
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An eco-friendly pathway to thermosensitive micellar nanoobjects via photoRAFT PISA: the full guide to poly(N-acryloylpyrrolidin)-block-polystyrene diblock copolymers†
Felix
Lauterbach
 and
Volker
Abetz
*
and
Volker
Abetz
*
Institute of Physical Chemistry, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: volker.abetzt@chemie.uni-hamburg.de
First published on 20th January 2020
Abstract
Spherical macromolecular assemblies, so-called latexes, consisting of polystyrene (PS) resemble a relevant class of synthetic polymers used for a plethora of applications ranging from coatings or lubricants to biomedical applications. Their synthesis is usually tailored to the respective application where emulsifiers, radical initiators, or other additives still play a major role in achieving the desired properties. Herein, we demonstrate an alternative based on the photoiniferter reversible addition–fragmentation chain transfer (RAFT) polymerization-induced self-assembly (PISA) of Poly(N-acryloylpyrrolidin)-block-polystyrene (PAPy-b-PS). This approach yields monodisperse nanospheres with tunable sizes based on an aqueous formulation with only two ingredients. These nanospheres are additionally thermosensitive, meaning that they change their hydrodynamic diameter linearly with the temperature in a broad range between 10 °C and 70 °C. Combined with the eco-friendly synthesis in pure water at 40 °C, the herein presented route constitutes an unprecedented pathway to thermosensitive diblock copolymer aggregates in short reaction times without any additives.
1 Introduction
The emulsion polymerization of styrene has historically been one of the most studied heterogeneous systems in polymer chemistry; first formulations were already invented in 1942 and played a significant role in understanding the mechanism of emulsion polymerization.1,2 Since then, waterborne polystyrene latexes have been widely used in academia for purposes like size calibration (e.g. in electron microscopy, cell counters,3 agarose gel electrophoresis,4etc.), agglutination tests,5,6 and phagocytosis experiments,7 as well as in industrial branches that deal with e.g. pigments,8 adhesives,9 or other coatings.10 For a long time, those latexes were made by free radical emulsion polymerization using surfactants as emulsifiers to control their size and surface activity.With the invention of controlled radical polymerization (CRP) mechanisms based on the reversible deactivation radical polymerization (RDRP) and their ability to produce block copolymers in a straightforward way, the use of surfactants became redundant as the required (macromolecular) chain transfer agent (CTA) could be used for both deactivating the growing polymer chains and stabilizing them in the surrounding aqueous medium.11–13 Further advantages of combining CRP techniques with emulsion polymerization to produce industrially relevant (block)copolymers are obvious: Waterborne processes are not only cheap, they moreover provide efficient heat transfer, a low viscosity, and renounce volatile organic compounds, while coincidently giving rise to well-defined polymer architectures.14–16
Several methods based on the RDRP mechanism have been developed, the most common ones being the atom transfer radical polymerization (ATRP),17,18 nitroxide-mediated polymerization (NMP),19 and RAFT polymerization.20,21 Besides the conventional thermal initiation, these methods allow control through external physical (light irradiation, ultrasonication)22–25 and chemical stimuli (metal-, redox-, enzyme-, Lewis acid-, or electrochemically-catalyzed),26–30 with light being lately one of the most prominent initiation sources.31 Using light as an external source allows highly resolved spatiotemporal control while making an adjustment of the radical concentration independent of the reaction temperature. By decoupling the concentration of active species from temperature, the use of thermoresponsive stabilizing blocks in polymerization-induced self-assembly (PISA) becomes further convenient. It is no longer required to make complex, hydrophilic stabilizers consisting of two or more monomer species,32,33 as one can simply tune the temperature so that the thermoresponsive polymer is still soluble. In the case of polymers with a lower critical solution temperature (LCST) the working temperature can be kept below the LCST so that the polymer does not collapse. It is worth noting that the rate of polymerization still depends on the thermal energy introduced to overcome the activation energy, however, deploying the advantages of light initiation makes PISA a more straightforward, universally applicable, and therefore industrially interesting process. Moreover, precise tailoring of the aggregate sizes and properties is of high interest, both from a fundamental and application point of view.
In here we present an unelaborate and cost-effective route to thermoresponsive diblock copolymer aggregates in aqueous solution via a two-step synthetic procedure (Fig. 1). The water-soluble PAPy block is polymerized via RAFT in the first step at 70 °C in a mixture of water and 1,4-dioxane in a continuous flow microreactor (see Fig. S1, ESI†). This is followed by an ab initio emulsion polymerization of styrene in batch conducted at 40 °C (see Fig. S2, ESI†) exhibiting particularly fast reaction kinetics – at such a low temperature – so that almost quantitative monomer conversions can be reached within less than five to eight hours. Additionally, three different trithiocarbonates (TTC) as CTA are studied to investigate the effect of endgroup functionality on the particle size distribution (PSD), stability of the emulsion, and reaction rate. The final diblock copolymer aggregates are furthermore studied regarding their thermosensitive behavior in solution via extensive dynamic light scattering (DLS) to proof the tunability of the aggregate size over a broad range of temperatures.
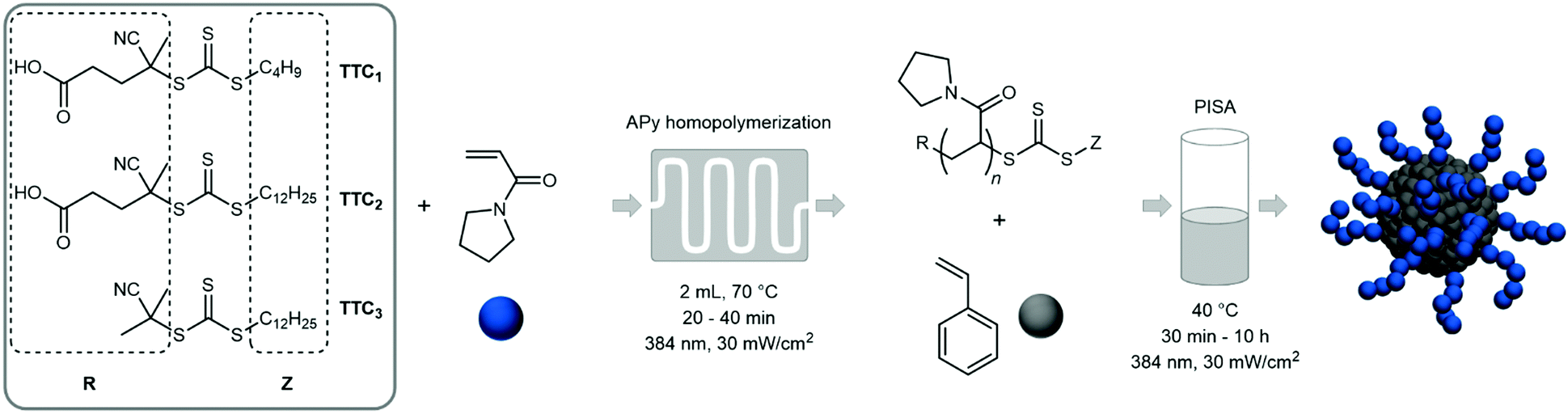 | ||
| Fig. 1 Two-step solution–emulsion photoiniferter RAFT polymerization of N-acryloylpoyrrolidin (APy) and styrene with various RAFT agents. APy is polymerized first in a continuous flow setup followed by the ab initio emulsion polymerization of styrene in water. | ||
2 Results and discussion
2.1 Homopolymerization of N-acryloylpyrrolidine
PAPy homopolymers and copolymers containing APy have been extensively investigated by our group in preceding studies.33–35 They have proven to exhibit a peculiar thermoresponsive behavior in aqueous solutions with an LCST of the homopolymer around 50 °C. For this study, five different PAPy homopolymers with phase transition temperatures (PTT) between 42 °C and 50 °C have been prepared that vary in molecular weight as well as endgroups with the scope to uncover the most feasible and universally applicable formulation for the subsequent PISA with styrene. The respective polymers as well as analytical data are depicted in Table 1.| Sample code | End groupa |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,theor.
