaning, super-hydrophobicity, thermal insulation, and others1 are commonly achieved through this approach. For example, particles of titanium dioxide are used for imparting photoelectric properties2,3 graphene oxide for conductivity,4–6 clay based coatings for thermal insulation,7 iron oxide for magnetic properties,8 chitosan for antibacterial properties,9,10 silver for conductivity11etc.The colloidal particles are however not used alone, as their poor physical adherence to the substrate would not grant durability to the coating and are therefore often combined with other polymers or binders. In the case of water-based formulations, a latex is often used12 and these dispersions are then applied to the substrate of interest, most often via spray coating, spin coating or dip coating.13 It has therefore become important to understand the governing parameters affecting the film forming tendency of such combination of particles, and the mechanism by which particles migrate towards the air-exposed surface, and eventually undergo a film formation process.
Prior research in this field of film formation process has primarily focused on understanding the drying process of complex latex systems. Keddie et al. for example, have demonstrated that non-film forming particles in a latex system cannot deform readily and hence accumulate to generate a rough surface compared to a dense surface morphology of a film forming latex.14 Trueman et al. have studied the stratification behavior i.e., the spatial arrangement of particles from the air surface of the film in the case of bimodal latex particles system with glass transition and film forming temperatures above room temperature. It was observed that in these systems, low initial volume fraction and a low concentration of the particles favored stratification i.e. gradual arrangement of constituent particles across the cross-section of the film.15 Keddie et al. has demonstrated that in concentrated regime, stratification was favored when the size ratio of the particles was at least 7.16 Furthermore, the experimental results were correlated with simulation and modeling of stratified films and it was concluded that higher particle size ratio favors stratification in dilute colloidal dispersions.17 Similarly, Zhou et al. has showed that stratification was favored by high particle size ratio and high Péclet number,18i.e. the ratio between mass transport by convection flow and diffusion of the particles.
Conventionally, in a binary mix of colloidal systems, the large particles are expected to be deposited at the top surface because of their lower diffusion constant and large Péclet number.19 Later, the concept of surface induced depletion was introduced in the previous models by Fortini and Sear to predict the formation of small particles enriched interface at the top of the film.16,20 Sear and Warren have reported that the drying of film in a mixture of colloids and polymers can create diffusiophoretic gradient of particle arrangement across the cross-section of the films. They define diffusiophoresis as the motion of one species in response to a gradient in the concentration of another. They observed stratification by detecting a layer of small polymer molecules on top of a layer of larger colloid particles. They also accounted for the solvent flow in their model which helped them to achieve a small on-top type of stratification.21,22
Cheng et al. varied the Péclet number and volume fraction of the particles in the system and by doing so they could achieve small-on-top or large-on-top stratification by involving the solvent explicitly.23 Yuri and Yurko used Monte Carlo simulation to understand stratification of hard colloid spheres by establishing a correlation between the evaporation rate to the arrangement of particles. They have observed that stratification is favored at lower evaporation rate,24i.e. when sufficient time is available for the particle arrangement. Later, Panagiotopoulos et al. have also observed small-on-top stratification from implicit-solvent molecular dynamic simulations under conditions similar to Fortini et al. They indicated that this type of stratification could occur under fast or moderate evaporation rate where the chemical potential gradient of particles supersedes the counteraction from the friction on the migration velocities of the particles.25
Francis et al. have reported stratification of small particles on top of the film with the larger particles at the bottom. They showed that stratification of smaller particles on top of air interface is favored if the smaller particles have repulsive forces which can help them pass through the contours of the large particles.26 Routh et al. went further and proposed a mechanism of stratification that takes into consideration the interparticle interaction. They stated that in a binary colloidal mixture if the big particles attract the big or small particles, the surface would be dominated by the presence of big particles. In the reverse scenario if the big particles repel the big and small particles, the film surface would be dominated by smaller particles which can accumulate together. In case where both big and small particles are attractive, both particles compete equally to diffuse towards the top surface of the film. In this case a marginal dominance of small particles could be observed on the film surface.27 Also, it was observed that increase in interactions between particles of same Péclet number can increase the segregation tendencies of the system whereas increase in Péclet number can desegregate them. This signifies that the film formation process involving particle migration is an intricate process, controlled by several factors including the particle dimensions, interactions, solvent system, processing parameters and others.28
Most of these studies focused on binary mixtures of latex dispersions with film forming temperature above room temperature which enabled to retain their original particle dimensions during the film forming process. In real scenario, most latex systems have a film forming temperature below room temperature to enable film formation without application of external heat. The situation is further intricated when the formulation is constituted of a complex mixture of latex with inorganic nanoparticles. In such cases besides the glass transition temperature of the latex particles, size ratios and interparticle interactions play an important role in the understanding of the film forming process, especially when the film forming process is conducted from concentrated suspensions. In this case the surface chemistry of the nanoparticle will be of central importance as it will directly influence the particles mobility during the drying process.
