
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the interaction of softwood hemicellulose with cellulose surfaces in relation to molecular structure and physicochemical properties of hemicellulose†
Polina
Naidjonoka
 *a,
Monica Arcos
Hernandez
b,
Gunnar K.
Pálsson
cd,
Frank
Heinrich
ef,
Henrik
Stålbrand
*g and
Tommy
Nylander
*ahi
*a,
Monica Arcos
Hernandez
b,
Gunnar K.
Pálsson
cd,
Frank
Heinrich
ef,
Henrik
Stålbrand
*g and
Tommy
Nylander
*ahi
aPhysical Chemistry, Department of Chemistry, Lund University, Lund, Sweden. E-mail: polina.naidjonoka@fkem1.lu.se; tommy.nylander@fkem1.lu.se
bPolymer & Materials Chemistry, Department of Chemistry, Lund University, Lund, Sweden
cDepartment of Physics & Astronomy, Uppsala University, Uppsala, Sweden
dInstitut Laue Langevin, Grenoble, France
eDepartment of Physics, Carnegie Mellon University, Pittsburgh, PA, USA
fNIST Center for Neutron Research, Gaithersburg, MD, USA
gBiochemistry and Structural Biology, Department of Chemistry, Lund University, Lund, Sweden. E-mail: henrik.stalbrand@biochemistry.lu.se
hNanoLund, Lund University, Lund, Sweden
iLund Institute of Advanced Neutron and X-ray Science LINXS, Lund, Sweden
First published on 7th July 2020
Abstract
The substantial part of the water-soluble hemicellulose fraction, obtained when processing cellulose to produce paper and other products, has so far been discarded. The aim of this work is to reveal the interfacial properties of softwood hemicellulose (galactoglucomannan, GGM) in relation to their molecular and solution structure. In this study the sugar composition of GGM was characterised by chemical analysis as well as 1D and 2D NMR spectroscopy. Previously it has been demonstrated that hemicellulose has high affinity towards cellulose and has the ability to alter the properties of cellulose based products. This study is focused on the interactions between hemicellulose and the cellulose surface. Therefore, adsorption to hydrophobized silica and cellulose surfaces of two softwood hemicellulose samples and structurally similar seed hemicelluloses (galactomannans, GMs) was studied with ellipsometry, QCM-D and neutron reflectometry. Aqueous solutions of all samples were characterized with light scattering to determine how the degree of side-group substitution and molecular weight affect the conformation and aggregation of these polymers in the bulk. In addition, hemicellulose samples were studied with SAXS to investigate backbone flexibility. Light scattering results indicated that GM polymers form globular particles while GGMs were found to form rod-like aggregates in the solution. The polysaccharides exhibit higher adsorption to cellulose than on hydrophobic surfaces. A clear correlation between the increase in molecular weight of polysaccharides and increasing adsorbed amount on cellulose was observed, while the adsorbed amount on the hydrophobic surface was fairly independent of the molecular weight. The obtained layer thickness was compared with bulk scattering data and the results indicated flat conformation of the polysaccharides on the surface.
Introduction
The interactions between hemicellulose and another major wood polysaccharide, cellulose, are of key importance for the organization and structure of the plant cell walls as well as for a range of applications. The major components of softwood hemicellulose are galactoglucomannans (GGM). Their content varies both in different parts of the tree, as well as between plant species with GGM, reaching up to 20% w/w of dry wood.1–4 Today a significant part of the softwood GGM is discarded together with the waste stream during processes like thermo-mechanical pulp production.5 However, it is now possible to recover this highly valuable polymer from such waste streams.6–8 While other mannan-based polysaccharides like Locust bean gum (LBG) and Guar gum (GG) are used in the industry, mostly in food related applications,9–12 GGMs from softwood are not yet fully exploited in spite of the large amount produced during pulp processing. However, numerous potential applications have been suggested and tested for GGMs in therapeutic reagents,13 as a raw material for biodegradable films14–16 and novel anticoagulants.17 Therefore, this study focuses on the interfacial interactions between cellulose and softwood hemicellulose and how it relates to the aggregation and molecular architecture in comparison to other mannan-based polysaccharides.Hemicellulose has a large number of side groups and their role for interaction with cellulose and to what extent they prevent the polysaccharide to assemble into larger aggregates is not clear. GGMs consist of a β-(1→4)-D-mannopyranose backbone that is partially interrupted by β-(1→4)-D-glucopyranose units.1,18,19 The backbone carries side-groups of α-(1→6)-D-galactopyranose and acetyl side groups.18 The degree of side-group substitution may vary between sources and preparations. Galactoglucomannans are mainly present in the secondary cell walls of conifer plants, however, small amounts are also found in dicot cell walls.3 Locust bean gum is produced in the seed endosperm of the carob tree Ceratonia silique L. and Guar gum in the seed of Cyamopsis tetragonoloba. Both LBG and GG contain only β-(1→4)-mannopyranose in the backbone and galactopyranose side-groups attached via α-(1→6) linkages.
In wood, cellulose forms microfibrils that are well organized with chains of hemicellulose located either on the surface of or in between the fibrils.20 The nature of the binding of hemicellulose is still under debate, however, it is believed that the polysaccharides interact with each other via hydrogen bonding.21,22 Benselfelt et al.23 showed that the adsorption of xyloglucan, the main hemicellulose in primary plant cell walls, onto cellulose surfaces is an entropy-driven process. An increase in entropy is caused by the release of water molecules from the cellulose layer as hemicellulose is adsorbed.
Several approaches have been used to reveal the nature of the interaction between cellulose and hemicellulose. Hayashi et al.20,24 studied the influence of the polymerization degree on the adsorption of xyloglucan on dispersed cellulose. They demonstrated that xyloglucans adsorb to the microfibrils of cellulose as a monolayer with highly branched parts extending into solution. The adsorption per weight unit was found to be larger on microcrystalline cellulose compared to amorphous cellulose. Similar results were obtained by Vincken et al.22 where xyloglucan was found to bind more efficiently to microcrystalline cellulose Avicel than to cellulose from cotton linters. Avicel has a 10 times smaller exposed surface-to-weight ratio than bacterial cellulose, but the per-area-adsorption of the polymeric xyloglucans is similar.25 Uhlin et al.26 used Acetobacter xylinum as a model system to produce cellulose in the presence of carboxymethyl cellulose (CMC), xylan, xyloglucan and ivory nut mannan. These polysaccharides were shown to affect cellulose structure in different ways. Xyloglucans and xylans showed the largest effect on the aggregation of cellulose due to a similarity in the backbone unit conformation with cellulose. Cellulose produced in the presence of mannans and CMC was found to have a very similar aggregation pattern but with a lower degree of cellulose crystallinity in comparison to a control grown without the polysaccharides. The charge of hemicellulose was found to be more crucial than the molecular weight for the adsorption to nanofibrillated cellulose (NFC). The adsorbed amount of Guar gum before and after hydrolysis showed comparable results in similar conditions. The adsorption of the anionic CMC showed strong dependence on the pH of the buffer, with irreversible adsorption at low pH but not at pH 8 where the carboxylic groups of both CMC and cellulose are deprotonated.27,28 The sorption of glucomannans to cellulose is affected by the presence of side groups that prevent a close contact of the polysaccharide with the cellulose fibre surface. In addition, by removing the side groups, solubility of the chains of the polysaccharides chain decreases29–31 and promotes binding to a surface. Deacetylation generates new hydroxyl groups that can take part in hydrogen bonding5,32 and at least 15 unsubstituted xylosyl units are required for adsorption.25
It is clear that the increasing use of hemicelluloses requires knowledge on how their molecular structure affect bulk solution behaviour and the adsorption to different types of surfaces. Here we compared the results of two types of GGMs, with different molecular weight and degree of side-group substitution, with those from seed hemicelluloses (LBG and GG). The LBG and GG samples are commonly used in industrial applications. Solution behaviour of the polysaccharides was characterized with light and small angle X-ray scattering, which gave information on the size, shape and stiffness, as well as the aggregation state of the polysaccharides. The knowledge gained from the polysaccharide molecular structure, conformation and aggregation was related to the results from adsorption studies on two relevant surfaces for applications, i.e. hydrophobic and spin-coated cellulose films. The combination of in situ ellipsometry, quartz crystal microbalance with dissipation (QCM-D) and neutron reflectometry (NR) allowed us not only to quantify the adsorption in terms of surface excess and adsorption kinetics, but by comparing the adsorption onto a cellulose surface with that on a hydrophobic surface we were able to gain insight into the nature of the surface interactions of hemicellulose.
Experimental
Materials
LvLBG was dissolved by wetting 1 g of powder with 2 mL of 95% ethanol followed by the addition of 90 mL sodium citrate buffer (0.05 M, pH 5.9) and stirring overnight at 4 °C. Thereafter, the solution was heated to 120 °C in a beaker covered with aluminium foil and under vigorous stirring with an IKA RCT basic magnetic stirrer. Stirring continued without heating for approximately 20 min until complete dissolution. The solution was then centrifuged (5338 × g, 10 min) to remove impurities and diluted with buffer to a final volume of 100 mL.38
GGM samples (10 mg mL−1) were dissolved directly in 0.05 M citrate buffer and stored at +4 °C for maximum 1 week until further use. Cellulose solutions were prepared according to Sczech et al.39 Microcrystalline cellulose powder (1 g) was first left to swell for 1 h in 10 mL of deionized water at 40 °C. Excess of water was then removed from the hydrated cellulose by centrifugation (2616 × g, 15 min). This was followed by an exchange with 10 mL of methanol, after which the mixture was incubated during stirring for 45 min, followed by centrifugation at 2616 × g for 15 min to remove the excess of the solvent. The same procedure was repeated once more with methanol and then twice with the anhydrous DMAc. After the last incubation, the mixture was centrifuged, and the cellulose pellet was then dissolved in 100 mL of 7% (w/v) LiCl/DMAc solution under stirring at room temperature for 15 h. The cellulose solution was then placed at 4 °C under stirring until the cellulose was completely dissolved.
