
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible membrane deformations by straight DNA origami filaments†
Henri G.
Franquelim
 *a,
Hendrik
Dietz
b and
Petra
Schwille
*a
*a,
Hendrik
Dietz
b and
Petra
Schwille
*a
aMax Planck Institute of Biochemistry, Martinsried near Munich, Germany. E-mail: hgfranq@biochem.mpg.de; schwille@biochem.mpg.de
bTechnical University of Munich, Garching Near Munich, Germany
First published on 6th May 2020
Abstract
Membrane-active cytoskeletal elements, such as FtsZ, septin or actin, form filamentous polymers able to induce and stabilize curvature on cellular membranes. In order to emulate the characteristic dynamic self-assembly properties of cytoskeletal subunits in vitro, biomimetic synthetic scaffolds were here developed using DNA origami. In contrast to our earlier work with pre-curved scaffolds, we specifically assessed the potential of origami mimicking straight filaments, such as actin and microtubules, by origami presenting cholesteryl anchors for membrane binding and additional blunt end stacking interactions for controllable polymerization into linear filaments. By assessing the interaction of our DNA nanostructures with model membranes using fluorescence microscopy, we show that filaments can be formed, upon increasing MgCl2 in solution, for structures displaying blunt ends; and can subsequently depolymerize, by decreasing the concentration of MgCl2. Distinctive spike-like membrane protrusions were generated on giant unilamellar vesicles at high membrane-bound filament densities, and the presence of such deformations was reversible and shown to correlate with the MgCl2-triggered polymerization of DNA origami subunits into filamentous aggregates. In the end, our approach reveals the formation of membrane-bound filaments as a minimal requirement for membrane shaping by straight cytoskeletal-like objects.
Introduction
Biological membranes are dynamic and bendable entities, whose shaping in the cellular context can be realized by a variety of widely conserved membrane-active proteins. The process of cellular membrane bending has been described to be primarily governed by the intrinsic curvature of the scaffolding subunits.1–3 Classical membrane shaping proteins, such as clathrin or BAR domains, have well-defined intrinsic curvatures and imprint their curved shapes onto membranes.4–6 Besides curvature effects, self-assembly of scaffolding subunits into higher-ordered oligomers has been thought to play a role during membrane remodelling. While the importance of protein curvature for membrane shaping is widely substantiated1–3 (especially for highly-curved scaffoldings, like BAR), the relative influence of protein self-assembly remains more difficult to gauge. For example, many cytoskeletal elements involved in membrane shaping, such as FtsZ, septins, actin or microtubules, self-assemble into ordered filaments and bundle once in their active states. FtsZ assembles into treadmilling ring-like filament bundles with a radius of ∼500 nm, at lower densities on the membrane,7,8 and into stable nematic meshes of crowded filaments of milder curvature, at higher densities on the membrane.7–9 Septins were also shown to pack into nematic meshes when bound to membranes, with individual filaments acquiring a preferred radius of curvature of around 1.4 μm.10,11 Microtubule filaments, on the contrary, do not have intrinsic curvature due to their high persistence length (5 mm).12,13 In the case of actin filaments, while these are mostly accepted to be straight (18 μm persistence length),12,13 shallow curvature may arise due to Arp2/3-mediated branching.14Cytoskeletal filaments are also non-equilibrium polymers in constant turnover with monomeric subunits in solution.15,16 Typically, these filament-forming proteins possess ATPase/GTPase activity, requiring nucleoside triphosphate (i.e. ATP or GTP) for multimerization.17–20 The assembly and disassembly of such filaments is therefore highly dynamic and, in addition, tightly regulated by a multiple of stabilizing and destabilizing effector proteins and motors.21–26 Indeed, many membrane remodelling functions, such as motility, cytokinesis and vesicle trafficking, rely on this controllable ability of the cytoskeleton to dynamically (de)polymerize at different timescales and cellular localizations.25–31 Notwithstanding the modest to non-existing intrinsic curvature displayed by the above-mentioned proteins, FtsZ, actin & Co. have been only described to remodel membranes in their active filamentous state, generating for instance wrinkled and/or tubular deformations when reconstituted with membrane model systems.9,32–38 How straight filaments, like actin or microtubules, are then able to bend membranes remains an open question. Very recently, Sain and colleagues39 used Monte Carlo simulations to infer the tubulation patterns on lipid vesicles arising from a coating with biofilaments. There, the authors modelled how the intrinsic curvature of filaments (i.e. nematic field), but also their bundling interactions (i.e. intermolecular processes) may drive tubulation. Interestingly, one of their predictions was that narrow tubular deformations may still emerge even in the absence of intrinsic curvature, due to the establishment of nematic interactions that allow the membrane to curve perpendicular to the filament's alignment. The authors then proposed the formation of filament bundles as a general driving force for membrane remodelling of vesicles coated with filaments, irrespective of their pre-exiting curvature.
To experimentally decipher the relative contribution of filament formation for the overall process of membrane deformation, here we mimic features of membrane-active cytoskeletal elements by synthetic membrane-active DNA origami objects capable of reversibly forming end-to-end interactions. This approach allows delineating the role of filament formation on membrane shaping from curvature effects by individual subunits.
The DNA origami folding method40–43 takes advantage of the unique and inherent building properties of DNA molecules, and has proven to be an extremely versatile engineering tool,44,45 especially when combined to model membranes.46–51 Seminal studies recently granted us better overview on how to efficiently attach cholesteryl-modified DNA origami to membranes52,53 and investigate diffusion.54–57 Membrane-interacting DNA origami58–65 and DNA tiles66,67 can moreover physically actuate on and deform lipid bilayers, as for instance demonstrated in our earlier work.68 There, we designed a set of curved DNA origami scaffolding subunits that mimic the intrinsic shapes of BAR domain proteins and are able to bend (e.g. tubulate) giant lipid vesicle, as a function of curvature, membrane affinity and surface density. Hence, DNA origami can be fruitfully employed as a modular toolkit for deciphering the physical–chemical foundations of membrane shaping and curvature generation.
In the present work, we take advantage of controllable self-assembly of DNA origami into higher-order objects based on basepair-stacking interactions69,70 to investigate the influence of subunit self-assembly and filament formation on membrane transformation. To this end, we designed a DNA origami subunit consisting of a 20-helix bundle without intrinsic curvature, able to engage self-assembly into filaments on top of lipid bilayers upon increasing the concentration of MgCl2 (and subsequent filament disassembly when the concentration of MgCl2 is reduced). Our measurements with giant unilamellar vesicles (GUVs) show that the DNA origami filaments thus formed are able to shape membranes into wrinkled and tubular deformations, and that such deformations are reversible only occurring if end-to-end interactions and consequent filament formation are triggered.
Considering the abundance of filament-forming motifs involved in cytokinesis and cell division, our biomimetic in vitro approach based on DNA origami provides robust physical–chemical evidences for the importance of controlled filament formation as a significant requirement for membrane remodelling.