In this study an attempt was made to understand the silica and binder particle migration during the non-equilibrium film forming process, with focus on the effect of the particle surface chemistry. For this purpose, we have used a combination of acrylic binder/latex and silica nanoparticles formulations. Silica nanoparticles can easily be surface modified to achieve various properties. Two types of surface chemistries have been considered; bare silica referred to as BSiO2 and bare silica surface modified with 3-glycidoxypropyltriethoxy silane referred to as MSiO2. This later surface modification imparts a resistance to gelling to the silica sol against e.g. salt, as illustrated Fig. 1. Factors governing the mobility and passage of binder and silica particles were evaluated with respect to formulation concentration and drying rate.
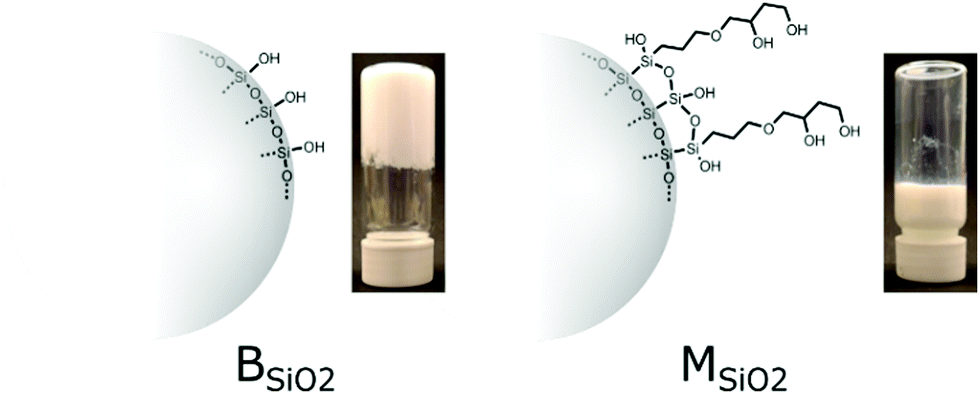 | ||
| Fig. 1 Surface chemistry of bare (BSiO2) and modified silica (MSiO2) and their respective gelation tendencies in presence of acrylic binder B1 (20 wt% silica, 5 wt% solid content). Gelation is induced by adding 0.1 g of potassium chloride to each one of 10 ml of 20 wt% of silica formulations with a total of 5 wt% solid content. | ||
Experimental
Materials
Sols of bare silica (BSiO2) and modified silica (MSiO2) i.e. silica nanoparticles modified with 3-glycidoxypropyltriethoxy silane (1 μmol m−2) were received from Nouryon, Bohus, Sweden. The corresponding chemical structures are shown in Fig. 1. AC2007 and AC2003, acrylic binders were procured from Alberdingk, Sweden, acrylic binder Primis AF1000 which contained acrylic coated silica binders along with some pre-dispersed silica particles was procured from Wacker chemicals, Germany, KCl was procured Merck, Sweden and Milli-Q water was used for preparation of the formulations.Methods
Results and discussions
Acrylate binders, bare and modified silica nanoparticles (BSiO2 and MSiO2) were selected for this study. The surface chemistry of silica nanoparticles is illustrated Fig. 1. The modified silica had a glycidoxypropyl moiety attached through silylation that spontaneously hydrolyses to form a diol. This surface functionalization provides a capping layer that increases the colloidal stability against salt, as compared to unmodified silica. This is demonstrated by adding 0.1 g of KCl to 10 ml of 20 wt% of silica formulations with a total of 5 wt% solid content. BSiO2 completely solidifies in 60 minutes whereas the MSiO2 is stable for over 30 days as shown in Fig. 1.Further gelation tests with higher binder content showed similar trend (see Fig. 1S, ESI†). The faster gelation of bare silica occurred due to formation of siloxane bonds at pH 8.5 which is prevented by the surface modification. KCl promoted this phenomenon by reducing the electrostatic repulsion between the silica nanoparticles. In that sense, the diol coverage also reduces binder–silica interactions.