Silicon wafers were cut into 2 × 1 cm pieces and cleaned according to Chang et al.40 Silica substrates were placed in a base mixture of 25% NH4OH, 30% H2O2, and H2O (1/1/5 by volume) at 80 °C for 5 min, rinsed with deionized water, and cleaned in an acid mixture of 32% HCl, 30% H2O2 and H2O (1/1/5 by volume) at 80 °C for 5 min. The substrates were thoroughly rinsed with water and ethanol, and stored in ethanol (99.7%) until further use.
Polished silicon substrates (Siltronix, Archamps-France) of 5 × 5 × 1.5 cm3 capped with a silicon oxide layer were used for Neutron Reflectometry (NR) measurements. Surfaces were cleaned as described above.
The substrates used for Quartz Crystal Microbalance with Dissipation (QCM-D, Q-Sense Analyzer, Biolin Scientific, Gothenburg, Sweden) measurements were quartz crystals (Q-Sense QSX 303) coated with gold and a top layer of SiO2 exposed to the solution. The fundamental frequency of the crystals was 4.95 MHz. The quartz QCM crystals were cleaned for 5 min in 2% (v/v) Hellmanex III® while sonicating and rinsed with water and 99.7% ethanol.
Hydrophobized silica substrates were prepared by first treating cleaned silica surfaces in an air plasma for 5 min at 0.02 mbar using a plasma cleaner (Harrick Scientific Corp, model PDC-3XG, New York, USA). The samples were then immediately placed in a desiccator under vacuum with 1 mL of DMOCS overnight. The hydrophobized silicon wafers were subsequently sonicated for 20 min in tetrahydrofuran and ethanol and stored in ethanol. Substrates were carefully rinsed with ethanol and water, and dried with nitrogen gas before each experiment.
Cellulose surfaces were prepared by spin-coating a few drops of clear cellulose solution on the silica surface (spin coater module LabSpin6/8, SUSS MicroTec SE, Germany) at 6000 rpm for 60 s. The spin-coated wafers were annealed at 100 °C for 10 min, cooled down to room temperature, placed into deionized water for 20 min, dried in a nitrogen flow and heated at 150 °C for 15 min. The cellulose covered substrates were used immediately. The cellulose film thickness was roughly 40 nm for all the substrates used for the ellipsometry measurements.
Light scattering data from GGM samples were recorded at a concentration of 1.5 mg mL−1 and from galactomannans at 3 mg mL−1 dissolved in 0.05 M citrate buffer (pH 5.9). All samples were filtered with a 0.45 μm-pore-size filter and transferred to clean borosilicate NMR tubes prior to the measurements. For SLS, angles from 40° to 140° were used with a step size of 2° for 6 s. The intensity autocorrelation functions were obtained for angles from 60° to 130° with steps of 10° for at least 300 s. All the measurements were performed at 25 °C.
For the static light scattering, the obtained scattering intensity I(q) was corrected for background scattering (ΔI(q)) and brought to an absolute scale according to eqn (1)41
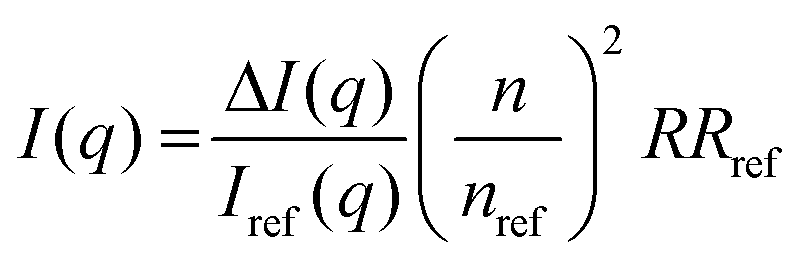 | (1) |
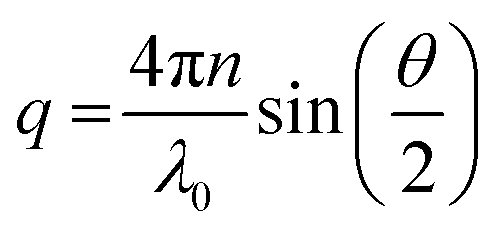 | (2) |
The decay rate (Γ) was obtained from the second order cumulative expansion,42 which was plotted at different scattering vectors. The slope of such a plot gives the translational diffusion coefficient D. The hydrodynamic radius RH was calculated according to the Stokes–Einstein equation:
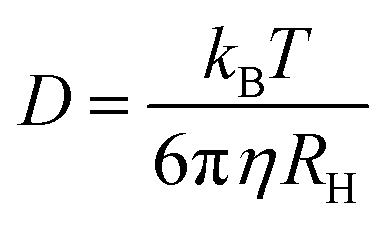 | (3) |
The optical properties of the silicon substrates were characterized before each experiment in two different media, air and aqueous buffer. To minimize the effect caused by imperfections of the optical components, the average positions of polarizer and analyzer in four zones were used to calculate the ellipsometric angles, ψ and Δ. Here, ψ represents the change in the relative amplitude and Δ, the phase shift of polarized light upon reflection at the interface. From the ellipsometric angles, the refractive index and layer thickness of the adsorbed layer were determined using a three or four layer optical model.47 The adsorbed amount Γ was calculated using the de Feijter equation48
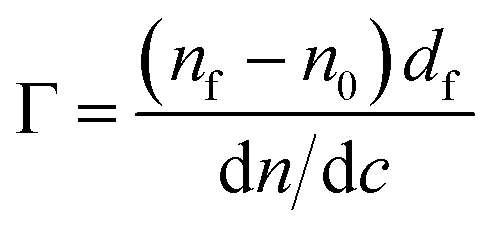 | (4) |
After the characterization of the bare surface, a polysaccharide solution was added to the ellipsometry cuvette to a final concentration of 0.02 mg mL−1. For the experiments with cellulose surfaces, pre-characterized silicon wafers were spin-coated with the cellulose solution before the addition of the sample.
As soon as the system reached an equilibrium, the cuvette was flushed with fresh buffer solution to remove the polysaccharide solution. This allowed to estimate the extent of reversible mannan binding to the substrate.
Hydrophobized crystals were placed in the flow cell and ethanol was passed through the system with peristaltic pump (Ismatec IPC-N 4, Zürich, Switzerland), followed by purging with 0.05 M citrate buffer (pH 5.9). The crystals were left to stabilize in the buffer for at least 30 min. The fundamental frequencies and dissipation energies for each overtone were determined before samples were injected at 150 μL min−1 at a concentration of 0.02 mg mL−1. All measurements were performed at 25 °C. The obtained data was treated and fitted with a Voigt viscoelastic model53 using Dfind software (QSense, Biolin Scientific).
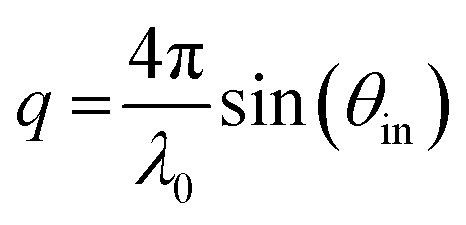 | (5) |
Here we used specular NR, i.e. the angle of reflection is equal to the incident beam angle.54 Measurements were performed at NIST Center For Neutron Research (NCNR, Gaithersburg, MD) on the NGD-MAGIK reflectometer over a q-range of 0.005–0.2 Å−1.55
Adsorption of TMP GGM was studied with NR on a hydrophobic surface. TMP was dissolved in deuterated 0.05 M citrate buffer (pH 5.9) at a concentration of 0.2 mg mL−1. First, the specular reflectivity of the bare surface immersed in deuterated (D2O) and protonated (H2O) buffer was measured versus q. The sample in D2O buffer was then injected and left for 1 hour to adsorb. Thereafter, two NR curves were measured after rinsing with D2O and H2O-based buffers, respectively. The raw data was reduced with the online data reduction service reductus.56 The reduced data was evaluated with the Motofit software, which uses the Abeles matrix method to calculate the reflectivity from a stratified interfaces.57,58 The best fit was obtained applying 4 layers as follows: Si–SiO2, hydrophobic layer, the transition layer containing acetyl groups of GGM interconnected with the hydrophobic layer and the top layer mainly consisting of the GGM sugar units. The layer thickness, roughness, solvent penetration and Scattering length density (SLD) were found for each layer by simultaneously fitting the model to the data from H2O and D2O contrasts. Attempt was also made to measure the interaction between GGM and the cellulose layer using neutron reflectometry. However, the poor SLD contrast between GGM and cellulose made modelling uncertain and we therefore prefer not to discuss these data. We are at the moment working on neutron reflectometry study where we use deuterated cellulose to enhance the SLD contrast.