Results and discussion
1. Design of self-assembling DNA origami structures
The core DNA origami design used throughout the current work (Fig. 1A and Fig. S1, ESI†) was the linear nanostructure named origami L (L for Linear), previously employed in ref. 68 as mimicry for a non-curved (“flat”) BAR domain. This 20 helix-bundle with approximate 110 nm × 16 nm × 8 nm dimensions displays additional functionalisation sites, notably (a) 7 top positions (termed T0–T6) where Alexa488-labeled DNA strands can be incorporated for fluorescence detection and (b) 3 bottom positions (termed B0, B3 and B6) where cholesteryl-modified DNA strands with tetraethylene glycol spacers (TEG-chol) can be added for membrane anchoring.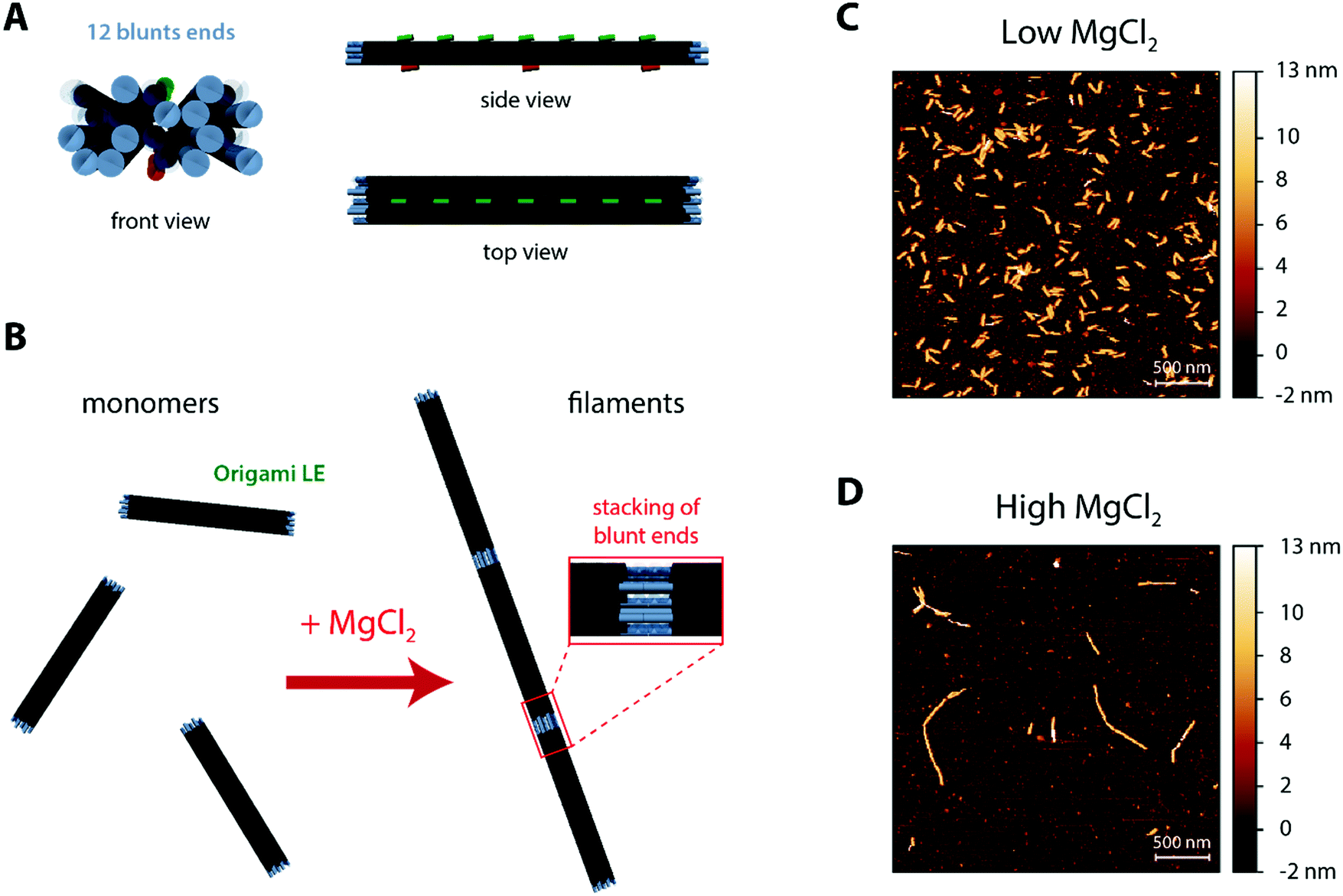 | ||
| Fig. 1 Design and structure of the filament-forming DNA origami LE. (A) Different views of the origami LE, depicting the added 12 matching blunt ends at both edges for end-to-end polymerization (blue), 7 top positions for fluorescence labels (green) and 3 bottom positions for membrane anchors (orange). (B) Representation of the MgCl2-triggered polymerization and respective stacking of blunt ends (inset). (C–D) Atomic force microscopy images on PLL-mica of DNA origami LE at a low MgCl2 (5 mM MgCl2 + 300 mM NaCl) and a high MgCl2 (70 mM MgCl2 + 187.5 mM NaCl) buffer. At low MgCl2 (C), origami LE is mostly monomeric; on the contrary upon increasing MgCl2 (D), origami LE self-assembles into longer filamentous structures. | ||
For the formation of DNA origami filaments (Fig. 1B), we have implemented a strategy analogous to the one previously used to generate arc-like oligomers from curved DNA origami subunits.68 Whereas each of the 20 helix-bundle edges would be usually kept as single-stranded segments, to avoid unnecessary oligomerization due to blunt-end interactions; some of these edges can be hybridized with complementary DNA staples and localized double-stranded blunt ends are formed. Throughout this work, we then intentionally added 12 matching blunt ends at both edges of defined helices on our DNA origami (named origami LE; E for Ends), in order to allow for stable intermolecular stacking interactions (Fig. 1B). Moreover, as blunt-end stacking can be further strengthen by elevating the total amount of Mg2+,69 MgCl2 can be here used as a controllable oligomerization trigger.
Hence, in order to characterize the ability of our designed origami LE to self-assemble, we pre-incubated origami samples in iso-osmolar buffer solutions containing low (5 mM) and high (70 mM) MgCl2 amounts for 15 minutes. Subsequently, we deposited the samples on top of freshly cleaved mica pre-coated with poly-L-lysine (PLL-mica)71 for visualization under atomic force microscopy (AFM). As seen in Fig. 1C, origami LE is mostly in a monomeric form at a low MgCl2 concentration (5 mM Tris–HCl, 1 mM EDTA pH 8.0 buffer with 300 mM NaCl and 5 mM MgCl2). Upon increasing the total MgCl2 concentration to 70 mM, (5 mM Tris–HCl, 1 mM EDTA pH 8.0 buffer with 187.5 mM NaCl and 70.625 mM MgCl2), blunt-end stacking interactions are favoured and μm-long DNA origami filaments made of lined-up LE subunits are formed, as seen in Fig. 1D.
By recapitulating other polymerization strategies,56,65,68,72 we further developed a DNA origami variant lacking blunt ends (origami L, Fig. S2A, ESI†) and another structure presenting 14 lateral single-stranded sticky TATATA extensions on both sides for side-to-side interactions (origami LS, S for Sides; Fig. S2B, ESI†). These control structures were subsequently deposited on PLL-mica and visualized under AFM. As seen in Fig. S3 (ESI†), whereas origami L, due to the absence of multimerizing strands, will stay monomeric at a high MgCl2 concentration (Fig. S3A, ESI†), origami LS on the contrary will form sheet-like oligomers resembling larger platforms (Fig. S3B, ESI†).
2. Formation of DNA origami filaments on supported lipid bilayers
After validating the functionality of our designed DNA origami in aqueous solution, we examined whether a membrane-bound variant of origami LE may freely self-assemble on top of lipid membranes. Hence, we started to assess (via laser scanning confocal microscopy) the interaction of origami L3E, a cholesteryl-modified version of origami LE displaying 3 × TEG-chol moieties for membrane anchoring and 7 × Alexa488 moieties for fluorescence detection, towards supported lipid bilayers (SLBs) composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) deposited on top of freshly-cleaved mica and doped with 0.01 mol% DiD for fluorescence detection. Since our goal was to trigger filament formation only once the DNA origami monomers were stably bound to membranes, all the initial attachment steps were performed at a low MgCl2 regime (5 mM MgCl2, 300 mM NaCl). As seen in Fig. 2A and Fig. S4A, B, Movie S1 (ESI†), the fluorescence signal of membrane-bound DNA origami L3E was homogenously distributed on top of the lipid bilayer (with rapidly diffusing membrane-bound particles). Fluorescence recovery after photobleaching (FRAP) (Fig. S5A and Movie S1, ESI†) further confirmed that the membrane-bound origami L3E was fully mobile on top of the supported bilayer (diffusion coefficient ≈0.2 μm2 s−1). From those results, we can then predict that our membrane-bound origami L3E is mostly in a monomeric (non-polymerized) state, at 5 mM MgCl2.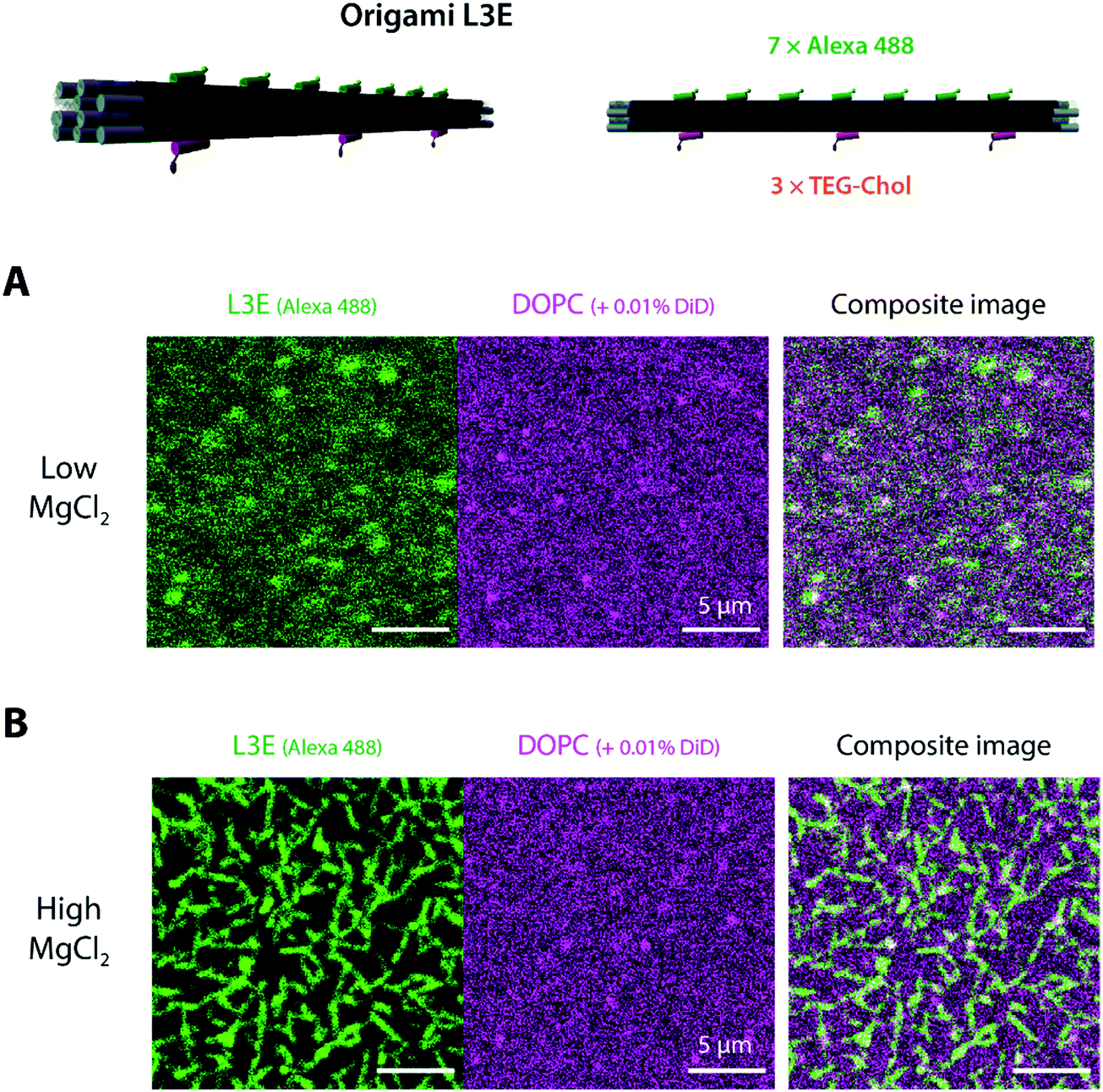 | ||
| Fig. 2 Attachment and polymerization of DNA origami L3E on top of supported lipid bilayers. Interaction of 0.5 nM Alexa488-labelled DNA origami L3E displaying 3 TEG-chol anchors (for membrane binding) and blunt ends (for end-to-end self-assembly) with DOPC SLBs (doped with 0.01% DiD) was followed using confocal microscopy. (A) At low MgCl2 (5 mM MgCl2 + 300 mM NaCl), a mostly homogenous distribution of origami L3E was observed on top of the lipid bilayers. (B) At high MgCl2 (70 mM MgCl2 + 187.5 mM NaCl), on the contrary, origami filaments were observed on top of the lipid bilayer. | ||
Generation of membrane-bound DNA origami filaments can be then triggered by increasing the total amount of MgCl2 in the buffer to 70 mM MgCl2. As seen in Movie S2 (ESI†), reorganization of the membrane-bound L3E subunits into long filaments happened instantaneously after thoroughly mixing MgCl2 into the imaging buffer. The density and length of membrane-bound filaments depends on the total L3E concentration used. Shorter and sparsely-distributed membrane-bound filaments were generated at 0.1 nM L3E (Fig. S4C, ESI†), whereas longer and more density-packed filaments/bundles were formed with 0.5 nM L3E (Fig. 2B and Fig. S4D, ESI†). FRAP experiments (Fig. S5B and Movie S3, ESI†) further revealed that origami L3E, once oligomerized, was largely immobile within the filaments (mobile fraction ≈10%); although lipid diffusion seemed mostly unaffected by the increase in MgCl2 concentration.