/kDa
n,theor.
/kDa |
N
rep.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) units units
|
Đ | PTTd/°C |
|---|---|---|---|---|---|
| a See Fig. 1. b Number averaged molecular weight and degree of polymerization calculated from monomer conversion which is given by 1H-NMR measurements (see ESI, eqn (S1) and (S3)). c Dispersity index determined by SEC in DMAc at 50 °C. d Phase transition temperature determined by visual turbidimetry in water at a concentration of 1% (w/w). | |||||
| PAPy13.8-TTC1 | TTC1 | 14.1 | 110 | 1.36 | 50.2 |
| PAPy3.1-TTC2 | TTC2 | 3.5 | 25 | 1.39 | 41.6 |
| PAPy8.2-TTC2 | TTC2 | 8.6 | 66 | 1.33 | 47.0 |
| PAPy23.6-TTC2 | TTC2 | 23.9 | 188 | 1.51 | 46.5 |
| PAPy9.7-TTC3 | TTC3 | 10.0 | 77 | 1.29 | 47.1 |
It becomes evident that the PAPy homopolymer which contains a butyl endgroup on one side of the polymer chain and a carboxylic acid (which is partially deprotonated at the resulting pH of 5) on the other (PAPy13.8-TTC1) shows the highest PTT. These functionalities make the whole polymer slightly more hydrophilic compared to PAPy homopolymers with similar chain length but a longer alkyl chain as an endgroup (compare PAPy8.2-TTC2). Consequently, the homopolymer with the shortest hydrophilic block and a dodecyl endgroup exhibits the lowest PTT. Surprisingly, exchanging the carboxylic acid on the α-terminal end with a non-ionizable residue does not significantly influence the solubility behavior (compare PAPy8.2-TTC2 and PAPy9.7-TTC3). The determination of the PTT by the onset of the clouding in visual turbidimetry, however, is sometimes operator-dependent and hence only indicative of the actual phase transition.36 Thus, we performed temperature-resolved DLS measurements of the PAPy homopolymers at the same concentration (1% (w/w)) in aqueous solution.
The resulting intensity correlation functions were fitted with the cumulant approach (see eqn (S4), ESI†) and evaluated via the Stokes–Einstein relation (eqn (S5), ESI†). They revealed some distinctive and peculiar thermoresponsive properties of the homopolymer and its aggregates in water as depicted in Fig. 2.
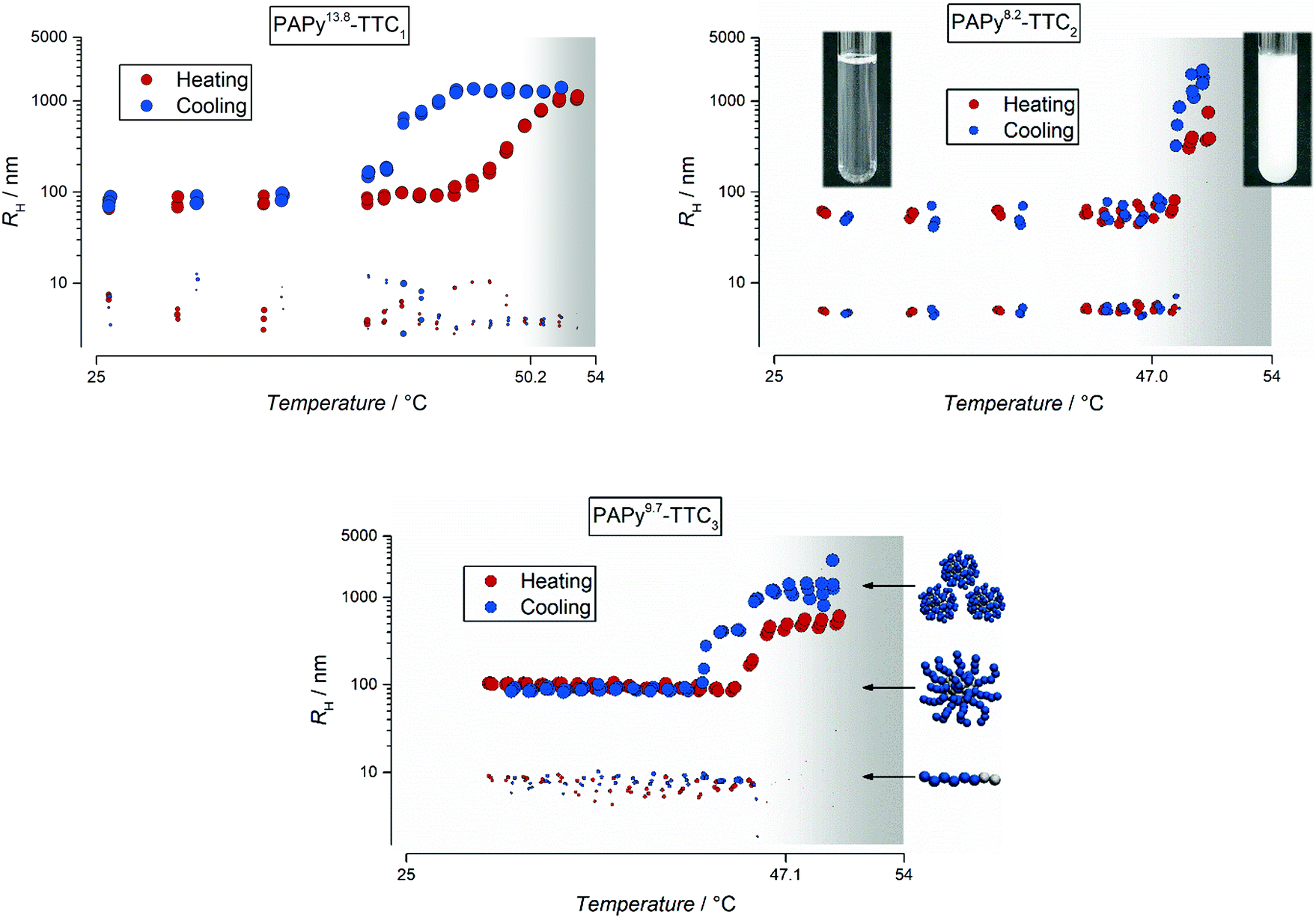 | ||
| Fig. 2 Temperature-dependent evolution of the hydrodynamic radii from three relevant PAPy homopolymers with different lengths and endgroups in aqueous solution (c = 1% (w/w)) obtained by DLS. The red dots depict the radii during heating of the polymer solution while the blue dots represent the respective radii during the cooling step. In all homopolymer samples the most suitable fit indicated two distinct aggregate species; both are shown in the graphs with different marker sizes, while the area of the marker represents the relative scattering intensity of the particular species. The optical cloud point observed via turbidimetry is indicated as the background (transition from white to grey). | ||
From the cumulant (Fig. 2) and more elaborate CONTIN analysis (see Fig. S12, S14, and S15, ESI†) it becomes obvious that the temperature-dependent and reversible aggregation of the PAPy homopolymers is by far more complex than usually assumed by just looking at the often deceptive courses of the scattering intensity with temperature (exemplarily shown in Fig. S15, ESI†). The scattering intensity is often considered solely for the investigation of an LCST behavior.37–39 However, we can show that fitting the intensity correlation function obtained from the DLS measurement with multiple exponential decays often yields deeper insights into the complex aggregation that takes places at the transition from a mostly dissolved to an agglomerated state.