Results and discussion
Chemical analysis
The results from the chemical analysis are summarized in Table 2. Assuming polymeric mannose, galactose, glucose and acetyl content comprise acetylated GGM (AcGGM), this component accounted for 77% and 83% of the dry-weight for the TMP-GGM and SP-GGM preparations, respectively. The SP-GGM preparation contained arabinoxylan, estimated from the acid released xylose and arabinose as described above (Table 2). No detectable arabinoxylan was present in TMP-GGM. The lignin content was estimated to be 3.0% (TMP-GGM) and 3.3% (SP-GGM) of the dry weight, based on simple UV light absorption measurements.| Sample | Molecular weight [kDa] | Molar ratio | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Man | Gal | Glc | Acetyl | Ara | Xyl | ||||
| Guar gum (GG) | 250.0 | 67 | 33 | 38 | |||||
| Locust bean gum | LBG | 556.0 | 78 | 22 | |||||
| LvLBG | 107.0 | 78 | 22 | ||||||
| Spruce galactoglucomannan (GGM) | TMP | 14.0 | 50 | 15 | 15 | 20 | 2, 6 and 33 | ||
| SP | 5.9 | 40 | 6 | 16 | 2 | 2 | 8 | ||
1D and 2D NMR spectroscopy
Expansions of the 2D-NMR data in the anomeric region of SP and TMP preparations are shown in Fig. 1. The proton NMR resonances in the anomeric region were assigned based on HSQC and confirmed by comparison with literature.18,19,59–61 The spectra recorded for both samples are similar confirming the similarity of the two samples. Hence, the SP is used as a template to describe assignments of the peaks. Table 3 shows the assignation in the anomeric area for SP using HSQC (the corresponding data for TMP are shown in the ESI†).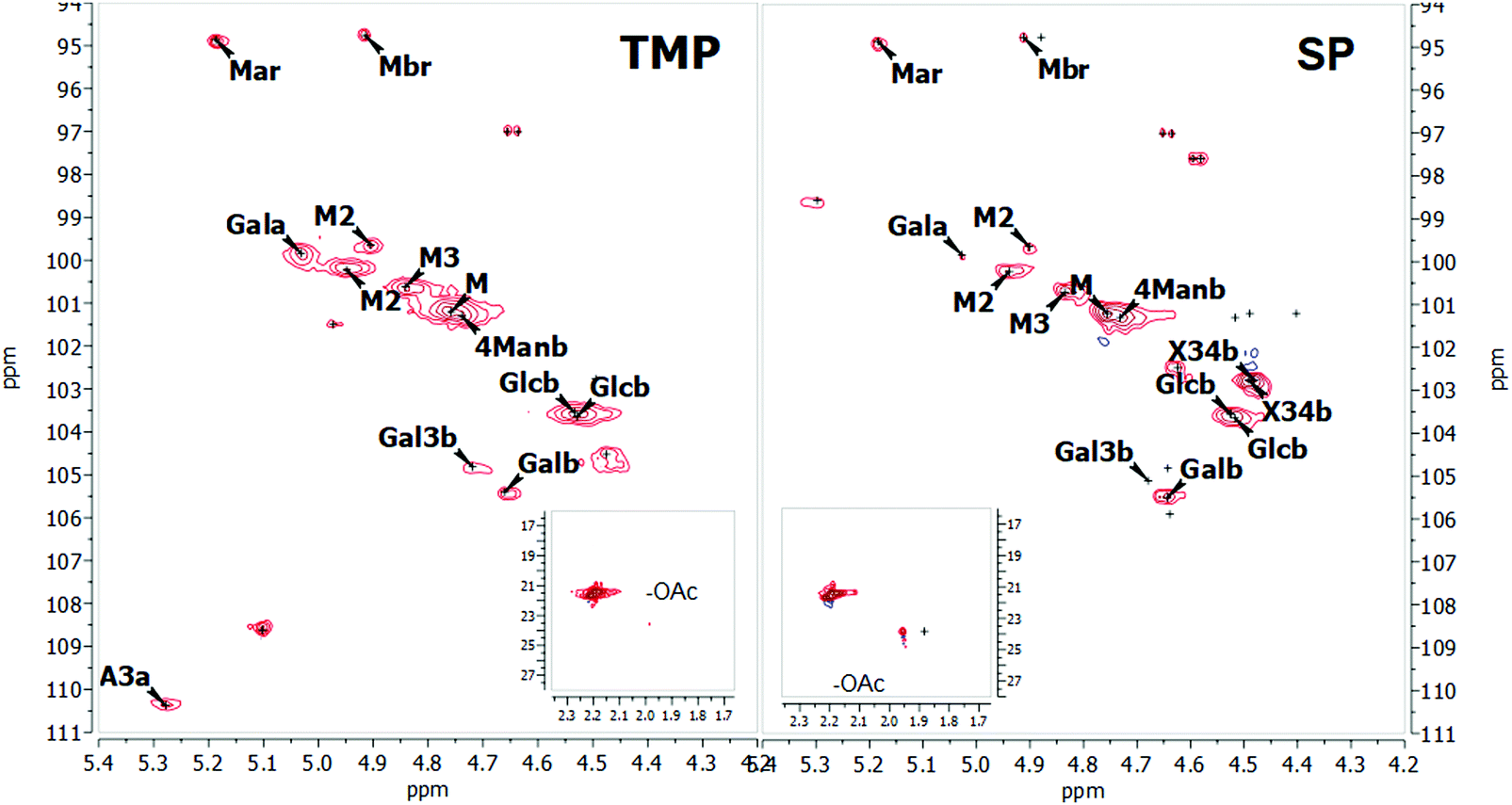 | ||
| Fig. 1 2D NMR spectra of the anomeric region for the TMP (on the left) and SP (on the right) samples at 25 °C. | ||
Spectra analysis showed that the studied hemicelluloses (TMP and SP) mainly contain mannose, glucose and galactose thus can be described as galactoglucomannan (GGM). HSQC showed no resonances in the aromatic region characteristic of lignin and these samples could be considered as fairly pure in terms of lignin contamination, which is also consistent with the UV spectroscopy analyses. However, a fair amount of arabino glucuronoxylan is also part of the SP sample as previously reported for a similar preparation by Palm and Zacchi.33 Sample TMP also contains arabino glucuronoxylan but in much lower amount. Traces of 4-O-methylglucoronic acid (4-O-Me-GlcA) were present in the SP sample, which was also reported by Palm and Zacchi.33
The 1D proton spectra of both samples at 70 °C is shown in Fig. 2 and corresponds to the general assignments in the anomeric region from Table 3. Apart from the anomeric signals assigned in the fingerprint region (4.4–5.5 ppm), signals at 2.1–2.24 ppm indicate presence of acetylated saccharides. This was further confirmed in HSQC, where typical cross peak for acetyl group is clearly seen in the insert of Fig. 1 at δH/δC 2.2–1.9/21.56–21.4. Of these acetylated saccharides, HSQC shows that they correspond to β-(1–4)-linked mannopyranosyl residues at C-2 and C-3 as previously reported.19,59,61 For both acetylated saccharides HSQC showed multiple cross peaks as well as several resonances in the 1D proton spectra (compare with inserts in Fig. 1). This reflects the random distribution of the acetylated residues along the backbone and was also observed by Lundqvist et al.18
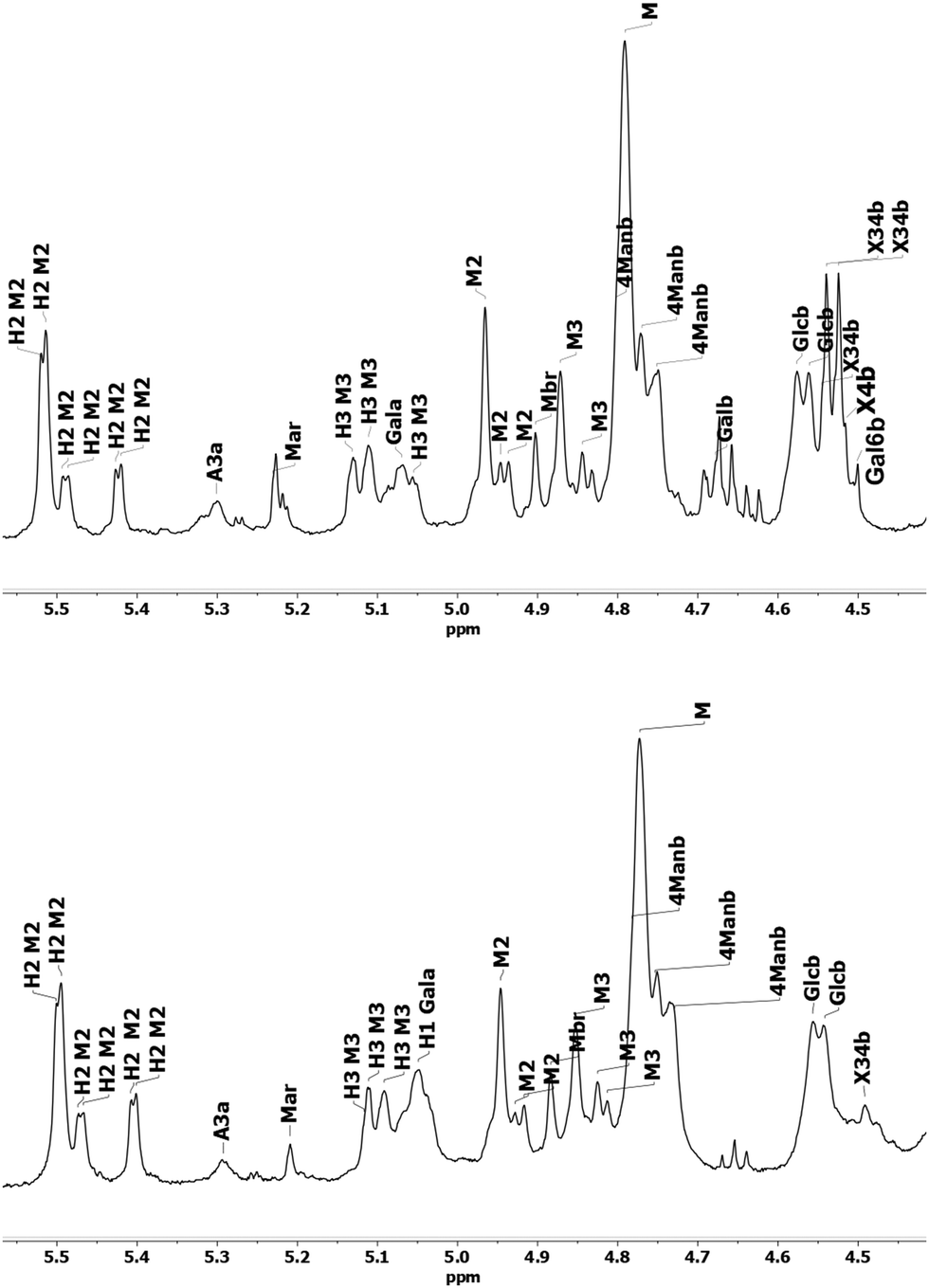 | ||
| Fig. 2 Anomeric region of 1H NMR spectra of O-acetyl-GGM SP (top) and TMP (bottom). Table 3 describes assignations. | ||
| Constituent | Annotation | Fig. key | 1H (ppm) | 13C (ppm) |
|---|---|---|---|---|
| a From 1H NMR. | ||||
| Mannose | α-ManpR | Mar | 5.18 | 94.87 |
| -4)-β-Manp-(1-, 2-O-Ac | M2 | 4.94 | 100.23 | |
| -4)-β-Manp-(1-, 2-O-Ac | M2 | 4.9 | 99.65 | |
| β-ManpR | Mbr | 4.88, 4.91 | 94.77 | |
| -4)-β-Manp-(1-, 3-O-Ac | M3 | 4.83 | 100.72 | |
| β-Manp-(1- | M | 4.76 | 101.21 | |
| -4)-β-Manp-(1- | 4Manb | 4.73 | 101.3 | |
| Glucose | -4)-β-Glcp-(1- | Glcb | 4.53 | 103.55 |
| -4)-β-Glcp-(1- | Glcb | 4.52 | 103.65 | |
| Galactose | αGalp-(1- | Gala | 5.03 | 99.84 |
| Other polysacharides | ||||
| Arabino-xylan | -4,3)-β-Xylp-(1- | X34b | 4.48, 4.49 | 102.77 |
| α-Araf-(1-3 | A3a | 5.28a | ||
| β-Galactan | -4)-β-Galp-(1- | Galb | 4.64 | 105.5 |
| Arabino-galactan | -3)-β-Galp-(1- | Gal3b | 4.68 | 105.11 |