3. Self-assembly of DNA origami oligomers on freestanding giant unilamellar vesicles
For the remaining sections, we used giant unilamellar vesicles (GUVs) as a freestanding membrane model system to further investigate the mechanism of action of filament-forming DNA origami.First, we allowed overnight binding of origami L3E, at different concentrations and in a low MgCl2 buffer, to GUVs composed of DOPC (and doped with 0.05% Atto 655-DOPE for fluorescence detection). As seen in Fig. 3A and Fig. S6, S7C (ESI†), GUVs were homogenously decorated with fluorescently-labelled origami L3E, independently of the origami concentration used. Also, no filamentous structures, patches or partners were observed; suggesting that origami L3E is mostly in a monomeric state at 5 mM MgCl2. In order to avoid GUV destabilization due to shear stress, addition of MgCl2 for triggering filament formation was performed by gently pipetting MgCl2 from the top of the chamber. Hence, as our triggering signal will be now diffusion-limited, formation of membrane-bound DNA origami filaments on GUVs required several minutes to occur. In this regard, we allowed a minimal 30 min equilibration time before imaging the GUVs at high MgCl2. As depicted in Fig. 3B, 4C and Fig. S7F, and Movies S4–S5 (ESI†), upon increasing the amount of MgCl2 in the system, stable membrane-bound DNA origami filaments were formed on top of GUVs, due to blunt-end stacking of origami L3E. To facilitate the identification of isolated DNA origami self-assembles (such as filaments) on the membrane, total origami concentrations were mostly kept ≤0.25 nM. While at low L3E concentrations (0.1 and 0.25 nM) individual filaments are easily detected on top of GUVs (Fig. 3B–D and 4C), at high L3E concentrations (1 nM), on the contrary, GUVs appeared fully covered by DNA origami and individual filaments were hardly distinguishable within the overall dense meshwork of bundles (Fig. 3E).
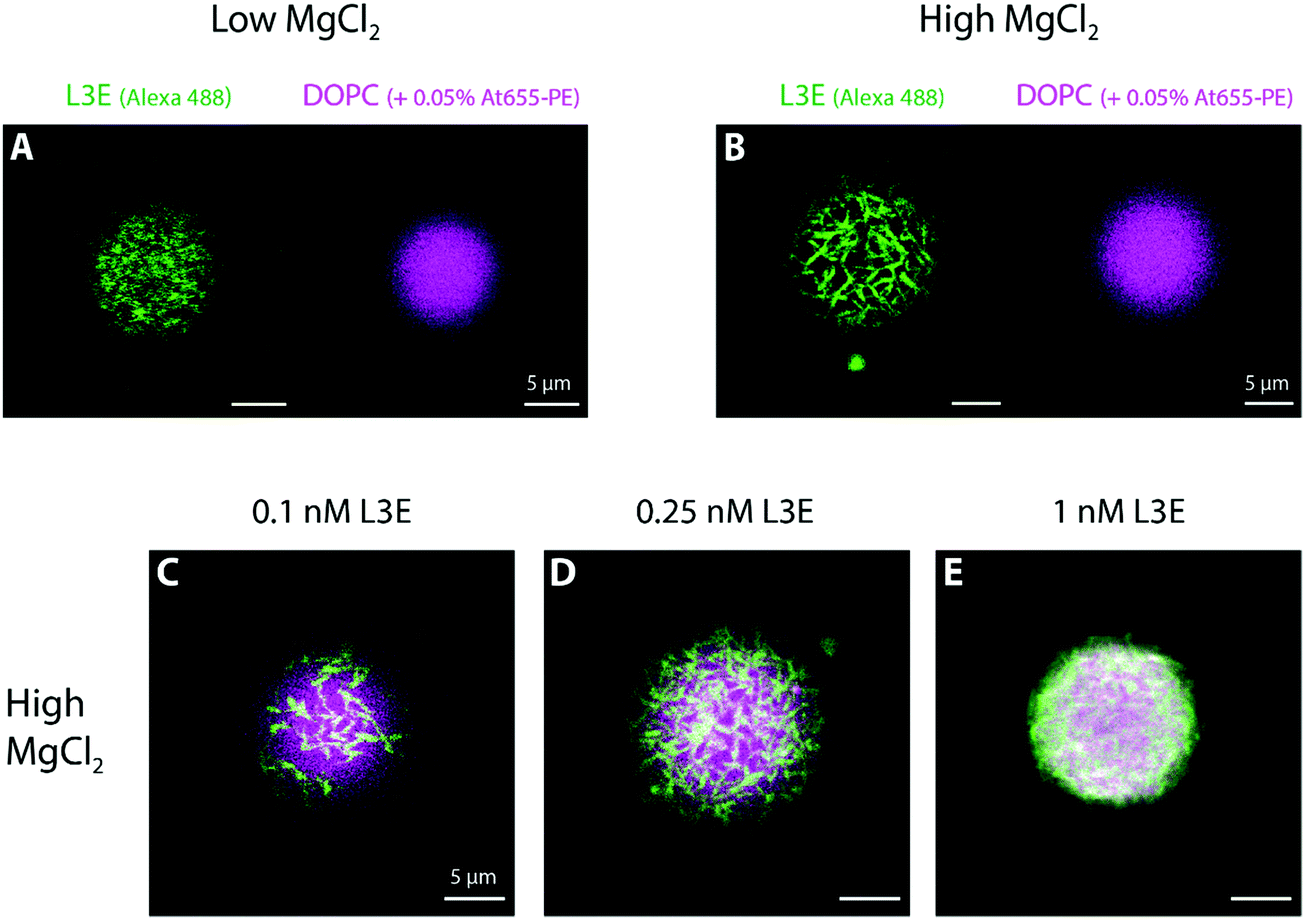 | ||
| Fig. 3 Self-assembly of DNA origami L3E on top of giant unilamellar vesicles. (A) Interaction of 0.25 nM Alexa488/TEG-chol-modified DNA origami L3E displaying blunt ends with DOPC GUVs (doped with 0.05% Atto655-DOPE) in the presence of low MgCl2 (5 mM MgCl2 + 300 mM NaCl). As depicted for the pole of selected GUVs, DNA origami was here homogenously distributed on top of the lipid vesicles. (B) At high MgCl2 (70 mM MgCl2 + 187.5 mM NaCl), origami L3E self-assembled into membrane-bound filaments. (C–E) Zoomed composite images of polymerized DNA origami L3E filaments on the pole of GUVs at different total concentration of origami (0.1–1 nM). As the overall concentration of DNA origami increases, a denser meshwork of origami filaments is observed. | ||
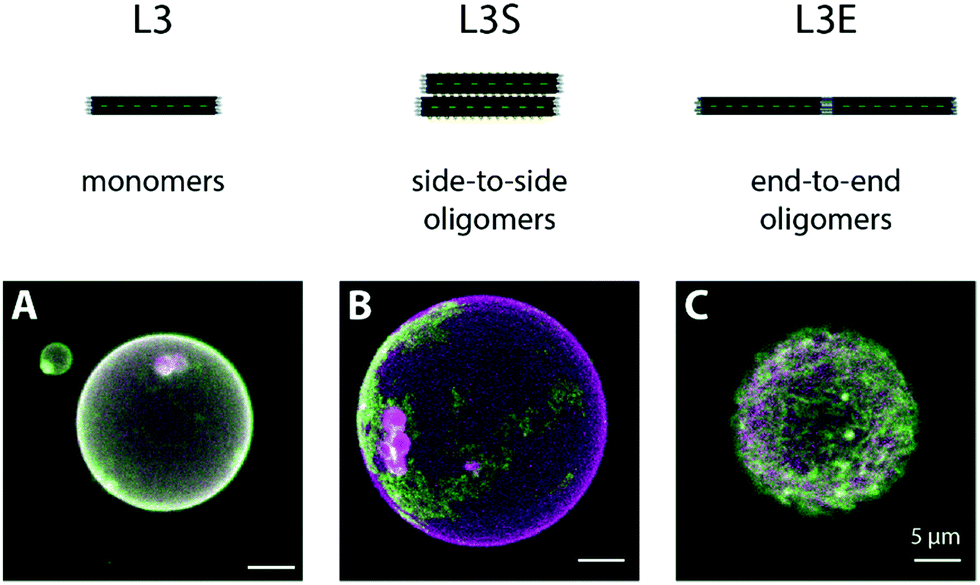 | ||
| Fig. 4 Polymerization of membrane-bound origami L3, L3S and L3E at high MgCl2. Organization of membrane-bound DNA origami L3 lacking blunt ends (A), L3S with lateral overhangs (B) and L3E displaying blunt ends (C) upon MgCl2-triggered self-assembly. DOPC GUVs were initially pre-incubated with 0.1 nM DNA origami at low MgCl2 (5 mM MgCl2 + 300 mM NaCl), where we observed a homogenous distribution of DNA origami on top of the GUVs (data not shown). Subsequently, the amount of MgCl2 was increased to 70 mM (and NaCl decreased to 187.5 mM). Membrane-bound DNA origami was allowed polymerize. Whereas origami L3 lacked the ability to self-assemble (A), origami L3S self-assembled into large membrane-bound platforms (B) and origami L3E into a filamentous meshwork (C). Green colour on confocal images corresponds to fluorescence signal of Alexa488-labelled DNA origami. Magenta colour corresponds to the signal obtained from the lipid membrane labelled with 0.05% Atto655-DOPE. | ||
In order to corroborate that the MgCl2-triggered formation of DNA origami filaments on membranes is specific to our L3E design, we examined the binding of an origami L variant (Fig. S2A, ESI†) displaying 3 TEG-chol anchors and lacking blunt-ends at the edges (edges kept as single stranded regions), named origami L3, as negative control. At a low MgCl2, origami L3 homogenously decorated GUVs (Fig. S7A, ESI†), similarly to what we observed for origami L3E in its “monomeric” state. When MgCl2 was subsequently increased to 70 mM, as opposed to origami L3E, no significant change in the membrane distribution of origami L3 was observed, nor filament formation (Fig. 4A and Fig. S7D, ESI†). Since origami L3 lacks blunt ends or other types of polymerizable strands, no DNA origami oligomers can hereafter be formed.