Silica sols and binder particles were characterized by dynamic light scattering and electrophoresis measurements; the corresponding values are noted in Table 1. The film forming and the glass transition temperatures of the binders, which are crucial for determining surface features of the films are also listed in Table 1.
| Sample code | Sample | Diameter (nm) | Zeta potential (mV) | T g* (°C) | Minimum film forming temperature (°C) |
|---|---|---|---|---|---|
| B1 | AC2007 binder | 110 ± 9 | −65.7 | 5 | 0 |
| B2 | AF1000 binder | 90 ± 5 | −55.6 | 22 | 9 |
| B3 | AC2003 binder | 120 ± 12 | −74.3 | 15 | 3 |
| BSiO2 | Bare silica | 12 ± 7 | −26.5 | NA | NA |
| MSiO2 | Modified silica | 13 ± 5 | −25.8 | NA | NA |
The morphology of the casted films was studied using scanning electron microscopy. Starting with a dilute formulation with a total solid content of 5 wt%, the silica content was gradually increased and the change in morphology of the films dried at room temperature i.e., 20 °C, 55% relative humidity was studied. The pH of the formulations was 8.5.
Effect of surface chemistry of silica on film forming behavior
Binary mixtures of BSiO2 and MSiO2 with acrylic binders B1, B2 and B3 were formulated. Starting with B1–MSiO2 formulations, the dispersions had 5 wt% of total solid content and of this 5 wt%, 10–90% in mass was silica. As can been in Fig. 2, the morphology of the films changed considerably with an increase in silica content. The surface topology of the sample with 10% silica (rest being binder, on a dry mass basis) was rather plain and flat, depicting encapsulation of silica particles within the matrix of the polymeric binder. This could be anticipated as the size of binder particles is 7 times the size of silica particles and the weight content of binder particles is high i.e., 90% compared to the silica content. | ||
| Fig. 2 Surface morphology of B1–MSiO2 films with a total concentration of 5 wt%, dried at room temperature. The % value indicates the silica mass fraction of the dried samples. The white bar indicates a scale of 100 nm. | ||
Also, the binder particles would tend to fuse to form a continuous film as the film forming temperature was below room temperature, i.e. below 20 °C. With an increase in silica content to 20%, spherical binder particles are assumed to be surrounded by strong silica networks as represented by dark circular zones in SEM images. The presence of silica at the grain boundaries of latex was also observed by Shiraz et al.29
The diameter of these circular zones is in correspondence to the diameter of binder i.e. 109 ± 9 nm, Fig. 2. Bare silica nanoparticles can form a network of interconnected particles when percolating, i.e. during water evaporation.30 This network is expected to encapsulate the binder particles, and this feature can become prevalent with increase in silica content. At 30% of silica, prominent pores are observed on the surface.
The pore formation relies on a mechanism based on polymer infiltration. Infiltration is the tendency of the polymer chains to move out and spread due to polymer chain mobility. This infiltration can occur as the film forming temperature (20 °C) ≫ Tg of binder (5 °C). During the drying process, smaller silica particles tend to surround the bigger latex particles and organize themselves into an interconnected network. Removal of solvent from the interstices of this silica network can create free spaces. The polymer chains of the binder can infiltrate through these interstices, leaving behind pores in their initial sites of positioning. This phenomenon of pore formation occurring due to polymer infiltration in dried films was also studied in detail by You et al. with latex particles having a Tg of 26 °C.31 This tendency of polymer infiltration of binders is visible above 10% silica, where the polymer domains are observed to be channeling across the cross-section of the films, as shown in Fig. 3a (sample labelled as *20 represents cross-section of film with 20% silica from a 5 wt% total solid content). With further increase in silica content to 40% the porous morphology resulting from binder infiltration became dominant. However above 60% of silica, fewer pores were observed.
 | ||
| Fig. 3 Cryo fractured cross-sectional morphology of B1–MSiO2 films with a total concentration of 5 wt%, dried at room temperature. The % value indicates the silica mass fractions. Sample labelled as *20 wt% shows lateral movement of binder particles across the cross section of the film potentially indicating polymer infiltration. The white bar indicates a scale of 100 nm, unless otherwise stated. | ||
At this point the number of silica particles are over 198 times the number of binder particles (Table 1S, ESI†) and theoretically at this concentration all the binder particles should be fully surrounded by silica particles.
With further increase in silica content the number of silica particles increased significantly compared to the number of binder particles. When reaching a ratio at which only a small quantity of binder particles was present, the binder particle could be considered to be distributed in a bed of silica matrix. Hence the number of pores resulting from infiltrated binders was reduced significantly. And this trend continued with increase in silica content to 90% where the morphology is similar to that of an entirely silica framework observed as a continuous silica film.