Ratios of the different sugars were estimated using a semi-quantitative approach based on the volume integration60,62 from HSQC as well as integration from 1D 1H. The limitation of using volume integrals from HSQC is that the phase in the directly detected dimension varies between the different cross-peaks, with different amounts of the dispersive component contributing, thereby affecting their integral. The disadvantage of using 1H is that with the overlap of peaks the accuracy of the integration is no more than 95% in some cases, this error would propagate when calculating ratios as they involve more than two integrals. Nevertheless, these methods are established and have been extensively used previously.35,59 Quantification of molar fraction an estimation of masses was also calculated as done in Rosengren et al.,36 using and external standard and using ERETIC 2 method as described previously. Given that the results from all the methods are comparable here we present the results from the ERETIC 2 based calculations in Tables 4 and 5. We have made the utmost effort to describe the correct assignation of all peaks.
Table 4 shows a summary of the composition estimates for both samples using NMR. The Man![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Glu:Gal was estimated to be 100:22:9 and 100:18:5 for TMP and SP, respectively. Both samples are fairly similar except in the galactose molar content. Here it should be noted that galactose content is calculated based on α-galactose, the presence of galactan would up this number. GGM in softwood is described usually as substituted with only αGalp units.19 Furthermore, the galactose peak is not well-resolved (cf.Fig. 2) and the integration is likely to substantial error. We also note that the chemical analysis (Table 2) gives a larger galactose content. The degree of acetylation (DSAc) was estimated by comparing the integrals of the acetylated regions and the GGM sugars in the anomeric region.60
:Glu:Gal was estimated to be 100:22:9 and 100:18:5 for TMP and SP, respectively. Both samples are fairly similar except in the galactose molar content. Here it should be noted that galactose content is calculated based on α-galactose, the presence of galactan would up this number. GGM in softwood is described usually as substituted with only αGalp units.19 Furthermore, the galactose peak is not well-resolved (cf.Fig. 2) and the integration is likely to substantial error. We also note that the chemical analysis (Table 2) gives a larger galactose content. The degree of acetylation (DSAc) was estimated by comparing the integrals of the acetylated regions and the GGM sugars in the anomeric region.60
Table 4 shows that the main difference between both samples is the presence of higher amounts of arabino glucurunoxylan constituents in the SP sample (see Table S2, detailed SP NMR peak assignments are shown in ESI†). The given values include the two major residues of arabino glucuronxylan-3,4)-β-Xylp-(1- and α-Araf-(1–3 because they are the major constituents identified unequivocally from the spectra. Other components related to other polysaccharides such as arabinogalactan were not possible to use for quantification given their small area, ambiguity in peak assignation and/or overlapping with other peaks. Based on the values given in Table 4, the ratio of Man:AraXyl of ∼1:0.24 was calculated for SP compared to 1:0.07 in TMP. Arabino glucuronoxylan is also a hemicellulose common in spent sulfite liquor (SSL) from spruce.62 It has been shown that small amounts of arabinose and xylose can be covalent constituents of GGM.63 Nevertheless, the presence of these polysaccharides may be due to hemicellulose contaminants. Other cross peaks related to arabino glucuronoxylan could be assigned to β-Xylp (4.13, 3.38/64.04) as well as the residue 3,4-Xylp in sample SP. Characteristic peaks of α-glucuronic acid were also detected. The cross peaks at 3.47/61.02 and 3.3/73.8 ppm indicate that a fraction of these residues carry the 4-O-methyl substituent.19
Table 5 shows the sugar composition in relation to the mannose content. The degree of acetylation (DSAc) was 0.15 in relation to the Manp units for both of the samples. As discussed above, the NMR spectra suggest that the acetylated residues are randomly distributed along the backbone. It is interesting to note that the molar fraction of non-acetylated mannose with galactose substituents is higher for the SP sample, however this can be related to the difficulties in determining the galactose content with NMR.