As additional control sample for DNA origami oligomerization, we further examined the lateral self-assembly of membrane-bound DNA origami into sheets using complementary base-pair interactions, analogously to what we had previously reported elsewhere.65 More precisely, we designed a variant of origami L3 (Fig. S2B, ESI†) displaying additional 14 single-stranded TATATA extensions at both sides, named origami L3S, and assessed its binding and self-assembly on top of freestanding membranes. The TATATA sequence is self-complementary, yet quite short with a melting temperature Tm = 5 °C (estimated using the Marmur–Doty formula73). Hence, we expect no significant self-hybridization to occur at low MgCl2. At high MgCl2, on the contrary, lateral self-assembly into sheets will be favoured, as observed under AFM for origami LS (Fig. S3B (ESI†); structure lacking cholesteryl-moieties). Indeed, at 5 mM MgCl2, we observed a homogeneous distribution of origami L3S on top of DOPC GUVs (Fig. S7B, ESI†), indicative of predominantly monomeric (or low oligomeric) structures. After increasing the amount of MgCl2 to 70 mM, we then observed the appearance of DNA origami patterns at the equatorial plane of GUVs, and large DNA origami platforms at the GUV poles (Fig. 4B and Fig. S7E, Movie S6, ESI†), as a direct consequence of the triggered lateral self-assembly into sheet-like polymers by origami L3S.
4. Membrane deformations induced upon polymerization of DNA origami filaments
As described for BAR-mimicking curved DNA origami,68 the density of membrane-bound DNA origami can act as threshold for the induction of curvature on giant vesicles. Indeed, only at high surface densities, membrane-bound DNA origami nanostructures have been reported to fully coat vesicles and sustain aberrant shapes56,65 or, in the case of curved structures, induce membrane tubulation.63,68 Henceforth in this section, we set out to investigate the macroscopic effects caused by the formation of DNA origami filaments on the shape of GUVs, at high DNA origami concentrations.As seen in Fig. S6 (ESI†), no significant membrane deformations were observed at 5 mM MgCl2 on GUVs decorated with origami L3E, even at higher concentrations (e.g. 1 nM). Upon triggering the formation of DNA origami filaments by setting the MgCl2 amount to 70 mM (Fig. 5A), GUVs with increased surface densities of membrane-bound origami L3E acquired a wrinkled and spike-like appearance, as seen in Fig. 5B–E. Overall, membrane remodelling by filaments seems to depend on the total concentration of origami L3E used, hinting for a key role of filament bundling and rearrangement of filaments into nematic phases.39,74–77 At 0.1 nM L3E, no significant membrane deformation by origami filaments was observed (Table S2 (ESI†); fraction deformed vesicles = 10.9 ± 10.1%, Ntotal = 174). At this concentration, the density of membrane-bound origami filaments was too low to fully cover vesicles (Fig. 4C). Upon increasing the concentration of origami L3E to 0.25 nM, a small yet significant fraction of GUVs started to display full coverage by DNA origami. As seen in Fig. 5F, these vesicles appeared to display wrinkled deformations after filament formation by MgCl2 (Table S2 (ESI†); fraction deformed vesicles = 37.1 ± 20.9%, Ntotal = 316). Vesicles pre-incubated with 0.5 nM and 1 nM origami L3E, on the other hand, were fully decorated with DNA origami (isolated filaments hardly observed) and the large majority of those GUVs (∼70%) presented wrinkled and even spike-like deformations, as seen in Fig. 5G–I and Fig. S8C–E, H–J, Movies S7 and S8 (ESI†) (Table S2 (ESI†); for 0.5 nM L3E: fraction deformed vesicles = 65.6 ± 12.1%, Ntotal = 401; for 1 nM L3E: fraction deformed vesicles = 70.2 ± 9.6%, Ntotal = 350). On that regard, recently published Monte Carlo simulations by Sain and colleagues39 further corroborate our observations with DNA origami L3E that tubular membrane deformations can be induced by filaments made of non-curved subunits. Taken into account their theoretical predictions, the membrane deformations observed for origami L3E may be driven by nematic interactions between adhering filaments at high surface densities. Such bundle-induced protrusions can be perceived as anisotropic membrane segments curved perpendicular to the filaments’ alignment,39 as depicted in Fig. 5A, that may arise at nematic defect locations, which will be then hotspots for membrane deformation.39
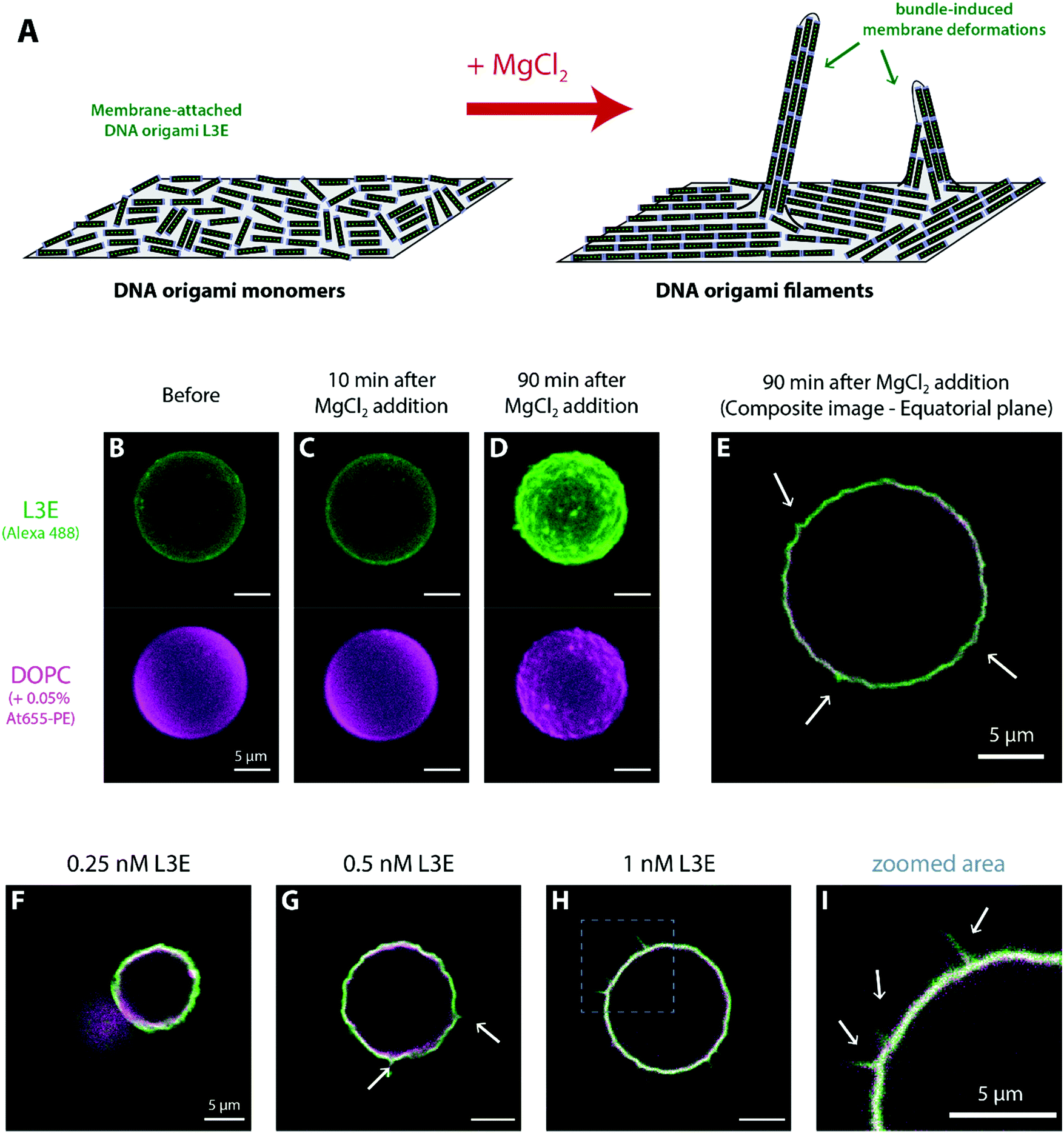 | ||
| Fig. 5 Membrane remodelling by DNA origami L3E filaments. (A) Schematic representation of the hypothetical bundle-induced membrane deformation process. At high densities of filaments, membrane can be curved perpendicular to the filaments’ alignment, leading to outward protrusions. (B–E) DOPC GUVs presenting high membrane-bound densities of DNA origami L3E acquired a rough and spike-like appearance after MgCl2-triggered polymerization of DNA origami into filaments. (F–I) In the presence of high MgCl2 (70 mM MgCl2 + 187.5 mM NaCl), some vesicles pre-incubated with 0.25 nM origami L3E presented significant deformations (B), while most vesicles pre-incubated with 0.5 nM (F) and 1 nM origami L3E (G) acquired well-defined rough and spike-like deformations, as depicted with more detail (arrows) in the zoomed area (H). | ||
To experimentally verify whether the filament formation by membrane-bound DNA origami subunits is indeed the driving force for the reported spike-like and wrinkled deformations on GUVs, we further assessed if (and how) the non-curved origami L3 (lacking the ability to form blunt-end stacking interactions and form filaments) and origami L3S (with the ability to self-assemble into lateral sheets) may deform membranes under similar experimental conditions. As seen in Fig. S8 (ESI†), no significant membrane deformations were observed for membrane-bound L3 (Fig. S8A and F, ESI†) and L3S (Fig. S8B and G, ESI†), even after 3 h at 70 mM MgCl2. For origami L3, as this structure is unable to establish intermolecular interactions (and considering previous results68), the reported absence of membrane deformations was to be expected. For origami L3S, on the contrary, while the lack of tubular membrane deformations was predictable, due to its ability to laterally self-assemble into sheets, we may have expected to observe flat vesicle deformations similar to those previously reported in ref. 65. Such difference may be here due to a shorter incubation time and tighter control of membrane tension (i.e. lack of osmotic shocks).