For all the homopolymer samples investigated in this study we found that already at temperatures below the LCST there are two different species present in aqueous solution. One species corresponds to well-dissolved unimers with hydrodynamic radii of typically below 10 nm; the other species consists of polymeric aggregates – potentially micelles – due to the already slightly amphiphilic character of the homopolymer. This may sound reasonable for the polymers with a dodecyl endgroup, yet unintuitive for the PAPy13.8-TTC1 that only has a butyl residue at the ω-terminal end. The shorter alkyl chain as the hydrophobic endgroup, thus, could be the reason for the slightly more pronounced hysteresis compared to the polymers with longer alkyl chains: reorganization from a higher agglomeration level needs probably more time when the amphiphilic character is less pronounced. This might be attributed to differently structured particles above the LCST – while more amphiphilic polymers could form micelles, less amphiphilic polymers probably build up more entanglements first which efficiently hampers dissolution upon cooling.
It becomes furthermore apparent that although the carboxylic acid at the α-terminal end does not significantly affect the position of the PPT, it nonetheless seems to influence the aggregation behavior at the PTT. Upon heating of the polymer solution, the pre-formed macromolecular aggregates agglomerate and grow further, additionally catching the unimers which are almost completely absent above the phase transition. During the cooling step – but still above the LCST – these aggregates that at this point have grown to the micrometer size still agglomerate further. Interestingly, no precipitates formed although the polymer–water system had enough time above the LCST to undergo macrophase separation. It indicates that there is still enough stabilization of the admittedly big aggregates by some soluble hydrophilic side groups of the PAPy homopolymer. Below the phase transition of PAPy9.7-TTC3, instead of smoothly and progressively disassembling into the stable aggregates as in the case of PAPy8.2-TTC2, this polymer shows a two-step aggregation that is distinctly visible upon cooling of the sample. This behavior is reproducible during three heating–cooling cycles with slow heating/cooling rates (compare Fig. S16, ESI†) proving that this is not a kinetic effect but rather a thermodynamically stable feature of the aggregation behavior. The three different sketches in Fig. 2 (unimers, micelles, and accumulated micelles) represent the three different species present in the temperature-dependent DLS measurements. Those species have been described for a different polymer in excellent detail by Eggers et al. and explanations especially for coil-to-globule transitions can be found there.40
That this two-step aggregation is not observable for the homopolymer with the carboxylic acid endgroup (TTC2-end-capped), might be attributed to the more hydrophilic corona of the pre-formed particle. Carboxylic acid functionalities could favor the almost instantaneous disassembly of the micrometer-sized agglomerates upon stretching of the PAPy chains below the LCST, at least if the PAPy block is long enough. For very short homopolymers (see Fig. S13, ESI†), the phase transition is generally broader and there is a visible hysteresis. Most probably this is due to the comparably large hydrophobic alkyl residue combined with a short hydrophilic polymer block. This pronounced amphiphilicity could favor the formation of agglomerates and demixing of polymer and solvent, thus hampering also the reverse process which can be additionally reinforced by the lower PTT of PAPy3.1-TTC2. It is worth mentioning at this point that the ratio between these two species is intensity weighted as obtained by the fit (and displayed here), meaning that it is hardly an exact representation of the actual volumetric and number fractions. Instead, one should keep in mind that the scattering intensity in any light scattering experiment is proportional to R6 (with R being the radius of the scattering particle). For the results in Fig. 2 and for our homopolymer solutions this means that the primarily present species is the well-dissolved unimer. Polymeric aggregates are still present but way less abundant. A ratio of 1:10 in Fig. 2 corresponds to at least 99:1 in terms of the scattering volume and 99999:1 in terms of the total number of scattering centers. Although this means that the smaller of the two species is always dominant, it is of special interest for us to show that LCST phenomena are not always as plain and simple as most literature suggests.
2.2 Chain extension of PAPy with styrene via PISA
The exemplary temperature studies show that the choice of the CTA itself plays a major role for the stability of the different species in aqueous solution. With this knowledge, first assumptions and predictions can be made for the subsequent emulsion polymerization with styrene. Since the size of the most abundant dissolved species of the pure PAPy homopolymer is below 10 nm and no further surfactant is added, the emulsion will be an ab initio emulsion polymerization rather than for instance a mini- or microemulsion.41,42 There are apart from that some expectable differences in the stabilization of the styrene droplets and the emerging micellar aggregates. As the polymer with the TTC1-endgroup should exhibit the lowest ability to stabilize styrene droplets due to the shortest alkyl chain which is not able to penetrate the styrene phase deeply, the polymers with TTC2 and TTC3-endgroups will certainly fulfill this requirement. This can be already concluded during preparation of the polymerization mixture. Upon addition of the styrene monomer to solutions of the different macromolecular CTAs (macroCTAs) it becomes obvious that the different components of the resulting emulsions are by far more likely to macroscopically separate again if a shorter alkyl chain endgroup is used.Yet, the emulsion polymerizations with PAPy13.8-TTC1 show still short inhibition periods for styrene of approximately 90 minutes and a remarkable degree of control indicated by the absence of dead homopolymer and dispersities below 1.3 (see Fig. S17, ESI†).43 Polymerization rates of the reactions with a total solids concentration of 10% (w/w) and 15% (w/w) are fast compared to literature block copolymer emulsions with PS as the hydrophobic block – that are usually prepared at more than 70 °C – delivering almost quantitative conversion within less than four hours after the start of the chain extension.33,44,45 The emulsion at 20% (w/w) solids content shows a slight rate retardation which is mainly attributed to the poor stabilization of the styrene droplets and micelles at higher concentrations of styrene. The SEC traces reveal, though, that the chain end livingness of the homopolymer is seemingly quantitative resulting in a successful chain extension with no residual homopolymer and dispersities well below 1.3 for the end products.