| M w [kDa] | R G [nm] | R H [nm] | R G/RH | ||
|---|---|---|---|---|---|
| GGM | TMP | 14.0 | 122 | 111 ± 8 | 1.10 |
| SP | 5.9 | 87 | 82 ± 6 | 1.07 | |
| Locust bean gum | LvLBG | 107.0 | 79 | 95 ± 14 | 0.75 |
| LBG | 556.0 | 111 | 210 ± 39 | 0.53 | |
| Guar gum | GG | 250.0 | 42 | 93 ± 16 | 0.61 |
The molecular weight and dimensions of the GGM monomer estimated from individual sugar groups are 0.18 kDa and 0.6 nm, respectively. Therefore, fully stretched linear polymer in a monodisperse sample with molecular weights of 14.0 kDa and 5.9 kDa, would be 47 nm and 20 nm long, respectively, which is almost three times lower than the obtained values. Thus, the GGM samples are likely to be aggregated. This conclusion is consistent with cryo-TEM images of TMP GGM at the same concentration (Fig. S4, in the ESI†) and with a recent study of a comparable GGM preparation that shows similar aggregated structures but larger in size,65 likely due to a higher molecular weight and solution concentration of the sample. The expected contour length of the locust bean gum and guar gum samples, calculated based on the molecular weight of the corresponding linear polymer, is about 10 times larger than the measured value. This suggests that these polysaccharides are not aggregated but form separate polymer chains in solution. Furthermore, both GGMs have a RG/RH slightly higher than 1 (Table 6) which suggests that the aggregates have an elongated shape. With a RG/RH of around 0.7, LBG and GG chains seems to adopt a globular shape in solution. The radius of gyration for the GG and LBG are similar to literature values.66,67
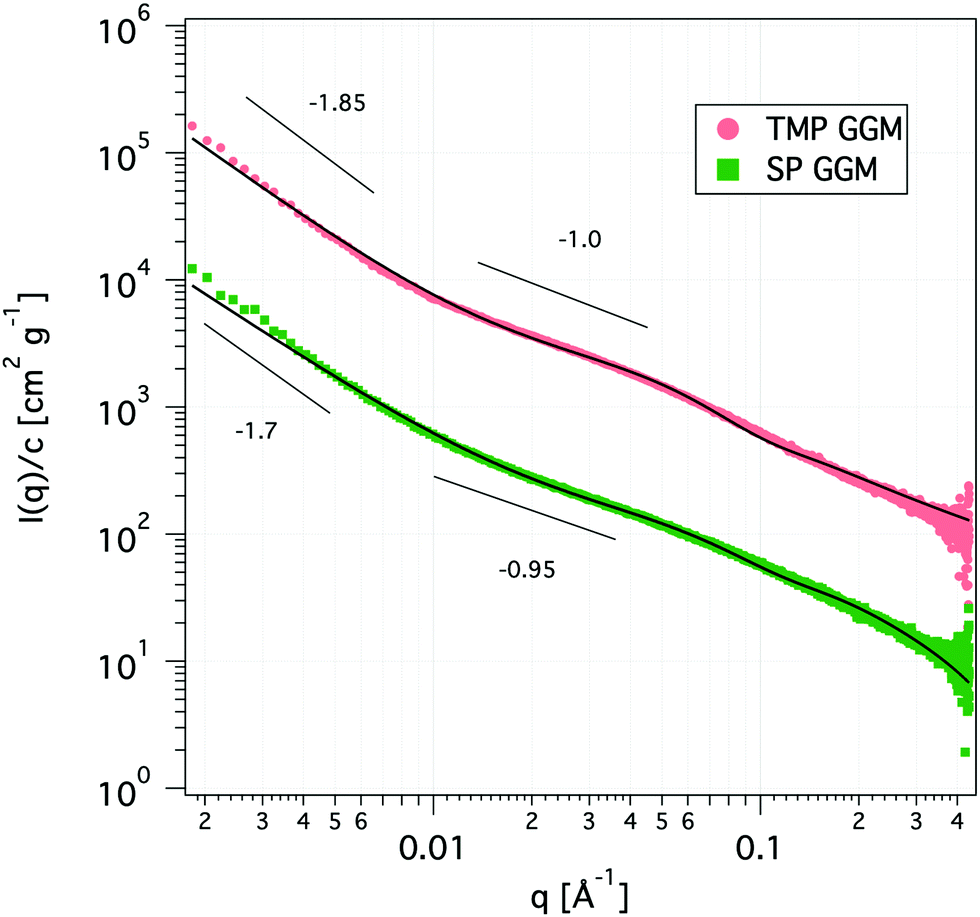 | ||
| Fig. 3 SAXS curves of the TMP (light red) and SP GGM (green) at a concentration of 10 mg mL−1. Solid lines are fits to the corrected Beaucage model. The scattering curve of the TMP was shifted upwards by a factor of 10 for clarity. | ||
The scattering curves show no distinct features other than a shift of the power-law decay from −1.85 at low q to −1.0 at high q for TMP GGM (−1.7 to −0.95 for SP GGM) as indicated in Fig. 3. The lack of a drastic change in the slope through the extended q-range indicates self-similarity or fractal-like behaviour of the polymer molecules in solution. The scattering from such a polymer chain can be described by using a mass fractal model, i.e. the scattering intensity, I(q) ∼ q−d. Such a model is valid as the exponents (−d) are between −1 and −3.45 The exponent of −1 at high q-values indicates rod-like shape of the polymer below its persistence length. Based on these observations, the scattering data was fitted to a corrected Beaucage model that describes fractal morphology with flexible cylinders as building blocks.45,46 This model gives two radii of gyration that were determined from the low-q region and the transition region between two slopes of the scattering curve indicated in Fig. 3. The slope at low-q, i.e. in the regime where the Guiner model is valid, RG represents the overall size of the polymer particle. However, in the case of (large) polymers, it can be challenging to reach the Guinier regime due to limited q-range of most instruments. The second RG, however, can be converted to a Kuhn length l by using the eqn (6)68,69
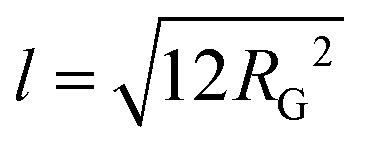 | (6) |
The Kuhn length is twice the persistence length and a measure of the polymer chain stiffness. For the data shown in Fig. 3, we obtained l = 8.4 and l = 11.2 nm for SP and TMP GGM, respectively. This demonstrates a decreased chain flexibility due to branching, as TMP GGM has a higher galactose substitution degree than SP GGM according to the chemical analysis results (Table 2). This is not shown by NMR though, but here as discussed above the quantification of galactose is a bit uncertain. Another factor that can affect the results is the presence of higher amounts of arabino glucurunoxylan constituents in the SP sample.
Due to a high density of short galactose branches on the polysaccharide backbone, we expect the GGMs to behave as comb-like polymers. A computational study showed that branching considerably stiffens the backbone of comb-like polymers and the Kuhn segment length increases with increasing side chain length due to excluded volume interactions between side chains.70
Films from dissolved cellulose were characterized with AFM in non-contact mode in air before the ellipsometry and neutron reflectometry experiments. Fig. 4 shows the topographical features of the spin-coated cellulose surface. The surface is uniformly covered with cellulose fibrils associated in random network giving a fairly smooth layer and the root mean square roughness of the film is 3 nm.
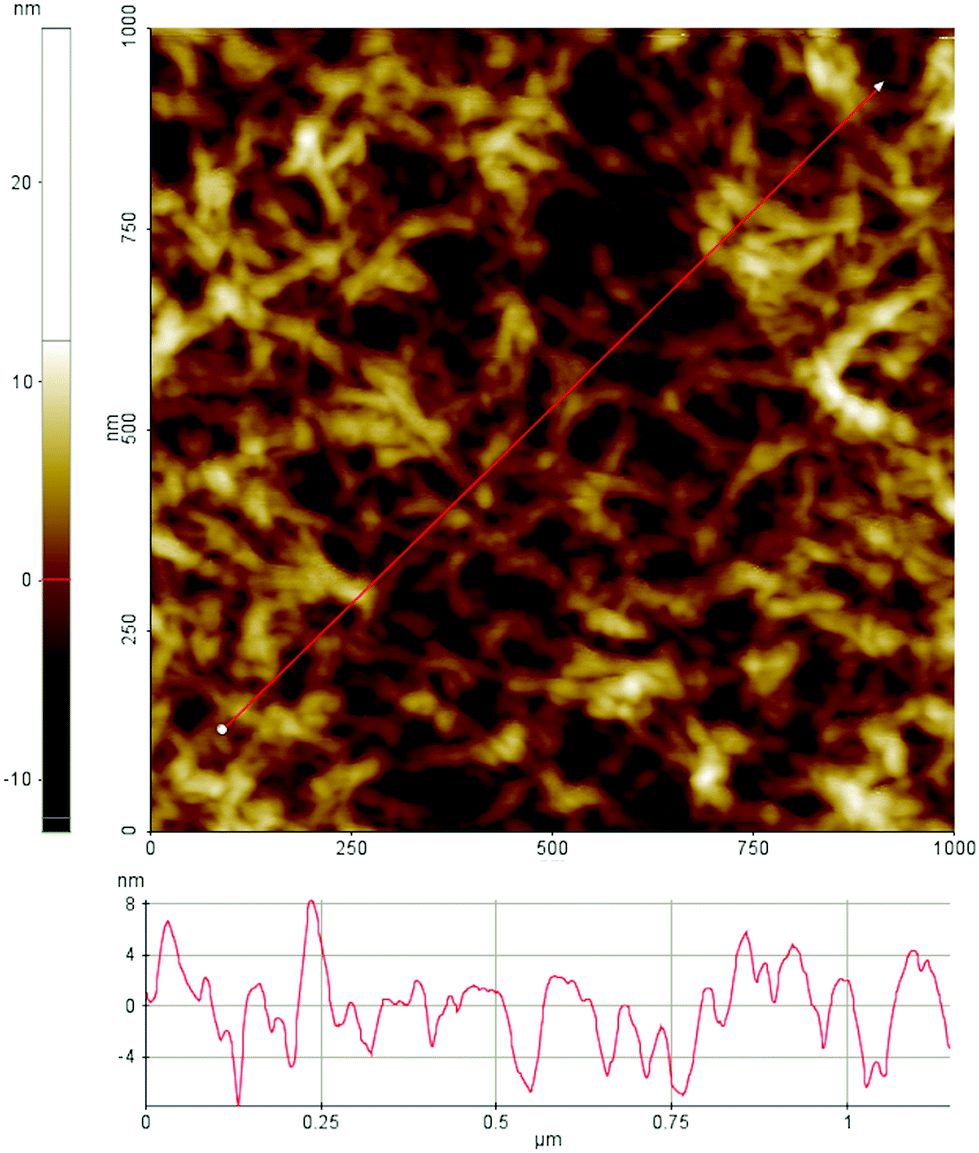 | ||
| Fig. 4 AFM topography image of a cellulose film spin coated on silica substrate. | ||
We note that the applied protocol gives high reproducibility when it comes to surface structures as observed with AFM in air. Unfortunately, it is not possible to determine the crystallinity of the film with AFM. It was also not possible to image the surfaces under water with the used AFM set-up. This is partly due to swelling of the surface layer as observed by ellipsometry and described further below. Swelling of the cellulose film in water has also been observed by Aulin et al.75
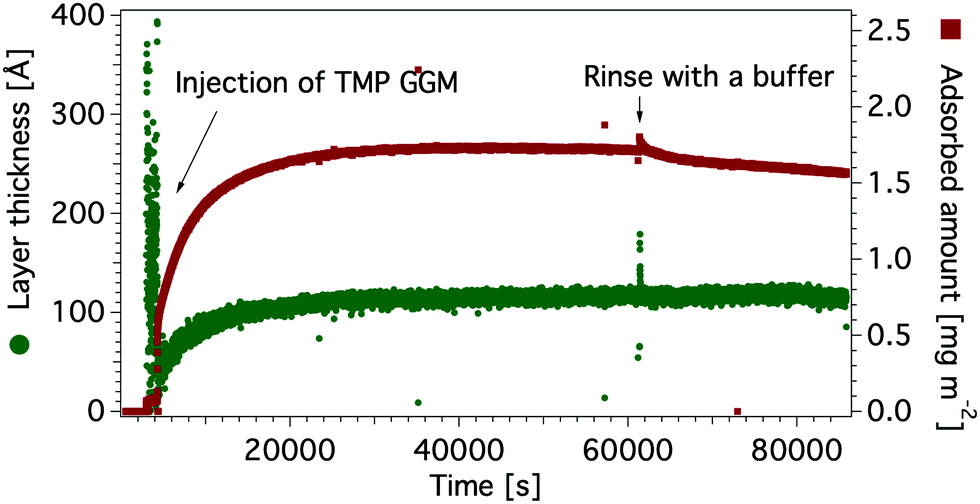 | ||
| Fig. 5 Adsorbed amount (red) and layer thickness (green) of TMP GGM on a hydrophobic silicon oxide surface. | ||
Fig. 6 summarizes ellipsometry results for mannans on hydrophobic and cellulose coated surfaces. The adsorption versus time data are shown in Fig. S6 and S7, in the ESI.† The adsorbed amount is higher on cellulose surfaces than on hydrophobic surfaces for all studied polysaccharides. This suggests a stronger attractive interaction of mannans to the cellulose surface. One might speculate that this is due to a specific interaction between the two types of polysaccharides, i.e. mannans and cellulose. It is known that simple sugar surfactants, i.e. hexadecyl maltosides, show strongly attractive head group interactions, which is different for different stereoisomers and have large effect on the assembly behaviour.76