Overall, as only the polymerized membrane-bound DNA origami L3E showed to promote significant vesicle shaping, our presented results put in evidence that linear filaments (and not lateral sheets) of scaffolding subunits may contribute more efficiently to the process of curvature generation and remodelling of lipid membranes.
Please note that membrane tension plays a fundamental role for the remodelling activity of membrane-shaping proteins, e.g. clathrin78 or BAR domains.79,80 Hyperosmotic imbalances can be used to lower membrane tension and putatively trigger membrane deformations, as previously reported for our curved DNA origami structures.68 Hence, in order guarantee that the observed membrane transformations are purely a consequence of the MgCl2-triggered polymerization of membrane-bound origami subunits, all the experiments performed throughout our present work were done in the absence of osmotic shocks, using osmotically balanced (iso-osmolar) solutions.
5. Reversible DNA origami polymerization and membrane deformations
Stacking interactions between and within DNA origami nanostructures are not only induced by the addition of MgCl2, but can be reversed with the reduction of the total MgCl2 in solution, as elegantly demonstrated by Gerling et al.69 Whereas this effect has been shown for DNA origami structures in aqueous solution, it has so far not been tested for membrane-bound DNA origami. Indeed, most assays on lipid membranes have focused on the reversible MgCl2-mediated adsorption/desorption of bare DNA origami structures to SLBs81,82 or MgCl2-dependent reversible sorting of DNA origami to fluid vs. rigid membrane phases.55,82 Although several studies showing (triggered) polymerization of membrane-bound DNA nanostructures via complementary single-stranded sticky or blunt-end stacking interactions are found in the literature,56,65,72,83,84 fully reversible oligomerization has so far only been documented by Suzuki et al.85 using photo-responsive DNA origami structures.Henceforth, we set out to ascertain in this section whether (a) MgCl2-triggered polymerization of membrane-bound origami L3E nanostructures can be reserved once the concentration of MgCl2 is lowered and (b) which effects would this cause on the shape of deformed vesicles. Such reversible (dis)assembly of membrane-bound DNA origami enabled us to mimic minimal dynamic polymerization and depolymerization properties of cytoskeletal filaments,28 using only a simple set of biochemical cues (i.e. cation exchange). As seen in Fig. 6, polymerization via blunt-end stacking interactions of 1 nM origami L3E bound to a DOPC supported lipid bilayer (Fig. 6A) can be strengthen with the addition of MgCl2 (Fig. 6B) and weakened with the addition of NaCl (Fig. 6C); demonstrating that the MgCl2-mediated formation of DNA origami filaments is indeed fully reversible on lipid bilayers (Fig. 6A–C). Subsequently, we performed similar MgCl2–NaCl exchange with iso-osmolar solutions (to avoid perturbations in membrane tension) on DOPC giant vesicles decorated with 1 nM origami L3E (Fig. 6D–F). At an initially low MgCl2 concentration, GUVs appear spherical and undeformed, as origami L3E is mostly monomeric (Fig. 6D). Upon increasing the amount of MgCl2 in solution, GUVs acquired a wrinkled and deformed appearance (Fig. 6E), as the polymerization of origami L3E into filaments was triggered. By reducing the concentration of MgCl2 with the addition of excess NaCl, GUVs interestingly regained their initial spherical appearance (Fig. 6F), which directly correlated with the disassembly of membrane-bound L3E filaments into L3E monomers.
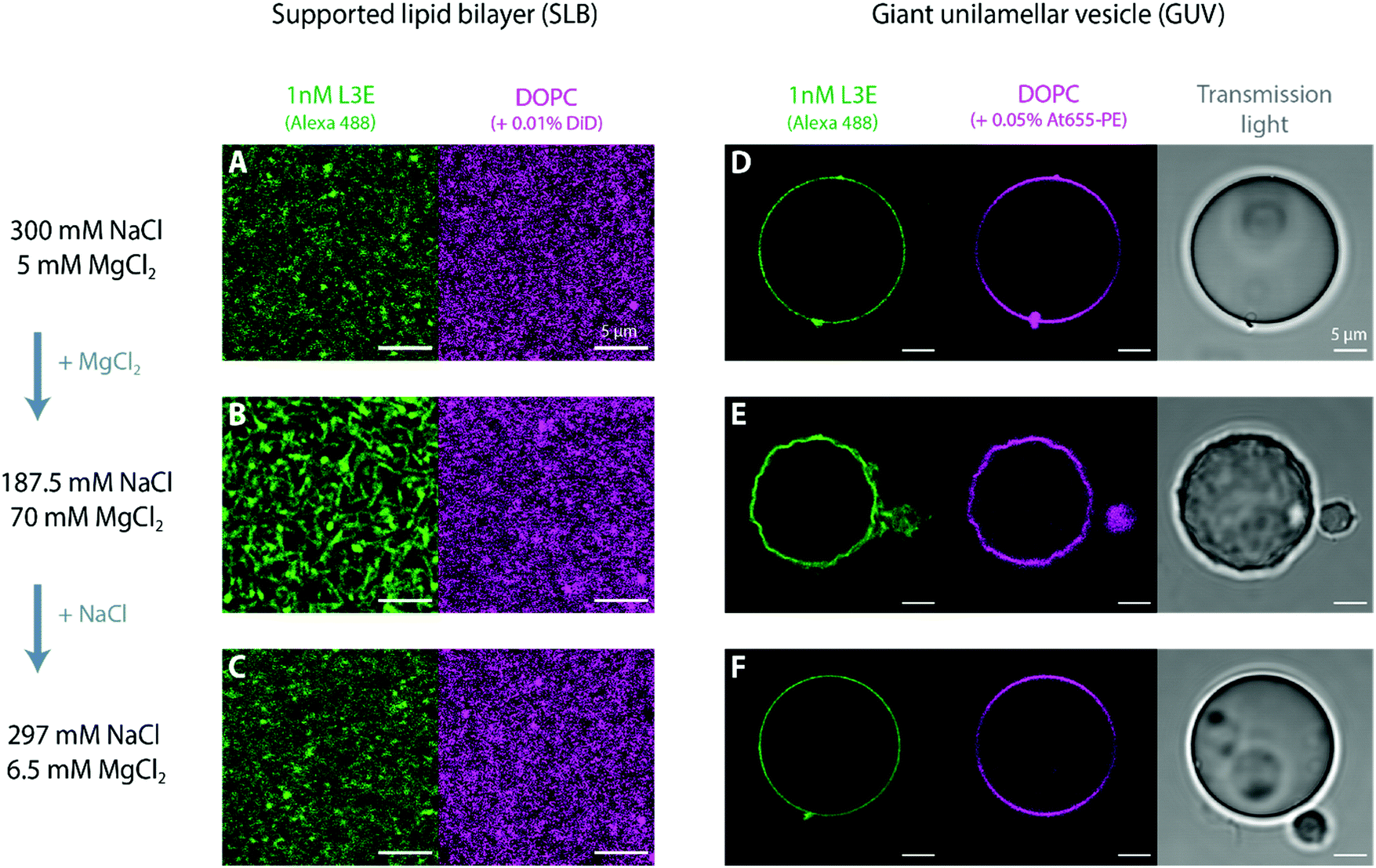 | ||
| Fig. 6 Reversible (de)polymerization of membrane-bound DNA origami L3E filaments. MgCl2-induced polymerization of membrane-bound DNA origami L3E (1 nM) can be reserved on top of a supported lipid bilayer (SLB) and giant unilamellar vesicle (GUV) with excess NaCl. At initially low MgCl2 amounts, membrane-bound origami L3E appears homogeneously distributed on top of SLBs (A), with GUVs remaining spherical/non-deformed (D). Subsequent addition of MgCl2 triggered polymerization of membrane-bound origami L3E into filaments, as seen on SLBs (B). MgCl2-mediated polymerization of DNA origami into filaments further promoted protruding membrane deformations on GUVs (E). Addition of NaCl and consequent reduction of MgCl2 triggered depolymerization of L3E filaments, as seen on SLBs (C). Such NaCl-mediated depolymerization of membrane-bound origami filaments ultimately led to a relaxation of the deformed GUVs into their original spherical shapes (F). Depicted images (A–C) correspond to the same SLB, 5 min prior/after the addition of MgCl2/NaCl. Depicted images (D–F) correspond to the same GUV, 90 min prior/after the addition of MgCl2/NaCl. | ||
Overall, our experiments prove that the polymerization of membrane-bound DNA origami subunits into filaments is reversible and that the generated forces can alter the morphology of freestanding membranes. These observations have important biophysical significance, as we were able for the first time to recapitulate the self-assembly and disassembly of cytoskeleton-like biomimetic filaments and respective membrane remodelling activity, simply by using DNA origami nanostructures and external cues. Strikingly, while the polymerization of membrane-bound DNA subunits into filaments can impose spatial constrains on membranes, forcing freestanding lipid bilayers to curve, membrane deformations can relax back once filaments depolymerize.