The emulsion polymerizations with the TTC2-polymer (PAPy8.2-TTC2) exhibit a significantly longer inhibition period and slightly slower reaction rates (compare Fig. S18–S20, ESI†). Shorter stabilizing blocks additionally increase the inhibition period further, presumably due to worse stabilization of the growing micelles. The inverse trend is observed when the length of hydrophilic macroCTA is increased, which coincidently accelerates the rate of the polymerization. Molecular weight dispersities of the final products range from 1.16 to 1.41, showing the tendency to be lower with a lower degree of polymerization (DP) of PS. It is worth mentioning, that via this synthetic pathway diblock copolymers can be prepared in moderate reaction times under sustainable reaction conditions (meaning in this case in water up to quantitative conversion with minimum energy consumption) that reach molecular weights up to 114 kDa. Especially in the case of the normally very slowly propagating styrene this constitutes an alternative route to diblock copolymers suitable for a plethora of applications that is unprecedented considering the reaction temperature of 40 °C.46,47
The most remarkable reaction kinetics can be found when using PAPy9.7-TTC3 as macroCTA (which does not have the carboxylic acid at the α-terminal end). Initially believed to facilitate the diffusion of the styrene monomer from the bigger monomer droplets into the smaller micelles where the reaction takes place, it not only provided high reaction rates and an inhibition of just about 60 minutes, it furthermore yielded stable emulsions up to 25% (w/w) solids content (see Fig. 3). The molecular weight distributions become slightly broader with final dispersities between 1.31–1.45, yet no residual homopolymer can be observed in the SEC traces indicating quantitative chain-end fidelity after the first reaction step. Interestingly, the highest reaction rate is found at highest solids content which is exactly the inverse trend compared to the homopolymer with carboxylic acid and butyl endgroups. This again highlights the use of TTC3 as CTA for the emulsion polymerization of styrene. In a nutshell, quantitative conversion can be reached at high solids content yielding diblock copolymers under good control in a PISA formulation without any initiator, surfactant, or other additive. It shows that the concept of light-initiation hence works for highly turbid systems where the penetration depth of the incident light is typically only a small fraction of the overall diameter of the reaction flask.
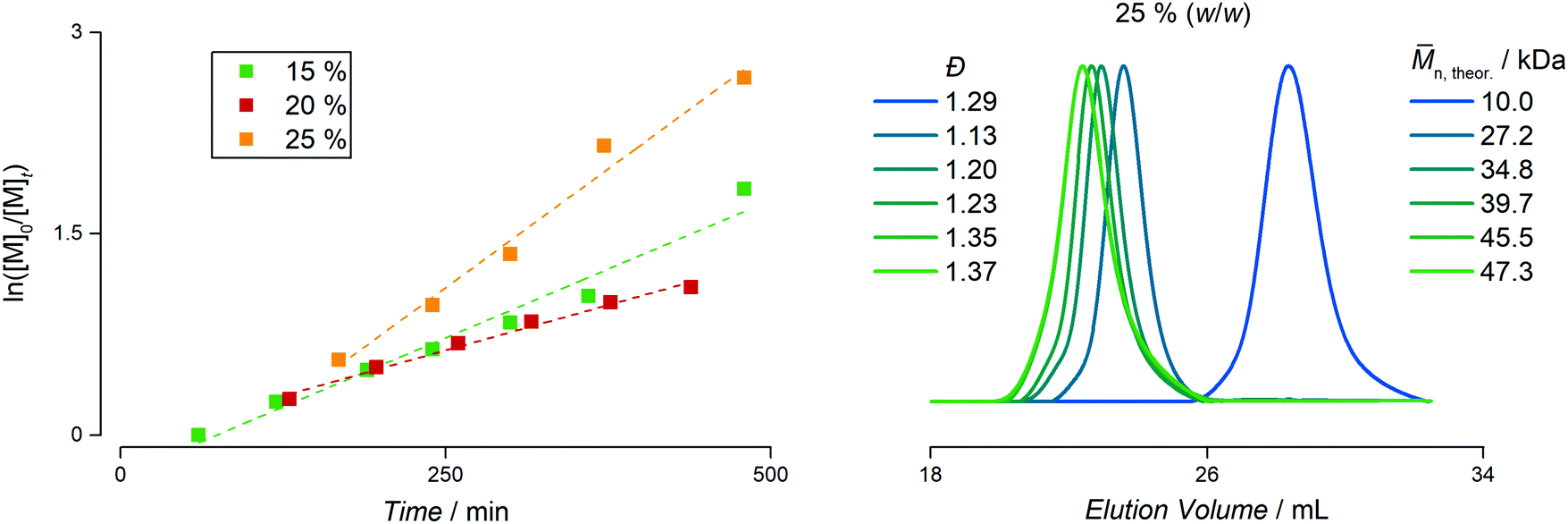 | ||
| Fig. 3 Left: Pseudo first-order kinetic plot of the styrene emulsion polymerizations with the PAPy9.7-TTC3 macroCTA at a total solids concentration of 15% (w/w), 20% (w/w), and 25% (w/w). Right: SEC traces for the samples taken from the kinetic study at 25% (w/w) solids content. The first SEC trace is the macroCTA itself, the remaining traces represent the diblock copolymer samples. The dispersities and overall theoretical molecular weights are given as well. | ||
Up to now we have shown the feasibility of the photoiniferter PISA polymerization to yield PAPy-b-PS diblock copolymers up to high solid contents and high molecular weights compared to other CRP methods. Those diblock copolymers constitute very interesting yet easily accessible sources for e.g. filtration membrane materials that to date are mainly synthesized via an elaborate anionic polymerization procedure.48–50
2.3 Aqueous dispersions of the PAPy-b-PS latexes
It would be regardless, though, to aim for just one purpose with this PISA formulation that almost incidentally yields already pre-formed, submicron-sized organic nanoparticles in an aqueous solution. The herein presented route does not need any initiator or surfactant, so that everything left in the reaction mixture is covalently bound to the nanoparticles. This makes unwanted migration of one of the reaction components in the final product less likely and therefore guarantees improved durability of the products properties.51–54 Additionally, the hydrophilic stabilizing block yields surface functionality to the nanoparticles without the need of further functionalization steps, which is hardly achievable in a conventional free radical emulsion polymerization.One of the aims of the herein presented PISA approach is tailoring the size and size distribution of the diblock copolymer aggregates in solution. The three most often observed morphologies of PISA polymers in water are spherical micelles, worm-like micelles, and vesicles (typically increasing in size in that order).55–57 This work specifically targets the micellar morphology, which has proven to be versatile in its use ranging from biomedical applications58–60 to coatings10 or adhesives.9 This aim is supported by the choice of PS as the hydrophobic core of the micelle due to its high glass transition temperature and hence low ability to undergo reorganization from spherical micelles to worm-like aggregates, especially at the low polymerization temperature of 40 °C.57,61,62
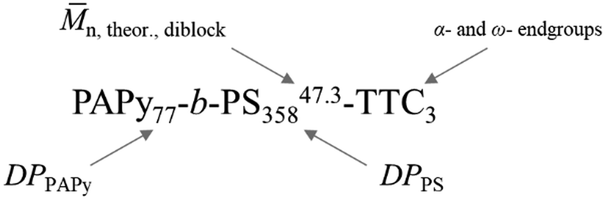
Fig. 4 shows exemplary transmission electron microscopy (TEM) images of three diblock copolymers prepared from three different homopolymers with varying endgroups, all with almost quantitative conversion during styrene polymerization. Nomenclature is chosen to be consistent with most other publications on PISA, where subscripts signify the degree of polymerization of the individual block. The superscript at the end stands for the overall theoretical number average molecular weight calculated from the conversion of both blocks and the last part of the name indicates the attached endgroups. Experimental data for the molecular weight (see Tables S2–S4, ESI†) is not meaningful since SEC data need suitable calibration, which is not available for the diblock copolymer.