Cryo fractured cross-section of the B1–MSiO2 samples were analyzed by SEM as shown in Fig. 3. Large pores were found throughout the bulk of the film, thus supporting the hypothesis of pore formation through polymer infiltration. The number of pores increased with an increase in silica content but with very high content of silica, i.e. above 60%, the numbers of pores gradually reduced. Very high silica concentrations can form early assembly of silica particles which can encapsulated the binder moieties and lower the free interstices through which the infiltration can occur. The surface and cross-section SEM images showed that simultaneous assembly of silica and polymer infiltration controlled and regulated the morphology of the composite films with similar trend of pore developments.
In a similar way, BSiO2 with similar particle size as MSiO2 was used and the formulations with binder B1 were casted as previously described. The surface morphology of these samples is shown in Fig. 4. The trend of surface morphology with increase in silica content at 5 wt% of total solid content resembles that of B1–MSiO2 silica samples, i.e. the surface pores became progressively prominent and then their number decreased above a silica content of 60%. The cryo-fractured cross-sections of these samples were also observed by SEM and are shown in Fig. 5. Unlike the B1–MSiO2 sample, no pores were visible throughout the sample cross-section.
 | ||
| Fig. 4 Surface morphology of B1–BSiO2 films with total solid content of 5 wt%, dried at room temperature. The % value indicates the silica mass fractions. The white bar indicates a scale of 100 nm. | ||
 | ||
| Fig. 5 Cross-section morphology of B1–BSiO2 films with total solid content of 5 wt%, dried at room temperature. The % value indicates the silica mass fractions. The white bar indicates a scale of 100 nm. | ||
The reason can be related to the gelling behavior of BSiO2 sol as compared to MSiO2, even in the presence of binder, which results in faster solidification and rapid structure formation, see Fig. 6b and c. This is particularly important during the drying of the film as the water evaporation tends to increase the ionic strength. Unmodified bare silica is prone to salt induced gelling as compared to modified silica, and hence will gel prematurely. Such gelling can result in early agglomeration of silica particles by lowering the interstitial spaces among them through which the polymer can infiltrate into, thus blocking the mechanism of pore formation. Hence the cross-section did not show any prominent pores.
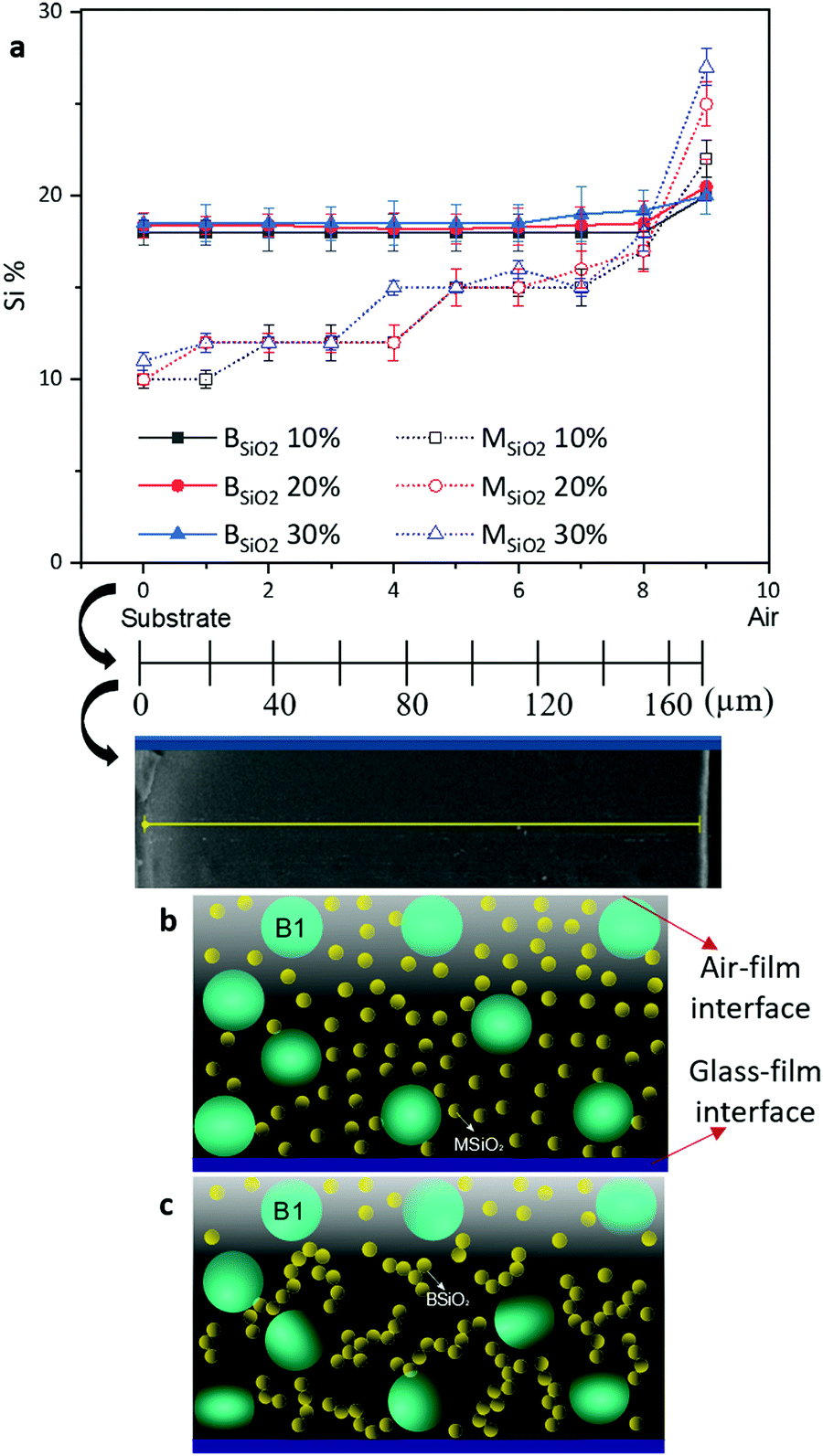 | ||