In the end, DNA nanotechnology bestows us with an unprecedent set of new tools to model key structure-functional properties of cytokinetic and membrane shaping proteins. For instance, with the help of DNA origami, we may be soon in the position to systematically comprehend how the mechanical and dynamic properties of biomimetic filaments can influence membrane bending or budding. Hence, our present work opens up new avenues for nanotech applications in the field of synthetic biology and structural biochemistry, helping us understand and mimic the physics underlying biological processes.
Conclusions
In this work, we developed and tested three variants of practically non-curved DNA origami with distinct inducible self-assembly properties: (a) origami L3E with blunt end edges able to polymerize into membrane-bound thin DNA origami filaments, (b) origami L3S with lateral single-stranded extensions able to generate large membrane-bound DNA origami sheets, and finally (c) origami L3 lacking the ability to polymerize. Polymerization of membrane-bound origami L3E into filaments (but also origami L3S into sheets) was triggered by increasing the overall MgCl2 concentration in solution. We showed that wrinkled and/or spiking membrane deformations can only be generated once membrane-bound DNA origami self-assembles into filaments, and that such membrane deformations can relax back once DNA origami filaments depolymerize by reducing MgCl2 in solution. Interestingly, while origami L3E was able to shape membranes when polymerized, origami L3S and L3, on the contrary, were not able to generate membrane deformations under similar experimental conditions.Altogether, our results provide clear evidences that membrane scaffolding subunits, even when lacking intrinsic curvature (e.g. actin and microtubules), may deform membranes once polymerized. At the end, our presented biomimetic approach adds exciting perspectives towards understanding the physical–chemical laws underlying vesicle shaping, validating the vital role of linear aggregation (end-to-end interactions) and controllable filament formation during the remodelling of biomembranes.
Experimental section
Materials
Single-stranded M13mp18 scaffold plasmid (p7249) and high purity salt free (HPSF) purified staple oligonucleotides required for DNA origami preparation were purchased from Bayou Biolabs (Metairie, LA, USA) and Eurofins Genomics (Ebersberg, Germany), respectively. 5′-Alexa488-functionalized oligonucleotides (HPLC-purified) needed for fluorescence detection were acquired from Eurofins Genomics. 5′-TEG-chol-functionalized oligonucleotides (also HPLC-purified) required for membrane binding were obtained from Sigma-Aldrich (Taufkirchen, Germany). Detailed list of functionalized oligonucleotides can be found in Table S1 (ESI†). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) utilized for producing supported and freestanding lipid bilayers was purchased from Avanti Polar Lipids (Alabaster, AL, USA). The fluorescent lipid Atto655-DOPE was obtained from AttoTEC GmbH (Siegen, Germany) and DiIC18(5) (DiD) from Thermo Fischer Scientifics (Waltham, MA, USA).Production of the DNA origami nanostructures
DNA origami produced throughout this work was based on a previous design (named origami L, Linear) described elsewhere.68 Briefly, folding of the DNA origami structures was performed in a one-pot reaction mix. 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nM staple oligonucleotides were mixed with 20nM p7249 plasmid in a folding buffer containing 5mM Tris–HCl, 1mM EDTA, 20mM MgCl2 and pH 8.0 (1 × FOB20). Thermal annealing was subsequently performed from 65 to 60°C in 1h and from 59 to 40°C in 40h, on a Eppendorf Mastercycle Pro (Hamburg, Germany) thermal cycler. Purification of the folded structures (in order to remove the excess of staple strands) was done using size-exclusion centrifugal filtration with Amicon Ultra 100kDa MWCO filters (Merck Millipore, Darmstadt, Germany) with an experimental buffer consisting of 5mM Tris–HCl, 1mM EDTA, 5mM MgCl2, 300mM NaCl, pH 8.0. Bulk concentrations of the purified fluorescently-labelled DNA origami structures were finally determined using a Jasco FP-8500 spectrofluorometer (Tokyo, Japan).
nM staple oligonucleotides were mixed with 20nM p7249 plasmid in a folding buffer containing 5mM Tris–HCl, 1mM EDTA, 20mM MgCl2 and pH 8.0 (1 × FOB20). Thermal annealing was subsequently performed from 65 to 60°C in 1h and from 59 to 40°C in 40h, on a Eppendorf Mastercycle Pro (Hamburg, Germany) thermal cycler. Purification of the folded structures (in order to remove the excess of staple strands) was done using size-exclusion centrifugal filtration with Amicon Ultra 100kDa MWCO filters (Merck Millipore, Darmstadt, Germany) with an experimental buffer consisting of 5mM Tris–HCl, 1mM EDTA, 5mM MgCl2, 300mM NaCl, pH 8.0. Bulk concentrations of the purified fluorescently-labelled DNA origami structures were finally determined using a Jasco FP-8500 spectrofluorometer (Tokyo, Japan).
Atomic force microscopy
Atomic force microscopy (AFM) was used to verify the correct assembly of the folded DNA origami nanostructures and infer the polymerization properties of the various structures at low and high MgCl2. To this end, 1 μL DNA origami (10 nM stock) were mixed with 50 μL of a 5mM Tris–HCl, 1mM EDTA, pH 8.0 buffer solution containing low (5mM MgCl2, 300 mM NaCl) or high (70mM MgCl2, 187.5 mM NaCl) MgCl2 amounts, and incubated for 15 min, before deposition on top of poly-L-lysine (PLL) functionalized mica (PLL-mica).71 For the preparation of PLL-mica substrates, 50 μL of a 0.01% PLL solution (Sigma-Aldrich) was deposited on top of freshly-cleaved mica for 10 min; then abundantly rinsed with ddH2O and imaging buffer.
Measurements were performed on a JPK Nanowizard 3 (Berlin, Germany) mounted on top of a Zeiss LSM 510 Meta microscope. AFM imaging was done in the QI mode (also known as Quantitative Imaging mode), after letting the DNA origami settle down on the positively-charged PLL-mica surface for 10 min, using BioLever Mini BL-AC40TS-C2 cantilevers (Olympus) with typical spring constants of 0.09–0.1 N m−1. Setpoint force was set to 200–250 pN, acquisition speed to 61.1 μm s−1, Z-length to 110 nm and image resolution to 256 × 256 pixels. Height, adhesion and slope images were recorded and line-fitted as required. Analysis of height images was performed using JPK SPM Data Processing (version 6.0.55) and Gwyddion (version 2.49).
Preparation of lipid membranes
Supported lipid bilayers (SLBs) were prepared via fusion of small unilamellar vesicles deposited on top of freshly cleaved mica previously glued on top of a glass coverslip, as described elsewhere86 with minor modifications. Briefly, 25 μL of bath-sonicated vesicles (4 mg mL−1 DOPC + 0.01 mol% DiD) were diluted into a final volume of 150 μL low MgCl2 buffer (5mM Tris–HCl, 1mM EDTA, pH 8.0, 5mM MgCl2, 300 mM NaCl). 75 μL of diluted vesicles (0.67 mg mL−1 lipid) were then incubated on top of freshly cleaved mica for 10 min, then rinsed with 1.5 mL low MgCl2 buffer. At the end, a total volume of 150 μL was kept in the chamber.
Giant unilamellar vesicles (GUVs) were mostly utilized throughout this work and prepared by electroformation in PTFE chambers with Pt electrodes, as previously described elsewhere87 with minor modifications. Briefly, 6 μL of a DOPC lipid mixture (2mg mL−1 in chloroform) doped with 0.05 mol% Atto655-DOPE were spread onto two Pt wires and dried in a desiccator for 30min. The PTFE chamber was filled with 350μL of an aqueous solution of sucrose with approximate 585 mOsm kg−1 osmolarity (iso-osmolar compared to the imaging buffer). An AC electric field of 2V (RMS) was applied at a frequency of 10Hz for 1.5h, followed by 2Hz for 0.75h.
Laser scanning confocal fluorescence microscopy
Fluorescence microscopy imaging was performed on a commercial laser scanning microscope LSM 780 with a ConfoCor3 unit and commercial laser scanning microscope LSM 800 (Zeiss, Jena, Germany) using a water immersion objective (C-Apochromat, 40×/1.2W UV-VIS-IR, Zeiss, Jena, Germany). On the LSM 780 system, samples were excited with the 488nm line of an Ar-ion-laser (for Alexa488 excitation) or with the 633nm line of a He–Ne laser (for Atto655 and DiD excitation); while on the LSM 800 system 488 nm and 640 nm laser diodes were used. Images were typically recorded utilizing a 1 Airy unit pinhole and 512 × 512 pixel resolution. Further image analysis was performed using the ImageJ software (http://rsb.info.nih.gov/ij/).