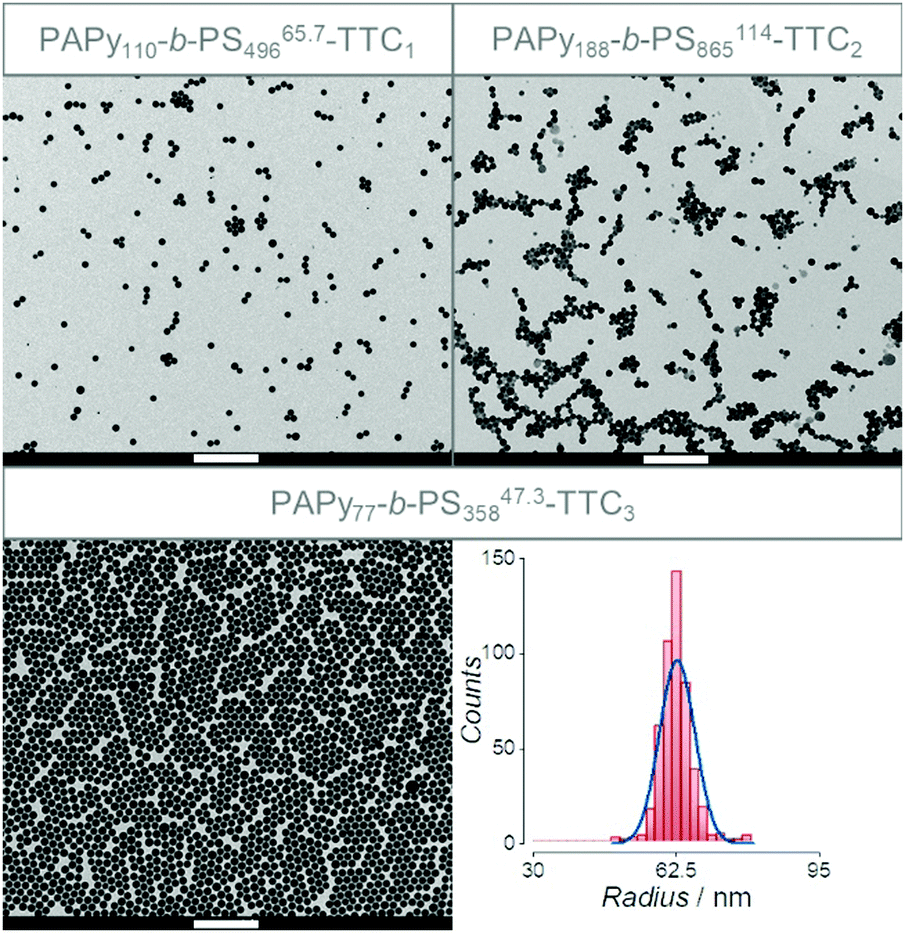 | ||
| Fig. 4 TEM images of the diblock copolymers prepared from PAPy13.8-TTC1 (PAPy110-b-PS49665.7-TTC1, top left), PAPy23.6-TTC2 (PAPy188-b-PS865114-TTC2, top right), and PAPy9.7-TTC3 (PAPy77-b-PS35847.3-TTC3, bottom left). All scale bars correspond to 1 μm. The size distribution of latex particles taken from analysis of the bottom left image is depicted on the bottom right. A Gaussian fit (blue line) gave a mean radius of 62.7 ± 4.0 nm. For more TEM images and all sized distributions see Fig. S22–S28 (ESI†). | ||
The TEM images in Fig. 4 clearly show that the sizes of the micellar aggregates in aqueous solution are in the same order of magnitude for all the used stabilizing blocks. The radii observed for all TEM measurements are well below 100 nm and by evaluating the size distributions of the latex particles a peculiar property becomes evident that fits to the kinetic theory of an ab initio emulsion polymerization: size distributions of the resulting micelles are broader if the inhibition period at the beginning of the emulsion polymerization is pronounced, being the case for PISA with PAPy-TTC2 macroCTAs. It is reasonable that this effect can be attributed to a process called secondary nucleation that in our case might be happening at the start of the rate enhancement.63–65 Due to a better solubilization that is already evident in Fig. 2, light-induced fragmentation of the RAFT endgroup, yielding macromolecular radicals, leads to a desorption of those radicals from the micelles into the aqueous phase. There they can either enter another micelle or add dissolved monomer until the increasing amphiphilicity leads once again to the collapse of the hydrophobic block, forming another micelle. However, further studies need to prove the dependence of size distributions and inhibition periods which is to the best of our knowledge only poorly described in current literature.66 Nevertheless, it is distinctive that those emulsion polymerizations with the shortest inhibition periods, overcoming nucleation much faster than the remaining growth needs, exhibit the narrowest size distributions (see Fig. 4, bottom right).
To furthermore highlight the potential of the herein presented PISA formulation with PAPy9.7-TTC3 to tailor the radius of the resulting spherical micelles while maintaining extraordinarily narrow size dispersities – fairly comparable to those prepared by emulsion polymerizations using surfactants – Fig. 5 depicts the radii of all emulsion polymerizations with this macroCTA measured by TEM as well as the hydrodynamic radii measured by DLS. It is remarkable that the micelles not only linearly increase in size, moreover the size seems to be completely independent of the concentration. In addition to the narrow aggregate size distributions as well as molecular weight distributions this once again emphasizes the potential of this formulation to tailor the properties of PAPy-b-PS diblock copolymers.