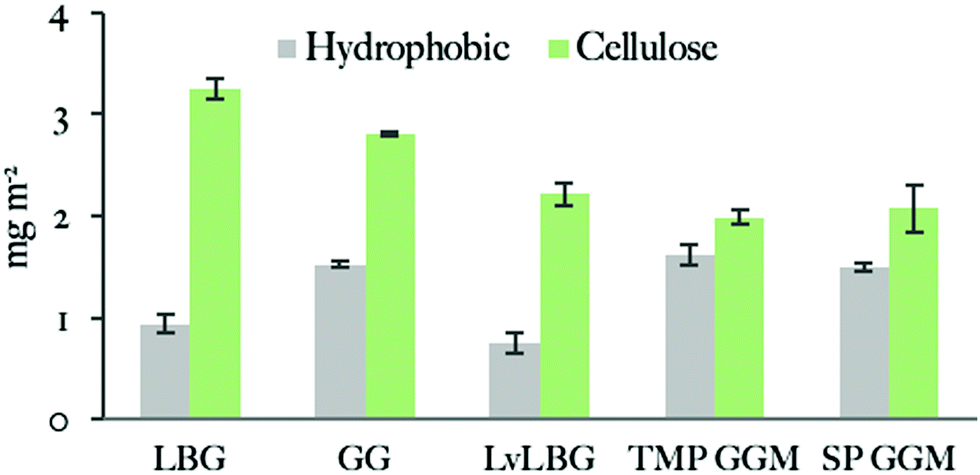 | ||
| Fig. 6 Adsorbed amounts of mannan-based polysaccharides on hydrophobic and cellulose surfaces studied by ellipsometry. Data is represented in the order of decreasing polysaccharide molecular weight. Measurements were repeated at least once. | ||
Similar tendency has also been observed with bacterial cellulose produced in the presence of mannan-based polysaccharides leading to a co-crystallisation of polysaccharides.26,32,77 A contributing factor could also be that the rougher cellulose substrate gives a larger effective area and hence a larger number of adsorption sites.
We note that the ellipsometry data has been analysed with a model comprised of individual homogenous layers, i.e. silicon, silicon oxide and a top layer consisting of cellulose that also includes mannans after adsorption. We have used an alternative model with a separate mannan layer on top of the cellulose layer (Table S3, ESI†). We would like to remind that the cellulose films prepared with the above described method is expected to have a semicrystalline structure. We also note from the AFM image in Fig. 4 that the film has a rather open structure and we can expect that such a structure can entrap a significant amount of water, leading to swelling of the film. This was also observed with ellipsometry (Fig. S5, in the ESI†). Since the mannan–cellulose surface interaction is more favourable than the water–cellulose surface interaction, mannan polymers adsorb on surface replacing the water molecules.23 Under such conditions we can regard the adsorption process as entropically driven. Compared to a flat and smooth hydrophobic surface the rougher cellulose surface has also a larger effective area to which hemicellulose can bind.
The adsorbed amount of the mannans to the cellulose surface is dependent on the molecular weight of polymer chains as the highest value was obtained with the 556 kDa Locust bean gum (3.4 ± 0.2 mg m−2) and the lowest one with the 5.9 kDa SP (2.1 ± 0.3 mg m−2) and 14 kDa TMP GGM (2.1 ± 0.1 mg m−2). The effect of the molecular weight on the adsorption of polysaccharides was previously noted by Kabel et al.25 who compared the adsorption of xylans with different molecular weights to bacterial cellulose. However, a study by Hannuksela et al.5 on the interaction between enzymatically modified guar gum and bleached kraft pulp found that the galactose substitution degree has a stronger impact on the adsorbed amount to cellulose than molecular weight. The highest adsorption was found for mannans with lower density of galactose units for both high and low molecular weight samples. The authors suggested that a close contact between mannan backbone and cellulose surface is necessary to obtain high adsorbed amount and a large density of side groups prevents this close contact.
We can conclude that all studied mannan polymers have fairly hydrophobic character based on the ellipsometry results that show significant adsorption on the hydrophobic surface. GGMs give higher adsorbed amount on hydrophobic surfaces than galactomannans. A plausible explanation is the acetylation carried by the GGM backbone but not by the galactomannans (Table 2).
The NMR spectra indicated that the acetylation is randomly distributed along the polysaccharide chain. An additional contributing factor may be the amphiphilic character of sugar monomers, displaying a hydrophobic surface that makes hydrophobic interactions favourable78 as described e.g. for cellulose.79,80 The hydrophobicity of mannoses is suggested to be similar or even slightly higher than for glucose.78 With the lower degree of galactosylation of the used GGM (Table 2) it could be argued that the GGM on average have longer unsubstituted backbone regions which potentially, in addition to acetyls, could be responsible for interactions with the hydrophobic surface. All the polysaccharides have higher adsorbed amount on the cellulose surface than on the hydrophobic surface. This is possibly due to attractive interaction between the carbohydrate groups of the adsorbing polymer and those in the cellulose surface. Although this cannot be the driving force of the hemicellulose attachment, it can possibly make the adsorption less reversible. In this respect, it can lead to an apparently higher adsorption. In addition, the more open structure of the cellulose film will lead to higher specific area as discussed above. The adsorption was found to increase with the molecular weight and in contrast to the study of Hannuksela et al.,5 we did not find a correlation between difference in branching as indicated by the chemical analysis (Table 2) and adsorption.
| LBG | GG | LvLBG | TMP GGM | SP GGM | ||
|---|---|---|---|---|---|---|
| Adsorbed amount (mg m−2) | QCM-D | 26 ± 6 | 30 ± 10 | 31 ± 4 | 28 ± 3 | 28 ± 1 |
| Ellipsometry | 0.94 ± 0.09 | 1.53 ± 0.03 | 0.8 ± 0.1 | 1.6 ± 0.1 | 1.50 ± 0.03 | |
| Solvent content [%] | 96 | 95 | 97 | 94 | 95 |
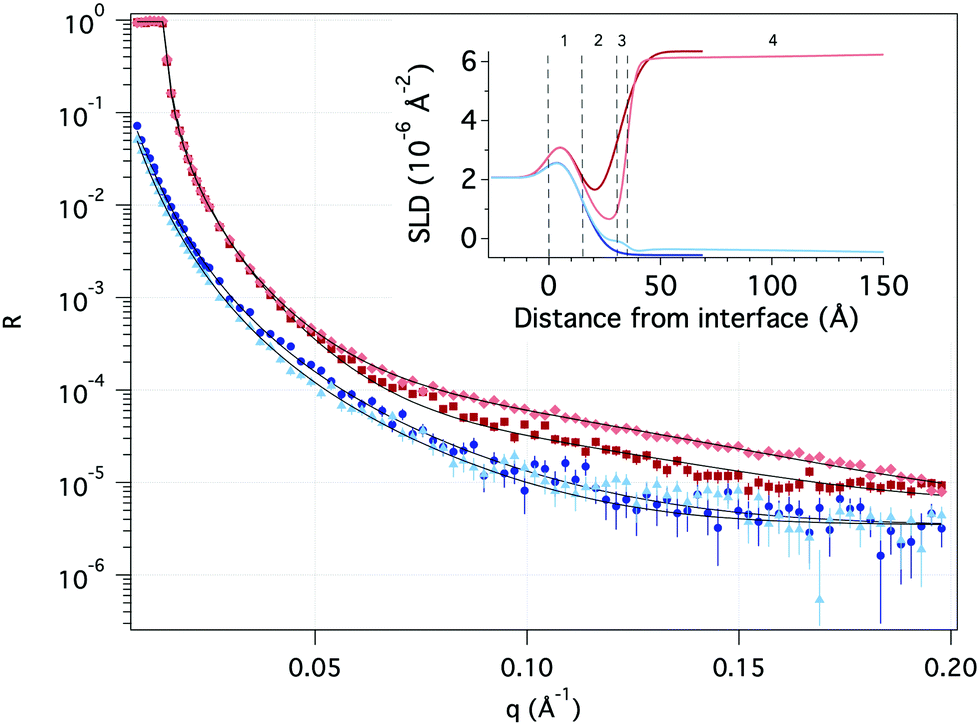 | ||
| Fig. 7 Neutron reflectivity as a function of momentum transfer (q) of pure hydrophobic surface (dark red in D2O and dark blue in H2O) and after adsorption of TMP (light red in D2O and light blue in H2O). Solid black lines represent the theoretical fit. The inset illustrates the scattering length density (SLD) profile as a function of distance from the Si surface based on the fitting with the numbers indicating layers described in Table 8. | ||
| 1 | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| SiOx | Hydrophobic layer | Transition layer | GGM | |||
| D2O | H2O | D2O | H2O | |||
| SLD [10−6 Å−2] | 3.47 | −0.44 ± 0.02 | 0.68 ± 0.08 | 0.5 ± 0.4 | 3.4 ± 0.1 | 1.9 ± 0.5 |
| Layer thickness [Å] | 15.6 ± 0.2 | 15.8 ± 0.1 | 4.0 ± 0.2 | 210 ± 11 | ||
| Solvent [v/v, %] | 8.7 ± 0.6 | 6.9 ± 0.3 | 14 ± 1 | 97.1 ± 0.1 | ||
| Roughness [Å] | 8.2 ± 0.1 | 8.3 ± 0.1 | 2.1 ± 0.2 | 97 ± 13 | ||
A significant change in the measured reflectivity is observed after the addition of TMP GGM compared to the bare surface, although the changes in the measurements with H2O solvent are smaller due to the comparatively low scattering contrast between H2O and TMP GGM.