Fluorescence recovery after photobleaching (FRAP) was carried out on the LSM 800. Two circular user defined regions of interest (ROI) with a radius (r) of 3 μm were measured during the experiment, one as reference and the other one corresponding to the photobleached area. Photobleaching was performed at full laser power (100%, 10 iterations). Images were acquired with a 512 × 512 μm pixel resolution, pixel dwell 0.85 μs, and scan time 521.31 ms. No line averaging was used. The mean fluorescence intensities of the ROIs were determined using Zen Blue 2.6 (Zeiss), normalized and corrected for possible drifts and bleaching during acquisition, and finally fitted in OriginPro 2015 using a modified equation derived by Soumpasis:88,89
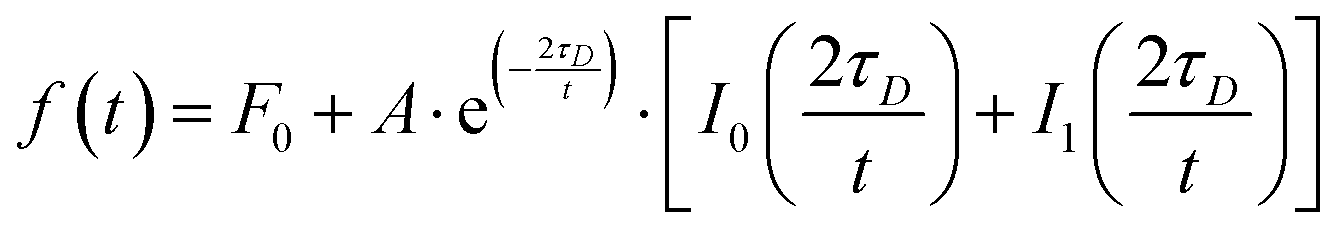
Interaction of DNA origami with model membranes
Experiments with SLBs were carried out in home-build 200 μL chambers, having freshly cleaved mica as substrate glued on top of coverslips with #1 glass bottom thickness. After the SLB was formed, 0.1–0.5 nM DNA origami diluted in low MgCl2 imaging buffer containing 5 mM MgCl2 and 300 mM NaCl was incubated inside the chamber for at least 30 min before fluorescence microscopy imaging.Experiments with GUVs were carried out in 35 μL SensoPlate 384-multiwell plates with # 1.5 glass bottom thickness (Greiner Bio-One, Kremsmünster, Austria). Prior usage, wells were freshly plasma cleaned, then passivated with PLL(20)-g[3.5]-PEG(2) (SuSoS AG, Dübendorf, Switzerland). 3μL of the GUV suspension (pre-diluted 1:10 in iso-osmolar sucrose solution) were mixed with 18μL DNA origami solution at a final 0.1–1nM concentration diluted in low MgCl2 imaging buffer containing 5 mM MgCl2 and 300 mM NaCl. Unless otherwise stated, samples were incubated overnight at 4 °C and let equilibrate at room temperature for 30 min before fluorescence microscopy imaging.
For both model membrane systems, increase of MgCl2 (and reduction of NaCl) was performed by adding few microlitres of a concentrated iso-osmolar 9 × FOB20 buffer solution in the chambers. Samples were then allowed to equilibrate at high MgCl2 (70 mM MgCl2, 187.5 mM NaCl) for several minutes (typically 90–180 min), at room temperature, before fluorescence microscopy imaging.
For the reversible (de)polymerization assays, SLBs and GUVs were prepared in home-made 40 μL chambers connected to an extra 2 mL reservoir. MgCl2 increase (and consequent NaCl reduction) was achieved by adding few microlitres of a concentrated iso-osmolar 9 × FOB20 buffer solution inside the 40 μL chambers. Subsequent addition of NaCl (and dilution of MgCl2), was achieved by filling the reservoir with excess iso-osmolar imaging buffer containing 5 mM MgCl2 and 300 mM NaCl (giving rise to a final salt concentration of 297 mM NaCl and 6.5 mM MgCl2).
Contributions
H. G. F. and P. S. initially conceived the project. H. G. F. carried out the experimental design, conducted experiments, analysed data and wrote the first manuscript draft. P. S. supervised the project and provided funding. H. D. supervised the initial design of DNA origami nanostructures. All authors contributed to revision of the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB-863 – Project ID 111166240. Additional support was provided by the Center for NanoScience (CeNS), Center for Integrated Protein Science Munich (CIPSM) and Nanosystems Initiative Munich (NIM). H. G. F. was awarded a Humboldt Research Fellowship (PTG/1152511/STP) from the Alexander von Humboldt Foundation. P. S. acknowledges the Max Planck Society for further support. The authors thank Jean-Philippe Sobczak for having previously established the strategies to polymerize DNA origami in solution and Alena Khmelinskaia for helpful discussions. Open Access funding provided by the Max Planck Society.References
- M. Simunovic, P. Bassereau and G. A. Voth, Curr. Opin. Struct. Biol., 2018, 51, 99–105 CrossRef CAS PubMed.
- M. Simunovic, G. A. Voth, A. Callan-Jones and P. Bassereau, Trends Cell Biol., 2015, 25, 780–792 CrossRef CAS PubMed.
- R. Lipowsky, Faraday Discuss., 2013, 161, 305–331 RSC.
- H. T. McMahon and J. L. Gallop, Nature, 2005, 438, 590–596 CrossRef CAS PubMed.
- A. Frost, V. M. Unger and P. De Camilli, Cell, 2009, 137, 191–196 CrossRef CAS PubMed.
- B. Qualmann, D. Koch and M. M. Kessels, EMBO J., 2011, 30, 3501–3515 CrossRef CAS PubMed.
- D. A. Ramirez-Diaz, D. A. Garcia-Soriano, A. Raso, J. Mücksch, M. Feingold, G. Rivas and P. Schwille, PLoS Biol., 2018, 16, e2004845 CrossRef PubMed.
- M. Loose and T. J. Mitchison, Nat. Cell Biol., 2014, 16, 38–46 CrossRef CAS PubMed.
- S. Arumugam, G. Chwastek, E. Fischer-Friedrich, C. Ehrig, I. Monch and P. Schwille, Angew. Chem., Int. Ed., 2012, 51, 11858–11862 CrossRef CAS PubMed.
- A. A. Bridges, M. S. Jentzsch, P. W. Oakes, P. Occhipinti and A. S. Gladfelter, J. Cell Biol., 2016, 213, 23–32 CrossRef CAS PubMed.
- A. Beber, C. Taveneau, M. Nania, F. C. Tsai, A. Di Cicco, P. Bassereau, D. Levy, J. T. Cabral, H. Isambert, S. Mangenot and A. Bertin, Nat. Commun., 2019, 10, 420 CrossRef CAS PubMed.
- P. Wisanpitayakorn, K. J. Mickolajczyk, W. O. Hancock, L. Vidali and E. Tüzel, bioRxiv, 2018, 252551, DOI:10.1101/252551.
- F. Gittes, B. Mickey, J. Nettleton and J. Howard, J. Cell Biol., 1993, 120, 923–934 CrossRef CAS PubMed.
- V. I. Risca, E. B. Wang, O. Chaudhuri, J. J. Chia, P. L. Geissler and D. A. Fletcher, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2913–2918 CrossRef CAS PubMed.
- M. Bezanilla, A. S. Gladfelter, D. R. Kovar and W. L. Lee, J. Cell Biol., 2015, 209, 329–337 CrossRef CAS PubMed.
- P. Sens and J. Plastino, J. Phys.: Condens. Matter, 2015, 27, 273103 CrossRef PubMed.
- H. Y. Kueh and T. J. Mitchison, Science, 2009, 325, 960–963 CrossRef CAS PubMed.
- P. de Boer, R. Crossley and L. Rothfield, Nature, 1992, 359, 254–256 CrossRef CAS PubMed.
- P. Ranjith, D. Lacoste, K. Mallick and J. F. Joanny, Biophys. J., 2009, 96, 2146–2159 CrossRef CAS PubMed.
- M. Abbey, M. Gaestel and M. B. Menon, Cytoskeleton, 2019, 76, 55–62 CrossRef CAS PubMed.
- S. Buracco, S. Claydon and R. Insall, F1000Research, 2019, 8(F1000 Faculty Rev), 197, DOI:10.12688/f1000research.18669.1.
- S. T. Sit and E. Manser, J. Cell Sci., 2011, 124, 679–683 CrossRef CAS PubMed.
- Sonal, K. A. Ganzinger, S. K. Vogel, J. Mucksch, P. Blumhardt and P. Schwille, J. Cell Sci., 2019, 132, jcs219899, DOI:10.1242/jcs.219899.
- S. K. Vogel, Z. Petrasek, F. Heinemann and P. Schwille, eLife, 2013, 2, e00116 CrossRef PubMed.
- K. A. Ganzinger and P. Schwille, J. Cell Sci., 2019, 132, jcs227488, DOI:10.1242/jcs.227488.
- S. Arumugam, Z. Petrasek and P. Schwille, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1192–E1200 CrossRef CAS PubMed.
- T. D. Pollard and G. G. Borisy, Cell, 2003, 112, 453–465 CrossRef CAS PubMed.
- W. Brieher, Mol. Biol. Cell, 2013, 24, 2299–2302 CrossRef CAS PubMed.
- L. Blanchoin, R. Boujemaa-Paterski, C. Sykes and J. Plastino, Physiol. Rev., 2014, 94, 235–263 CrossRef CAS PubMed.