| Fig. 6 (a) Line scan EDS analysis of cross-sections of B1–MSiO2 and B1–BSiO2 films with a total solid content of 5 wt%, with various silica %. On the x-axis, the origin is located at the substrate surface. (b) Schematic describing the spatial loci of silica particles in B1–MSiO2 and (c) B1–BSiO2 formulation during the film formation process. | ||
The cross-sectional silica morphology confirmed the hypothesis that the gelling of silica can prevent particles mobility during the film formation process. As the binder particles had a film forming temperature below room temperature and a Tg of 5 °C, leading to that the binder particles were present in form of a continuous film and could not retain their original spherical shape in the films, which rendered their localization impossible. The small size of the silica particles used (13 ± 5 nm) also constituted a challenge to accurately localize the silica using SEM. Line scan EDS analysis of the cross-sections was therefore performed using silicon abundance to study the spatial arrangement of the silica nanoparticles across the film. The results are shown in Fig. 6a.
As can be observed from the EDS analysis of B1–MSiO2 samples, the silicon (Si) content increased from the glass surface towards the air film interface, i.e. towards the outer surface in all samples, irrespective of the amount of silica present in various formulated films. However, in the case of B1–BSiO2, the silica content was almost constant across the cross-section of the films, except of a slight increase close to the surface exposed to air.
This difference in silica migration supports the fact that silica nanoparticle network formation plays a central role in the development of film morphology.
In fact, the observations can be explained by the following theoretical framework. In the film forming process, evaporation of the solvent and diffusion of particles are the two main processes affecting the particle migration tendency. This is usually described by the Péclet number (Pé), which is a dimensionless value coming from the ratio between the mass transfer due to convection flow and the mass transfer resulting from the diffusion of the particles. The characteristic time of diffusion (td) for a particle during film formation process is proportional to the square of the height (H) of the film and inversely related to the diffusion coefficient (D): td = H2/D. The time for the liquid in the film to evaporate (te) is proportional to the initial film thickness (H) and inversely related to the evaporation rate (E): te = H/E. Pé helps to determine the dominant process (evaporation or diffusion) during the film formation process.17,32,33 The diffusion coefficient (D) of a particle is given by D = kBT/(6ηπR), where kB is the Boltzmann constant, T is the absolute temperature, R is the radius of the particle and η is the viscosity of the solvent. Hence the Péclet number can be depicted as expressed in eqn (1).