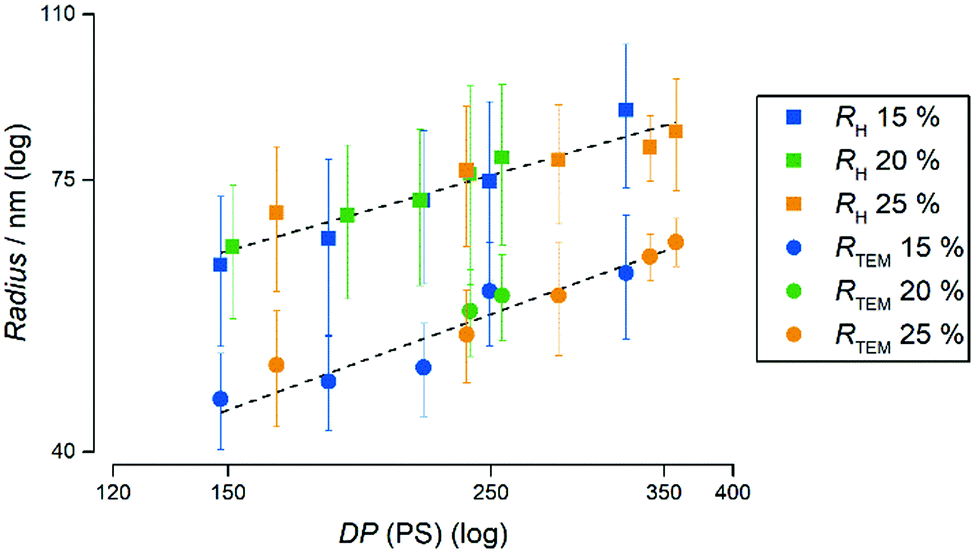 | ||
| Fig. 5 Evolution of the radii of all polymers made from the PAPy9.7-TTC3 macroCTA at 15% (w/w), 20% (w/w), and 25% (w/w) solids concentration compared to the degree of polymerization (DP) of PS. The radii measured by TEM are depicted as circles and the hydrodynamic radii calculated from DLS measurements are depicted as squares. The respective PDI values can be found in Fig. S35 (ESI†). | ||
After examining the size of the particles at room temperature, detailed studies of the solubility behavior have been conducted depending on the temperature. Therefore, dilute samples of the final diblock copolymer micelles were analyzed by DLS in a temperature range from 10–70 °C. Fig. 6 exemplarily shows an evolution of the hydrodynamic radii of PAPy77-b-PS35847.3-TTC3 at 0.02% (w/w) solids concentration.
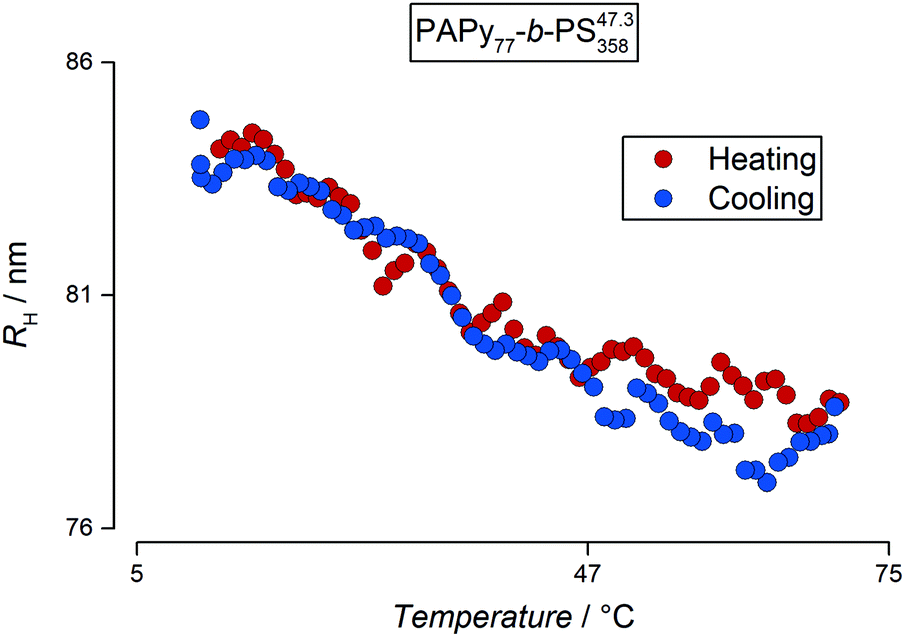 | ||
| Fig. 6 Evolution of the hydrodynamic radii of PAPy77-b-PS35847.3-TTC3 at 0.02% (w/w) solids concentration with temperature calculated from DLS measurements. The red dots represent the radii during heating of the polymer dispersion and the blue dots represent the respective radii during the cooling step. The most probable fit solution yielded one distribution of particle sizes. The PTT of the used homopolymer is also depicted on the temperature axis. | ||
The DLS data for the diblock copolymers were fitted with the same routine as the homopolymer solutions. For all PISA samples, however, the most probable fit solution as well as CONTIN analysis indicated that the analyzed dispersions only consisted of one particle species in the range of a few ten to one hundred nanometers depending on the DP as already depicted in Fig. 5. This leads to the conclusion that there are no unimers or otherwise dissolved polymer chains left that are not aggregated into micelles. These micelles in turn exhibit an interesting behavior: upon heating of the particle dispersion, the hydrodynamic radius decreases as an almost linear function of the temperature with a slight decline in the shrinking rate towards the end of the heating ramp between 60 °C and 70 °C; a behavior typically observed in microgels.67,68 It is worth mentioning that the reduction in size is only attributed to the collapse of the corona block. Assuming a fully stretched PAPy chain, this reduction correlates to almost 50% of the overall contour length of this block. The decrease in size is fully reversible until the aggregate size reaches its initial value in the cold state. Most importantly, no coagulates were observable which is in strong contrast to the observations of the PAPy homopolymers. Particle aggregation above the LCST of the homopolymer resulted in a sharp increase of the hydrodynamic radius; when the system was given a few hours to equilibrate, these aggregates precipitated from solution.
This does not happen for the diblock copolymer aggregates made by PISA in water. The same observation was made for a more complex system in an earlier work by Eggers et al.33 In contrast to other previous studies for a similar diblock copolymer system prepared by forced self-assembly after the polymerization, the PISA-made micelles stay stabilized in water and tolerate much higher temperatures than the actual phase transition.69 The lack of agglomeration can be attributed to two major reasons; the first one is the high glass transition temperature (Tg) of the PAPy block (142 °C of the dry homopolymer; collapsed PAPy in water might show a lower Tg due to partial hydration)35 that makes the micelle behave like a solid sphere rather than an aggregate with a dense core and a soft corona which potentially entangles with the corona of adjacent micelles. The second reason could be the comparatively low DPs of the herein used homopolymers which are perhaps below or in the range of the entanglement molecular weight of PAPy. Both reasons – a high Tg and short DPs – attribute to only weak interaction between different micelles and hence inhibit agglomeration. With reasonable certainty we can exclude charge stabilization effects that might arise in the case of TTC1 and TTC2 end-capped polymers due to dissociated carboxylic acids at a solution pH of 5. Since all TTC3 end-capped micelles (that do not carry any charges) are also stable at elevated temperatures, we hypothesize that DP and Tg are the most influential parameters.