- S. Kretschmer, K. A. Ganzinger, H. G. Franquelim and P. Schwille, BMC Biol., 2019, 17, 43 CrossRef PubMed.
- A. W. Bisson-Filho, Y. P. Hsu, G. R. Squyres, E. Kuru, F. Wu, C. Jukes, Y. Sun, C. Dekker, S. Holden, M. S. VanNieuwenhze, Y. V. Brun and E. C. Garner, Science, 2017, 355, 739–743 CrossRef CAS PubMed.
- D. A. Ramirez-Diaz, A. Merino-Salomon, M. Heymann and P. Schwille, bioRxiv, 2019, 587790, DOI:10.1101/587790.
- M. Osawa, D. E. Anderson and H. P. Erickson, EMBO J., 2009, 28, 3476–3484 CrossRef CAS PubMed.
- R. Kusters, C. Simon, R. Lopes Dos Santos, V. Caorsi, S. Wu, J. F. Joanny, P. Sens and C. Sykes, Soft Matter, 2019, 15, 9647–9653 RSC.
- F. C. Keber, E. Loiseau, T. Sanchez, S. J. DeCamp, L. Giomi, M. J. Bowick, M. C. Marchetti, Z. Dogic and A. R. Bausch, Science, 2014, 345, 1135–1139 CrossRef CAS PubMed.
- C. Simon, R. Kusters, V. Caorsi, A. Allard, M. Abou-Ghali, J. Manzi, A. Di Cicco, D. Levy, M. Lenz, J. F. Joanny, C. Campillo, J. Plastino, P. Sens and C. Sykes, Nat. Phys., 2019, 15, 602–609 Search PubMed.
- K. Durre, F. C. Keber, P. Bleicher, F. Brauns, C. J. Cyron, J. Faix and A. R. Bausch, Nat. Commun., 2018, 9, 1630 Search PubMed.
- E. Loiseau, J. A. Schneider, F. C. Keber, C. Pelzl, G. Massiera, G. Salbreux and A. R. Bausch, Sci. Adv., 2016, 2, e1500465 Search PubMed.
- G. Kumar, N. Ramakrishnan and A. Sain, Phys. Rev. E, 2019, 99, 022414 CrossRef CAS PubMed.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D. N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- K. F. Wagenbauer, F. A. S. Engelhardt, E. Stahl, V. K. Hechtl, P. Stommer, F. Seebacher, L. Meregalli, P. Ketterer, T. Gerling and H. Dietz, ChemBioChem, 2017, 18, 1873–1885 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- H. Ramezani and H. Dietz, Nat. Rev. Genet., 2020, 21, 5–26 CrossRef CAS PubMed.
- A. Czogalla, H. G. Franquelim and P. Schwille, Biophys. J., 2016, 110, 1698–1707 CrossRef CAS PubMed.
- S. Mishra, Y. Feng, M. Endo and H. Sugiyama, ChemBioChem, 2019, 20, 1–13 CrossRef.
- Y. Dong and Y. Mao, ChemBioChem, 2019, 20, 2422–2431 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, J. List and F. C. Simmel, Acc. Chem. Res., 2014, 47, 1807–1815 CrossRef CAS PubMed.
- W. Bae, S. Kocabey and T. Liedl, Nano Today, 2019, 26, 98–107 CrossRef CAS.
- Q. Shen, M. W. Grome, Y. Yang and C. X. Lin, Adv. Biosyst., 2019, 1900215 Search PubMed.
- A. Khmelinskaia, H. G. Franquelim, E. P. Petrov and P. Schwille, J. Phys. D: Appl. Phys., 2016, 49, 194001 CrossRef.
- A. Khmelinskaia, J. Mücksch, E. P. Petrov, H. G. Franquelim and P. Schwille, Langmuir, 2018, 34, 14921–14931 CrossRef CAS PubMed.
- A. Czogalla, D. J. Kauert, R. Seidel, P. Schwille and E. P. Petrov, Nano Lett., 2015, 15, 649–655 CrossRef CAS PubMed.
- A. Czogalla, E. P. Petrov, D. J. Kauert, V. Uzunova, Y. Zhang, R. Seidel and P. Schwille, Faraday Discuss., 2013, 161, 31–43 RSC.
- S. Kocabey, S. Kempter, J. List, Y. Xing, W. Bae, D. Schiffels, W. M. Shih, F. C. Simmel and T. Liedl, ACS Nano, 2015, 9, 3530–3539 CrossRef CAS.
- S. Kempter, A. Khmelinskaia, M. T. Strauss, P. Schwille, R. Jungmann, T. Liedl and W. Bae, ACS Nano, 2019, 13, 996–1002 CAS.
- Y. Dong, Y. R. Yang, Y. Zhang, D. Wang, X. Wei, S. Banerjee, Y. Liu, Z. Yang, H. Yan and D. Liu, Angew. Chem., Int. Ed., 2017, 56, 1586–1589 CrossRef CAS PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed.
- Y. Yang, J. Wang, H. Shigematsu, W. Xu, W. M. Shih, J. E. Rothman and C. Lin, Nat. Chem., 2016, 8, 476–483 CrossRef CAS PubMed.
- Z. Zhang, Y. Yang, F. Pincet, M. C. Llaguno and C. Lin, Nat. Chem., 2017, 9, 653–659 CrossRef CAS PubMed.
- J. Fu, S. W. Oh, K. Monckton, G. Arbuckle-Keil, Y. Ke and T. Zhang, Small, 2019, 15, e1900256 CrossRef.
- M. W. Grome, Z. Zhang and C. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 22987–22992 CrossRef CAS.
- M. W. Grome, Z. Zhang, F. Pincet and C. Lin, Angew. Chem., Int. Ed., 2018, 57, 5330–5334 CrossRef CAS.
- A. Czogalla, D. J. Kauert, H. G. Franquelim, V. Uzunova, Y. Zhang, R. Seidel and P. Schwille, Angew. Chem., Int. Ed., 2015, 54, 6501–6505 CrossRef CAS PubMed.
- O. Birkholz, J. R. Burns, C. P. Richter, O. E. Psathaki, S. Howorka and J. Piehler, Nat. Commun., 2018, 9, 1521 CrossRef PubMed.
- K. Gopfrich, T. Zettl, A. E. Meijering, S. Hernandez-Ainsa, S. Kocabey, T. Liedl and U. F. Keyser, Nano Lett., 2015, 15, 3134–3138 CrossRef CAS PubMed.
- H. G. Franquelim, A. Khmelinskaia, J. P. Sobczak, H. Dietz and P. Schwille, Nat. Commun., 2018, 9, 811 CrossRef PubMed.
- T. Gerling, K. F. Wagenbauer, A. M. Neuner and H. Dietz, Science, 2015, 347, 1446–1452 CrossRef CAS PubMed.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS PubMed.
- M. Bussiek, N. Mucke and J. Langowski, Nucleic Acids Res., 2003, 31, e137 CrossRef PubMed.
- A. Johnson-Buck, S. Jiang, H. Yan and N. G. Walter, ACS Nano, 2014, 8, 5641–5649 CrossRef CAS.
- J. Marmur and P. Doty, J. Mol. Biol., 1962, 5, 109–118 CrossRef CAS.
- L. M. Traub, Dev. Cell, 2015, 35, 664–666 CrossRef CAS.
- N. A. McDonald, C. W. Vander Kooi, M. D. Ohi and K. L. Gould, Dev. Cell, 2015, 35, 725–736 CrossRef CAS PubMed.
- M. Simunovic, A. Srivastava and G. A. Voth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20396–20401 CrossRef CAS PubMed.
- M. Simunovic, A. Saric, J. M. Henderson, K. Y. C. Lee and G. A. Voth, ACS Cent. Sci., 2017, 3, 1246–1253 CrossRef CAS PubMed.
- M. Saleem, S. Morlot, A. Hohendahl, J. Manzi, M. Lenz and A. Roux, Nat. Commun., 2015, 6, 6249 CrossRef CAS PubMed.
- Z. Shi and T. Baumgart, Nat. Commun., 2015, 6, 5974 CrossRef CAS PubMed.
- M. Simunovic and G. A. Voth, Nat. Commun., 2015, 6, 7219 CrossRef CAS PubMed.
- Y. Suzuki, M. Endo and H. Sugiyama, Nat. Commun., 2015, 6, 8052 CrossRef CAS PubMed.
- Y. Sato, M. Endo, M. Morita, M. Takinoue, H. Sugiyama, S. Murata, S. I. M. Nomura and Y. Suzuki, Adv. Mater. Interfaces, 2018, 5, 1800437 CrossRef.
- N. Avakyan, J. W. Conway and H. F. Sleiman, J. Am. Chem. Soc., 2017, 139, 12027–12034 CrossRef CAS PubMed.
- C. M. A. Journot, V. Ramakrishna, M. I. Wallace and A. J. Turberfield, ACS Nano, 2019, 13, 9973–9979 CrossRef CAS PubMed.
- Y. Suzuki, M. Endo, Y. Yang and H. Sugiyama, J. Am. Chem. Soc., 2014, 136, 1714–1717 CrossRef CAS PubMed.
- H. G. Franquelim, D. Gaspar, A. S. Veiga, N. C. Santos and M. A. Castanho, Biochim. Biophys. Acta, 2013, 1828, 1777–1785 CrossRef CAS PubMed.
- A. J. Garcia-Saez, D. C. Carrer and P. Schwille, Methods Mol. Biol., 2010, 606, 493–508 CrossRef CAS PubMed.
- D. M. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS PubMed.
- H. Eto, N. Soga, H. G. Franquelim, P. Glock, A. Khmelinskaia, L. Kai, M. Heymann, H. Noji and P. Schwille, ACS Appl. Mater. Interfaces, 2019, 11, 21372–21380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm00150c |
| This journal is © The Royal Society of Chemistry 2021 |