| Pé = td/te = HE/D = (6ηπRHE)/kBT | (1) |
In our systems water was used as a dispersant with η value of 1 × 10−3 Pa s at room temperature. The value of E is reported as E = 1.1 × 10−7 ms−1 at room temperature conditions of evaporation by Utgenannt et al.34 So, the calculated Péclet number values for silica and the binder particles are 7.5 and 51, respectively, at room temperature. The reason for the accumulation of small particles at the top in binary mixture of particles with Péclet number greater than 1 is explained in detail by Sear et al.20 According to eqn (1), a system with Pé ≫ 1, signifies that evaporation rate is dominant over diffusion.15,16 Pé ≫ 1 for both particles would result in competition between them to accumulate at the free surface of the descending interface due to the faster evaporation compared to diffusion.35–40 As the interface goes down during the drying process, both the smaller silica and the larger binder particles tend to accumulate below this moving interface and this accumulation region forms and grows with time, provided that the particles can move freely. This is a region where the density of both particles is highest at the air–film interface and there is a density gradient below this accumulation interface towards the glass surface. Some of the silica and binder particles are expected to get trapped at this descending interface.20 This layer continues to grow as long as the small particles can continuously filter through the loose network of the large binder particles. During this, the binder particles also start to coalesce as their Tg and minimum film forming temperature is far below 20 °C. This coalescence immobilizes them, so that despite their higher Pé number, the binder ceases to move while Si particles can still move towards the surface due to Pé > 1. Eventually this growth stops at higher volume fractions due to the slowing of the dynamics and the jamming of the small particles. This is in accordance with the EDS data for B1–MSiO2 samples which showed an increase in silica content on the film surface with an increase in Si% at a constant total solid content of the formulation.
While the Pé number was able to predict the distribution of silica particles in B1–MSiO2 films, it was of less significance in directly postulating the arrangement of silica particles in the B1–BSiO2 films where the silica particles show faster gelation tendencies (Fig. 6). In this case, the rapid buildup of viscosity would induce a continuous change of the Péclet number values as a function of time. Formation of early silica agglomerates can restrict their migration towards the top surface by seeping through the contours of the binders as one can expect in case of B1–MSiO2. This resulted in an almost even distribution of silica across the cross-section of the films as shown in the EDS data. Thus, in our systems, the main assumption is that there will be no network formation unless reaching highly concentrated films, i.e. when the particles start to percolate. Then the film forming occurs with polymer chain infusion, etc., while binder and silica particles are locked in a given spatial position. If the gelling occurs prematurely as in the case of BSiO2, the eqn (1) does not provide a good description of the system based on the initial data and the final morphology is mostly dictated by the gelling behavior rather than solely by the Péclet numbers of the system.
Effect of rate of evaporation on film forming behavior
B1–MSiO2 samples were observed to be resistant against early gelation which helped the silica particles to migrate toward the air interface during the film formation process. Faster removal of solvent can result in crowding of particles and initiate early detainment of the structure. To study the influence of temperature on morphology and the distribution of silica across the films, rapid drying of the samples with B1–MSiO2 with 5 wt% solid content was carried out at 50 °C and 100 °C respectively. The SEM pictures of the surface morphology are shown in Fig. 7. | ||
| Fig. 7 Surface morphology of B1–MSiO2 films with total solid content of 5 wt% dried at 50 °C (left column) and 100 °C (right column). The % value indicates the silica mass fraction. The white bar indicates a scale of 100 nm. | ||
Compared to the morphology of samples dried at room temperature (Fig. 2), a significant lowering of surface pores was observed against any given silica content. Faster removal of solvent by increasing the drying temperature can result in congestion of silica particles and this compacting is expected to lower the interstitial space between them. The binder particles entrapped within the silica matrix would experience free spaces through which infiltration can occur. This can lower the pore formation process. The EDS analysis of cross sections of these samples did not show any trend in silicon content variation across the cross section of the films (data not shown here) indicating the absence of any stratification. This faster solidification of structure by increasing the drying rate resulted in formation of fused morphology by limiting the stratification and infiltration. These results are in agreement with the previous experiments, as here the faster drying induced loss of mobility through percolation, similar to the previously described gelling process of B1–BSiO2.
Effect of type of binders on the morphology of films
Fig. S2 (ESI†) displays the surface morphology of the samples with MSiO2 formulated with binders B2 and B3, at 5 wt% solid content. B2 and B3 are also acrylic based binders with slight variation in particle sizes. B2 is an acrylic binder coated onto a silica core with an average particle size of 90 ± 5 nm. B3 has an average particle size of 110 ± 9 nm and is purely acrylic. The trend of morphology variation is similar as that with formulations with B2–MSiO2 against gradual increase in silica content. The pore sizes of B1–MSiO2, B2–MSiO2 and B3–MSiO2 are 95 ± 12 nm, 80 ± 7 nm and 101 ± 9 nm respectively, which correlates with the size of binders. The interesting outcome of this study is that irrespective of the type of binder used, and provided that Tg and film forming temperatures are close or below room temperature, the change of surface morphology with increase in silica content follows the similar trend i.e. gradual engulfment of the binder particles in the silica matrix instead of localized phase separation of the two systems.Conclusions
Whereas, the mechanisms of film formation is well understood for binary mix of colloids where the particles retain their structure after film formation,22,41–43 this paper discusses the influence of silica surface chemistry, formulation constituents, and temperature on the film forming behavior of silica–binder systems where the binder forgoes its initial morphology after the film formation process. Two types of silica nanoparticles were considered, one that could undergo gelling during the film forming process (bare silica, BSiO2), and one that was modified to resist salt induced gelling (MSiO2).Overall, the nature of silica appeared to play a central role in dictating the morphology of the films by controlling the particle migration during the film formation process. The main finding of this paper is that is possible to control both surface and bulk properties of films through the regulation of surface chemistry of colloidal silica and film forming temperature of the binder. Faster drying rate or gelation, as was observed for bare silica, was found to affect the morphology by inhibiting the formation of intricate silica network through which polymer infiltration could occur resulting in lower pores across the cross-sections of the films.