The best fit to the data with a χ2 of 3.6 was obtained by using a simple optical model where the TMP GGM layer is divided into two. Here, the layer closest to the hydrophobic surface is expected to have more of GGM moieties with acetyl groups and parts of the hydrophobic layer i.e. the transition layer. The top layer is expected to be much more hydrated with more of the non-acetylated sugar moieties. The roughness takes care of the fact there are no sharp borders between the layers, but rather a (Gaussian) distribution of matter.
The transition layer is around 4 Å with the scattering length density (SLD) equal to 0.7 × 10−6 Å−2 in D2O and 0.5 × 10−6 Å−2 in H2O. This suggests that there is very little water in this layer. The top layer is 210 Å with the SLD of 3.4 × 10−6 Å−2 and 1.9 × 10−6 Å−2 in D2O and H2O, respectively. According to Raghuwanshi et al.82 every glucose unit in cellulose contains three hydrogens that are instantly exchanged with deuterium, therefore SLD of the polysaccharide is different in D2O and H2O. The top layer is much thinner than expected from the hydrodynamic radius (see Table 6), indicating that the polymer takes a flat conformation on the hydrophobic surface. As we have shown previously by combining the results obtained with ellipsometry and QCM-D, the top layer is highly hydrated with the solvent amount of 97%.
Conclusions
The nature of the interactions between hemicellulose and cellulose is important for both understanding the processes inside plant cell walls, as well as a tool for development of various sustainable materials to replace fossil-based ones. Although these interactions have been studied since 1980s, the nature of the interactions are still not fully understood due to lack of access to well-defined samples and challenges when it comes to preparation of samples and characterisation techniques.The main aim of this work was to reveal the structural properties of softwood hemicellulose that controls their affinity to cellulose surfaces. The adsorption of softwood hemicellulose (GGM) and similar galactomannans was studied with ellipsometry, QCM-D and neutron reflectometry. To provide further understanding of the forces that control the interaction between hemicellulose and cellulose, the adsorption was also performed on the hydrophobised silica. The combined results from these measurements show:
1. All polymers showed a higher adsorbed amount on the cellulose surface than on the hydrophobic one. This can be related to the larger specific area of the cellulose layer. In addition, attractive interaction between the adsorbing polymer sugar groups and corresponding groups in the cellulose layer might contribute to anchoring the polymer to the surface.
2. The substantial adsorption to the hydrophobic surface indicates that the polysaccharides have a fairly hydrophobic character. The results also suggest that GGM samples are more hydrophobic as shown by NR possibly due to the presence of acetyl side groups in the GGM structure that interact with the hydrophobic surface.
Cellulose films prepared in this study are expected to have a semi-crystalline nature with amorphous regions entrapping large amount of water molecules. The interaction between mannan and cellulose molecules are more favourable than cellulose–water interaction due to relatively hydrophobic nature of both polymers. We therefore expect that increase of entropy due to the release of water upon adsorption is one of the driving forces.
Mannans form a diffuse monolayer with parts extending into the solution as observed by QCM-D and NR. This is supported by the SAXS data showing that GGMs are quite stiff, likely due to bulky galactose side groups. DLS, SLS and Cryo-TEM indicate that polymers are present in the solution in the aggregated state. However, the observed thickness of the layer is smaller, which indicates that they rearrange and take flat conformation at the interface.
The adsorption to cellulose coated surface increases with the molecular weight, however, no clear impact of the galactose substitution degree was found.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research in this work was financed by BIOFUNC research project (supported by the Swedish Foundation for Strategic Research RBP14-0046) and FORMAS. The authors thank the NIST Center for Neutron Research for allocating beam time. We are grateful to Brian Maranville and Zhenhuan (Michael) Zhang at NIST Center for Neutron Research for the support and help. We are grateful to Marie Skepö for giving an opportunity to perform measurements at SOLEIL. We acknowledge SOLEIL for the provision of synchrotron radiation facilities and we would like to thank Javier Perez for assistance in using the SWING beamline. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No. 654000. The National Center for High Resolution Electron Microscopy, Lund University, is gratefully acknowledged for providing experimental resources. We also thank Anna Carnerup at the Physical Chemistry Department, Lund University, for the support provided during the cryo-TEM measurements. We thank prof. Karin Schilleń for the help with DLS and SLS data treatment and interpretation as well as Göran Carlström for help in interpreting the NMR data. We are grateful to Basel Al-Rudainy for the assistance with the NMR analysis. Certain commercial materials, equipment and instruments are identified in this work to describe the experimental procedure as completely as possible. In no case does such an identification imply a recommendation or endorsement by NIST, nor does it imply that the materials, equipment, or instrument identified are necessarily the best available for the purpose.References
- E. Sjostrom, Wood chemistry: fundamentals and applications, Gulf professional publishing, 1993 Search PubMed.
- J. Lundqvist, A. Jacobs, M. Palm, G. Zacchi, O. Dahlman and H. Stålbrand, Carbohydr. Polym., 2003, 51, 203–211 CrossRef CAS.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef.
- T. Hannuksela, M. Tenkanen and B. Holmbom, Cellulose, 2002, 9, 251–261 CrossRef CAS.
- A. Andersson, T. Persson, G. Zacchi, H. Stålbrand and A.-S. Jönsson, Appl. Biochem. Biotechnol., 2007, 137, 971–983 CrossRef PubMed.
- B. Al-Rudainy, M. Galbe and O. Wallberg, Sep. Purif. Technol., 2017, 187, 380–388 CrossRef CAS.
- B. Al-Rudainy, M. Galbe, H. Schagerlöf and O. Wallberg, Holzforschung, 2018, 72, 839–850 CAS.
- B. Ünal, S. Metin and N. D. Işikli, Int. Dairy J., 2003, 13, 909–916 CrossRef.
- P. K. Soma, P. D. Williams and Y. M. Lo, Front. Chem. Eng. China, 2009, 3, 413–426 CrossRef CAS.
- M. A. Cerqueira, A. I. Bourbon, A. C. Pinheiro, J. T. Martins, B. W. S. Souza, J. A. Teixeira and A. A. Vicente, Trends Food Sci. Technol., 2011, 22, 662–671 CrossRef CAS.
- S. Parija, M. Misra and A. K. Mohanty, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 2001, 41, 175–197 CrossRef.
- A. Ebringerová, Z. Hromádková, V. Hříbalová, C. Xu, B. Holmbom, A. Sundberg and S. Willför, Int. J. Biol. Macromol., 2008, 42, 1–5 CrossRef PubMed.
- K. S. Mikkonen, M. I. Heikkilä, H. Helén, L. Hyvönen and M. Tenkanen, Carbohydr. Polym., 2010, 79, 1107–1112 CrossRef CAS.
- J. Hartman, A. C. Albertsson, M. S. Lindblad and J. Sjöberg, J. Appl. Polym. Sci., 2006, 100, 2985–2991 CrossRef CAS.
- J. Hartman, A. C. Albertsson and J. Sjöberg, Biomacromolecules, 2006, 7, 1983–1989 CrossRef CAS PubMed.
- A. Doliška, S. Willför, S. Strnad, V. Ribitsch, K. S. Kleinschek, P. Eklund and C. Xu, Holzforschung, 2012, 66, 149–154 Search PubMed.
- J. Lundqvist, A. Teleman, L. Junel, G. Zacchi, O. Dahlman, F. Tjerneld and H. Stålbrand, Carbohydr. Polym., 2002, 48, 29–39 CrossRef CAS.
- T. Hannuksela and C. Herve du Penhoat, Carbohydr. Res., 2004, 339, 301–312 CrossRef CAS PubMed.
- T. Hayashi, K. Ogawa and Y. Mitsuishi, Plant Cell Physiol., 1994, 35, 1199–1205 CrossRef CAS.
- Å. Linder, R. Bergman, A. Bodin and P. Gatenholm, Langmuir, 2003, 19, 5072–5077 CrossRef.
- J.-P. Vincken, A. de Keizer, G. Beldman and A. G. J. Voragen, Plant Physiol., 1995, 108, 1579–1585 CrossRef CAS PubMed.