However, this can hardly be the reason for the gradual size transition that the micellar aggregates undergo upon heating. A more plausible explanation lies in the free volume of the solubilized corona and the hydrophobicity of the adjacent PS core. Due to densely packed PAPy chains on the surface of the micelle, the water molecules are not able to penetrate the whole corona which lowers the solubility slightly and shifts the onset of the phase transition to lower temperatures. Additionally, these closely packed chains cannot collapse freely and repel the water molecules out of the initially solubilized corona progressively, making the collapse a rather gradual feature compared to the analogous homopolymers (Fig. 7). Given that the thermoresponse is not stepwise but stretched over a wide range of temperatures, yet reversible with no hysteresis, the terminology stipulates the use of ‘thermosensitive’ instead of ‘thermoresponsive’.
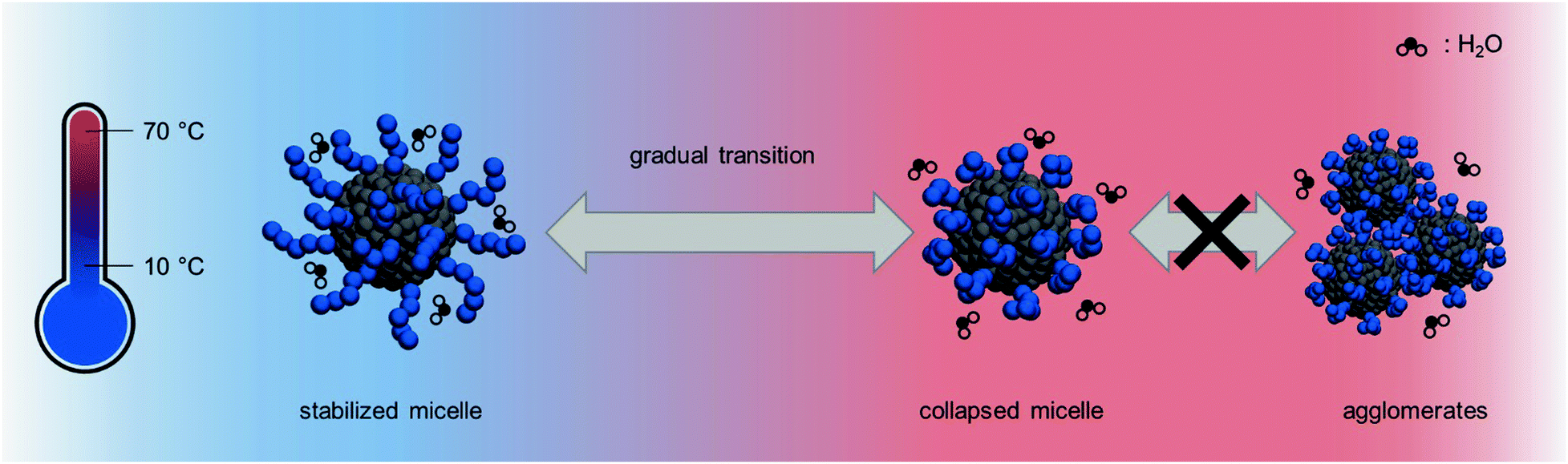 | ||
| Fig. 7 Molecular scheme of the thermosensitivity of PAPy-b-PS spherical micelles prepared via PISA. At low temperatures, the hydrophilic corona is solubilized by water. Upon heating, water molecules are repelled from the surface and the PAPy block collapses, but the aggregate is still stabilized. No agglomeration takes place due to unfavorable interaction among different micelles. | ||
The last thing to prove for our PISA system is that the sensitivity is not a onetime feature. Therefore, we conducted a series of DLS measurements where we calculated the hydrodynamic radii during three heating–cooling cycles. The result for an exemplary sample (this time for a diblock copolymer with a TTC1 endgroup) is depicted in Fig. 8. The evolution of the hydrodynamic radii during repeated cycles of heating and cooling indeed shows that the thermosensitivity is a repetitive property. The fact that the minima and maxima of the hydrodynamic radius are reached at similar temperatures while no precipitates have formed during more than 28 hours of heating to 70 °C and cooling to 10 °C furthermore underlines the predictability and durability of the examined latex dispersions.
 | ||
| Fig. 8 Evolution of the hydrodynamic radii of PAPy110-b-PS49665.7-TTC1 at 0.02% (w/w) solids concentration with temperature during three heating–cooling cycles calculated from DLS measurements. The red dots represent the radii during heating of the polymer dispersion and the blue dots represent the respective radii during the cooling step. The times marked on the x-axis represent the intervals of heating and cooling. | ||
3 Conclusion
We presented the photoiniferter two-step solution–emulsion polymerization of PAPy-b-PS diblock copolymers with three different endgroups. During the first step, PAPy is formed in a continuous microfluidic process and its peculiar thermoresponsiveness is evaluated depending on the endgroup of the homopolymer. It becomes evident that the aggregation behavior strongly depends on the amphiphilicity and chain length of the used PAPy while endgroups with carboxylic acids additionally promote thermal reorganization. In the second step, we demonstrated the feasibility of the photoiniferter emulsion polymerization of styrene in water at 40 °C yielding fast reaction rates with no inhibition period and reaching quantitative conversion within five to eight hours by using the most suitable endgroup (TTC3). This chain extension constitutes a remarkable example of how conventional emulsion formulations can be replaced by novel methods that work without surface-motile low molecular weight surfactants, volatile organic solvents, and even initiators, paving the way for greener and economically advantageous reactions. The resulting micellar nanoobjects obtained from this PISA approach can be straightforwardly tuned in size by controlling the DP of the PS block. Additionally, TEM images prove that this surfactant-free pathway yields spherical micelles with exceptionally narrow size distributions while the individual polymer chains forming these micelles are also uniform in composition and molecular weight, respectively. The thermosensitivity of the diblock copolymer latexes in aqueous solutions was proven to be predictable and remarkably linear over repeated cycles. A typical size decrease of almost 15% was observed upon heating of the dispersion up to 70 °C, with complete reversibility and no formation of precipitates. The PAPy-b-PS diblock copolymers prepared via the herein presented minimum-ingredient and economical pathway are potential candidates for a plethora of applications like filtration membranes, temperature-sensitive coatings, or adhesives (besides many more) due to the straightforward tunability of the reaction and hence product properties. Molecular weights can be altered from a few ten kDa to more than 100 kDa while the size of the spherical micelles formed by PISA can be coincidently tailored in the range from a few ten nanometers to more than 100 nm. Additionally, long-term tests of the concentrated dispersions prove colloidal stability up to six months after preparation.4 Methods
4.1 Materials
Styrene (Sigma Aldrich, 99%) was deinhibited over activated basic alumina prior to each polymerization. The RAFT agents TTC2 (ABCR, 97%) and TTC3 (ABCR, 97%) were used as received. The chemicals used as reactants, acryloyl chloride (ABCR, 96%, stabilized with phenothiazine), pyrrolidine (Acros, 99%), butanethiol (Sigma Aldrich, 99%), potassium hydroxide (Merck, >85%), carbon disulfide (Merck, 99%), p-tosyl chloride (Merck, 98%), and 4,4′-azobis(4-cyanopentanoic acid) (Sigma Aldrich, 98%) were used as received. All solvents for reactions, purifications, and NMR, dichloromethane (Acros, 99.9%), 1,4-dioxane (Grüssing, 99%), dimethylformamide (VWR, 99.5%), acetone (Merck, 99%), methyl acetate (Merck, 99%), n-hexane (VWR Chemicals, 95%), chloroform-d1 (Euriso-Top, 99.8%), and tetrahydrofuran-d8 (Euriso-Top, 99.5%) were used without further purification. All other chemicals, sodium chloride Grüssing (99%), KHSO4 (Grüssing, 99%), NaHCO3 (Grüssing, 99%), Mg2SO4 (Grüssing, 99%), and activated basic aluminum oxide (Merck, 99%, grain size between 0.063–0.200 mm) were also used as received.4.2 Synthesis of APy