These observations were discussed considering the concept of Péclet number (Pé). In the case of modified silica, stratification and morphology were according to expectations.20 In the case of bare silica, the gelation occurred at an earlier stage, resulting in hindered particle stratification.
These results help in rationalizing the mechanism of stratification of nanoparticles. Moreover, an interplay of polymer infiltration along with silica stratification controlled the surface and bulk morphology of the films. These results put a specific emphasis on the importance of the surface chemistry of the nanoparticles in the film forming process, which is of particular importance for applications where coatings with controlled pore distributions are needed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in part at the Chalmers Material Analysis Laboratory, CMAL. The authors wish to thank the VINNOVA, Sweden's innovation agency for financial support (grant # 2017-03724).References
- ed. W. C. Smith and Textile Institute, Smart textile coatings and laminates, CRC Press, Boca Raton, Fla., 2010 Search PubMed.
- B. Li, L. Huang, N. Ren and M. Zhou, Appl. Surf. Sci., 2014, 290, 80–85 CrossRef CAS.
- A. Ibrahim, Z. A. Hamid and A. A. Aal, Mater. Sci. Eng., A, 2010, 527, 663–668 CrossRef.
- S. Halevy, Y. Bochlin, Y. Kadosh, A. Kaplan, H. Avraham, A. Nissim, R. Ben Hamo, T. Ohaion-Raz, E. Korin and A. Bettelheim, Sci. Rep., 2017, 7, 4987 CrossRef CAS PubMed.
- Q. Liang, S. A. Hsie and C. P. Wong, ChemPhysChem, 2012, 13, 3700–3706 CrossRef CAS PubMed.
- R. Karthick, M. Brindha, M. Selvaraj and S. Ramu, J. Colloid Interface Sci., 2013, 406, 69–74 CrossRef CAS PubMed.
- H. H. Murray, Appl. Clay Sci., 2000, 17, 207–221 CrossRef CAS.
- D. K. Kim, M. Mikhaylova, Y. Zhang and M. Muhammed, Chem. Mater., 2003, 15, 1617–1627 CrossRef CAS.
- M. Z. Elsabee and E. S. Abdou, Mater. Sci. Eng., C, 2013, 33, 1819–1841 CrossRef CAS PubMed.
- E. Avcu, F. E. Baştan, H. Z. Abdullah, M. A. U. Rehman, Y. Y. Avcu and A. R. Boccaccini, Prog. Mater. Sci., 2019, 103, 69–108 CrossRef CAS.
- H. Zhao, M. Tian, Z. Li, Y. Zhang, S. Zhu, X. Zhang, S. Chen and L. Qu, Mater. Lett., 2019, 240, 5–8 CrossRef CAS.
- J. L. Keddie and A. F. Routh, Fundamentals of Latex Film Formation, Springer Netherlands, Dordrecht, 2010, pp. 1–26 Search PubMed.
- P. Nguyen-Tri, T. A. Nguyen, P. Carriere and C. Ngo Xuan, Int. J. Corros., 2018, 1–19 Search PubMed.
- J. L. Keddie, P. Meredith, R. A. L. Jones and A. M. Donald, Macromolecules, 1995, 28, 2673–2682 CrossRef CAS.
- R. E. Trueman, E. Lago Domingues, S. N. Emmett, M. W. Murray, J. L. Keddie and A. F. Routh, Langmuir, 2012, 28, 3420–3428 CrossRef CAS PubMed.
- D. K. Makepeace, A. Fortini, A. Markov, P. Locatelli, C. Lindsay, S. Moorhouse, R. Lind, R. P. Sear and J. L. Keddie, Soft Matter, 2017, 13, 6969–6980 RSC.