- T. Benselfelt, E. D. Cranston, S. Ondaral, E. Johansson, H. Brumer, M. W. Rutland and L. Wågberg, Biomacromolecules, 2016, 17, 2801–2811 CrossRef CAS PubMed.
- T. Hayashi, T. Takeda, K. Ogawa and Y. Mitsuishi, Plant Cell Physiol., 1994, 35, 893–899 CrossRef CAS PubMed.
- M. A. Kabel, H. van den Borne, J.-P. Vincken, A. G. J. Voragen and H. A. Schols, Carbohydr. Polym., 2007, 69, 94–105 CrossRef CAS.
- K. I. Uhlin, R. H. Atalla and N. S. Thompson, Cellulose, 1995, 2, 129–144 CrossRef CAS.
- S. Ahola, P. Myllytie, M. Osterberg, T. Teerinen and J. Laine, BioResources, 2008, 3, 1315–1328 Search PubMed.
- P. Eronen, K. Junka, J. Laine and M. Österberg, BioRes, 2011, 6, 4200–4217 CAS.
- S. Gao and K. Nishinari, Biomacromolecules, 2004, 5, 175–185 CrossRef CAS PubMed.
- S. Gao and K. Nishinari, Colloids Surf., B, 2004, 38, 241–249 CrossRef CAS PubMed.
- J.-y. Liu, H.-c. Wang, Y. Yin, N. Li, P.-l. Cai and S.-l. Yang, Carbohydr. Polym., 2012, 89, 158–162 CrossRef CAS PubMed.
- T. Iwata, L. Indrarti and J. I. Azuma, Cellulose, 1998, 5, 215–228 CrossRef CAS.
- M. Palm and G. Zacchi, Sep. Purif. Technol., 2004, 36, 191–201 CrossRef CAS.
- F. Örså, B. Holmbom and J. Thornton, Wood Sci. Technol., 1997, 31, 279–290 CrossRef.
- R. Schröder, P. Nicolas, S. J. Vincent, M. Fischer, S. Reymond and R. J. Redgwell, Carbohydr. Res., 2001, 331, 291–306 CrossRef.
- A. Rosengren, S. J. Butler, M. Arcos-Hernandez, K.-E. Bergquist, P. Jannasch and H. Stålbrand, Green Chem., 2019, 21, 2104–2118 RSC.
- G. Wider and L. Dreier, J. Am. Chem. Soc., 2006, 128, 2571–2576 CrossRef CAS PubMed.
- Megazyme, https://www.megazyme.com, accessed December 2017.
- R. Sczech and H. Riegler, J. Colloid Interface Sci., 2006, 301, 376–385 CrossRef CAS PubMed.
- D. P. Chang, M. Jankunec, J. Barauskas, F. Tiberg and T. Nylander, ACS Appl. Mater. Interfaces, 2012, 4, 2643–2651 CrossRef CAS PubMed.
- T. Zemb and P. Lindner, Neutron, X-Rays and Light. Scattering Methods Applied to Soft Condensed Matter, North Holland, 2002 Search PubMed.
- D. E. Koppel, J. Chem. Phys., 1972, 57, 4814–4820 CrossRef CAS.
- SasView, https://www.sasview.org/, accessed February 2019.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- B. Hammouda, J. Appl. Crystallogr., 2010, 43(6), 1474–1478 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1996, 29, 134–146 CrossRef CAS.
- F. Tiberg and M. Landgren, Langmuir, 1993, 9, 927–932 CrossRef CAS.
- J. A. De Feijter, J. Benjamins and F. A. Veer, Biopolymers, 1978, 17, 1759–1772 CrossRef CAS.
- G. Robinson, S. B. Ross-Murphy and E. R. Morris, Carbohydr. Res., 1982, 107, 17–32 CrossRef CAS.
- C. Xu, A. Pranovich, L. Vähäsalo, J. Hemming, B. Holmbom, H. A. Schols and S. Willför, J. Agric. Food Chem., 2008, 56, 2429–2435 CrossRef CAS PubMed.
- Y. Ono, T. Ishida, H. Soeta, T. Saito and A. Isogai, Biomacromolecules, 2016, 17, 192–199 CrossRef CAS PubMed.
- M. V. Voinova, M. Jonson and B. Kasemo, Spectroscopy, 2004, 18(4), 537–544 CrossRef CAS.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391 CrossRef CAS.
- J. Penfold and R. Thomas, J. Phys.: Condens. Matter, 1990, 2, 1369–1412 CrossRef CAS.
- J. A. Dura, D. J. Pierce, C. F. Majkrzak, N. C. Maliszewskyj, D. J. McGillivray, M. Lösche, K. V. O’Donovan, M. Mihailescu, U. Perez-Salas, D. L. Worcester and S. H. White, Rev. Sci. Instrum., 2006, 77(7), 74301–7430111 CrossRef PubMed.
- B. Maranville, W. Ratcliff II and P. Kienzle, J. Appl. Crystallogr., 2018, 51, 1500–1506 CrossRef CAS.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- A. Nelson, J. Phys.: Conf. Ser., 2010, 251, 012094 CrossRef.
- B. Al-Rudainy, M. Galbe, M. Arcos Hernandez, P. Jannasch and O. Wallberg, Polymers, 2019, 11, 35 CrossRef PubMed.
- J. Berglund, S. Achar, M. Lawoko, M. Lindström, F. Vilaplana, J. Wohlert and G. Henriksson, Cellulose, 2019, 26, 2155–2175 CrossRef CAS.
- H. Lindqvist, J. Holmback, A. Rosling, K. Salminen, B. Holmbom, M. Auer and A. Sundberg, BioResources, 2013, 8, 994–1010 CrossRef.
- B. Al-Rudainy, M. Galbe, F. Lipnizki and O. Wallberg, Membranes, 2019, 9, 99 CrossRef CAS PubMed.
- I. M. Sims, D. J. Craik and A. Bacic, Carbohydr. Res., 1997, 303, 79–92 CrossRef CAS PubMed.
- W. Burchard, Laser light scattering in biochemistry, 1992, pp. 3–22 Search PubMed.
- S. Kishani, F. Vilaplana, W. Xu, C. Xu and L. Wågberg, Biomacromolecules, 2018, 19, 1245–1255 CrossRef CAS PubMed.
- D. R. Picout, S. B. Ross-Murphy, N. Errington and S. E. Harding, Biomacromolecules, 2001, 2, 1301–1309 CrossRef CAS PubMed.
- D. R. Picout, S. B. Ross-Murphy, K. Jumel and S. E. Harding, Biomacromolecules, 2002, 3, 761–767 CrossRef CAS PubMed.
- B. W. Mansel, C.-Y. Chu, A. Leis, Y. Hemar, H.-L. Chen, L. Lundin and M. A. K. Williams, Biomacromolecules, 2015, 16, 3209–3216 CrossRef CAS PubMed.
- P. Hernandez-Cerdan, B. Mansel, A. Leis, L. Lundin and M. A. K. Williams, On the Structural Analysis of Polysaccharide-Networks by Transmission Electron-Microscopy: Comparison with SAXS, 2018 Search PubMed.
- Y. Nakamura and T. Norisuye, Polym. J., 2001, 33, 874–878 CrossRef CAS.
- C. Zhang, R. Liu, J. Xiang, H. Kang, Z. Liu and Y. Huang, J. Phys. Chem. B, 2014, 118, 9507–9514 CrossRef CAS PubMed.
- A. L. Da Róz, F. L. Leite, L. V. Pereiro, P. A. P. Nascente, V. Zucolotto, O. N. Oliveira and A. J. F. Carvalho, Carbohydr. Polym., 2010, 80, 65–70 CrossRef.
- H. A. Krässig, Cellulose, structure, accessibility and reactivity, Gordon and Breach Publishers, Philadelphia, PA, 1993 Search PubMed.
- C. J. Garvey, I. H. Parker and G. P. Simon, Macromol. Chem. Phys., 2005, 206, 1568–1575 CrossRef CAS.
- C. Aulin, S. Ahola, P. Josefsson, T. Nishino, Y. Hirose, M. Österberg and L. Wågberg, Langmuir, 2009, 25, 7675–7685 CrossRef CAS PubMed.
- J. Larsson, A. Sanchez-Fernandez, N. Mahmoudi, L. C. Barnsley, M. Wahlgren, T. Nylander and S. Ulvenlund, Langmuir, 2019, 35(43), 13904–13914 CrossRef CAS PubMed.
- S. E. C. Whitney, J. E. Brigham, A. H. Darke, J. S. G. Reid and M. J. Gidley, Carbohydr. Res., 1998, 307, 299–309 CrossRef CAS.
- M. Janado and Y. Yano, J. Solution Chem., 1985, 14, 891–902 CrossRef CAS.
- C. Yamane, T. Aoyagi, M. Ago, K. Sato, K. Okajima and T. Takahashi, Polym. J., 2006, 38, 819–826 CrossRef CAS.
- B. Lindman, G. Karlström and L. Stigsson, J. Mol. Liq., 2010, 156, 76–81 CrossRef CAS.
- P. Eronen, M. Österberg, S. Heikkinen, M. Tenkanen and J. Laine, Carbohydr. Polym., 2011, 86, 1281–1290 CrossRef CAS.
- V. S. Raghuwanshi, J. Su, C. J. Garvey, S. A. Holt, W. Raverty, R. F. Tabor, P. J. Holden, M. Gillon, W. Batchelor and G. Garnier, Cellulose, 2017, 24, 11–20 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm00264j |
| This journal is © The Royal Society of Chemistry 2020 |