- M. Schulz and J. L. Keddie, Soft Matter, 2018, 14, 6181–6197 RSC.
- J. Zhou, Y. Jiang and M. Doi, Phys. Rev. Lett., 2017, 118, 108002 CrossRef PubMed.
- R. E. Trueman, E. Lago Domingues, S. N. Emmett, M. W. Murray and A. F. Routh, J. Colloid Interface Sci., 2012, 377, 207–212 CrossRef CAS PubMed.
- A. Fortini, I. Martín-Fabiani, J. L. De La Haye, P.-Y. Dugas, M. Lansalot, F. D’Agosto, E. Bourgeat-Lami, J. L. Keddie and R. P. Sear, Phys. Rev. Lett., 2016, 116, 229901 CrossRef PubMed.
- R. P. Sear and P. B. Warren, Phys. Rev. E, 2017, 96, 062602 CrossRef PubMed.
- R. P. Sear, J. Chem. Phys., 2018, 148, 134909 CrossRef PubMed.
- Y. Tang, G. S. Grest and S. Cheng, Langmuir, 2018, 34, 7161–7170 CrossRef CAS PubMed.
- Y. Reyes and Y. Duda, Langmuir, 2005, 21, 7057–7060 CrossRef CAS PubMed.
- M. P. Howard, A. Nikoubashman and A. Z. Panagiotopoulos, Langmuir, 2017, 33, 3685–3693 CrossRef CAS PubMed.
- H. Luo, C. M. Cardinal, L. E. Scriven and L. F. Francis, Langmuir, 2008, 24, 5552–5561 CrossRef CAS PubMed.
- A. K. Atmuri, S. R. Bhatia and A. F. Routh, Langmuir, 2012, 28, 2652–2658 CrossRef CAS PubMed.
- A. J. Carr, W. Liu, K. G. Yager, A. F. Routh and S. R. Bhatia, ACS Appl. Nano Mater., 2018, 1, 4211–4217 CrossRef CAS.
- H. Shiraz, S. J. Peake, T. Davey, N. R. Cameron and R. F. Tabor, J. Colloid Interface Sci., 2018, 528, 289–300 CrossRef CAS PubMed.
- B. You, N. Wen, Y. Cao, S. Zhou and L. Wu, Polym. Int., 2009, 58, 519–529 CrossRef CAS.
- B. You, L. Shi, N. Wen, X. Liu, L. Wu and J. Zi, Macromolecules, 2008, 41, 6624–6626 CrossRef CAS.
- A. F. Routh and W. B. Russel, Ind. Eng. Chem. Res., 2001, 40, 4302–4308 CrossRef CAS.
- A. F. Routh and W. B. Russel, Langmuir, 1999, 15, 7762–7773 CrossRef CAS.
- A. Utgenannt, R. Maspero, A. Fortini, R. Turner, M. Florescu, C. Jeynes, A. G. Kanaras, O. L. Muskens, R. P. Sear and J. L. Keddie, ACS Nano, 2016, 10, 2232–2242 CrossRef CAS PubMed.
- A. F. Routh, Rep. Prog. Phys., 2013, 76, 046603 CrossRef PubMed.
- J.-P. Gorce, D. Bovey, P. J. McDonald, P. Palasz, D. Taylor and J. L. Keddie, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 421–429 CrossRef CAS PubMed.
- P. Ekanayake, P. J. McDonald and J. L. Keddie, Eur. Phys. J.-Spec. Top., 2009, 166, 21–27 CrossRef.
- C. M. Cardinal, Y. D. Jung, K. H. Ahn and L. F. Francis, AIChE J., 2010, 56, 2769–2780 CrossRef CAS.
- Y. Reyes, J. Campos-Terán, F. Vázquez and Y. Duda, Modell. Simul. Mater. Sci. Eng., 2007, 15, 355–368 CrossRef CAS.
- S. Cheng and G. S. Grest, J. Chem. Phys., 2013, 138, 064701 CrossRef PubMed.
- W. Liu, A. J. Carr, K. G. Yager, A. F. Routh and S. R. Bhatia, J. Colloid Interface Sci., 2019, 538, 209–217 CrossRef CAS PubMed.
- N. I. Lebovka, Y. Yu. Tarasevich and N. V. Vygornitskii, Phys. Rev. E, 2018, 97, 022136 CrossRef CAS PubMed.
- A. F. Routh and W. B. Zimmerman, Chem. Eng. Sci., 2004, 59, 2961–2968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm00317d |
| This journal is © The Royal Society of Chemistry 2020 |