The synthesis of N-acryloylpyrrolidine was adapted from the synthesis of N-acryloylpiperidine as described by Jo et al.:70 a solution of acryloyl chloride (7.5 mL, 92 mmol, 1 eq.) in CH2Cl2 (50 mL) was added dropwise during 30 minutes to a solution of pyrrolidine (15.0 mL, 183 mmol, 2 eq.) in CH2Cl2 (100 mL) at 0 °C under N2 atmosphere and subsequently stirred for another two hours. During the reaction the flask was permanently flushed with N2 to take away the emerging HCl.The solution was then washed with brine (100 mL), an aqueous solution of KHSO4 (1 M, 100 mL), brine (100 mL), an aqueous solution of NaHCO3 (5% (w/w), 100 mL), and again brine (100 mL). The organic phase was dried over Mg2SO4, evaporated to dryness and filtered over activated basic alumina to yield a light-yellow oil (5.84 g, 50.7%).
4.3 Synthesis of TTC1
The synthesis of 4-cyano-4-(butylsulfanylthiocarbonyl)sulfanylpentanoic acid was adapted from the synthesis of 4-cyano-4-(propylsulfanylthiocarbonyl)sulfanylpentanoic acid as described by Xu et al.:71 1-butanethiol (5.00 mL, 467 mmol, 1 eq.) was added dropwise to a solution of potassium hydroxide (3.2834 g, 585 mmol, 1.25 eq.) in water (11.0 mL). After stirring for 30 minutes, carbon disulfide (2.80 mL, 464 mmol, 1 eq.) was added. The mixture was stirred vigorously at room temperature for 1 hour and subsequently cooled to 0 °C. A solution of p-tosyl chloride (4.4350 g, 233 mmol, 0.5 eq.) in acetone (22.5 mL) was added dropwise for 20 minutes and stirred for another 2 hours. The excess acetone was removed on a rotary evaporator and the remaining red solution was extracted with dichloromethane (3 × 50 mL). The combined organic phases were washed with water (50 mL), dried over Mg2SO4 and evaporated to dryness to yield solid bis(butylsulfanylthiocarbonyl)disulfide (12.4623 g, 81%).A solution of bis(butylsulfanylthiocarbonyl)disulfide (5.0156 g, 152 mmol, 1 eq.) and 4,4′-azobis(4-cyanopentanoic acid) (5.3098 g, 189 mmol, 1.25 eq.) in methyl acetate (65 mL) was heated to reflux for 24 hours. The solution was evaporated to dryness and the product was purified by silica gel column chromatography with methyl acetate:n-hexane (1:2 (v/v)) as mobile phase to yield 4-cyano-4-(butylsulfanylthiocarbonyl)sulfanylpentanoic acid (5.9142 g, 67%).
4.4 PhotoRAFT polymerization of APy
In a typical photoRAFT polymerization of APy, TTC2 (0.0498 g, 0.123 mmol, 1 eq.) and APy (1.4358 g, 11.5 mmol, 93 eq.) were dissolved in a mixture of water (3.37 g) and 1,4-dioxane (5.06 g). Dimethylformamide (0.3 mL) was added as an internal reference for 1H-NMR analysis and an initial sample was taken. The solution was connected to the isocratic pump of the millireactor and purged with N2 for 10 minutes at 0 °C. The glass chip millireactor (2 mL, static mixer) was heated to 70 °C and the flowrate was set to 0.1 mL min−1 yielding a residence time of 20 minutes. A UV-LED (OmniCure® AC450) with self-made beam-widening (10 × 10 cm) and a thin plexiglass shield to mitigate the intensity to 30 mW cm−2 was used as light source 5 cm above the chip surface. A crude 1H-NMR sample in chloroform-d1 was taken after the reaction to determine the monomer conversion. The product was precipitated three times in cold n-hexane, filtered and dried in vacuo overnight.For the polymerizations with TTC1, the flowrate was set to 0.05 mL min−1 to give a residence time of 40 minutes. In the case of TTC3, pure 1,4-dioxane was used as solvent to ensure better solubility of the less hydrophilic CTA.
4.5 PhotoRAFT emulsion polymerization of styrene
In a typical photoRAFT emulsion polymerization of styrene in batch, PAPy9.7-TTC3 (0.0502 g, 5.02 μmol, 1 eq.) was dissolved in Milli-Q® water (1.0 mL) and styrene (0.2004 g, 1.92 mmol, 383 eq.) was added.‡ The resulting emulsion was homogenized for 30 minutes at 1000 rpm, subsequently purged with N2 for 10 minutes at 0 °C and immersed into an oil bath at 40 °C. The same lamp used for the homopolymerizations was directed at the sample in a distance of 5 cm, the sample was stirred at 600 rpm and the intensity was set to 30 mW cm−2. Crude polymer samples were taken from the reaction mixture at different times under N2 protection. These samples were used for 1H-NMR analysis in tetrahydrofuran-d8, GPC measurements, and diluted to 0.2% (w/w) for TEM and 0.02% (w/w) for DLS, respectively.4.6 Analytics
Temperature-dependent DLS measurements of the homopolymers were conducted in temperature steps of 1 °C with three measurements per temperature. The polymer solutions were prepared 24 hours prior to the measurements at a concentration of 10 mg mL−1 by dissolving the polymer in Milli-Q® water that was pre-filtered through microporous regenerated cellulose filters (average pore diameter: 200 nm). The maximum temperature was chosen to be approximately 3 °C above the cloud point estimated by visual turbidimetry.
Temperature-dependent DLS measurements of the diblock copolymer aggregates were conducted in temperature steps of 5 °C with three measurements per temperature. The polymer emulsions were withdrawn directly from the polymerization mixture and diluted with pre-filtered Milli-Q® water to 0.2 mg mL−1 to avoid multiple scattering. The temperature range was set to 10–70 °C (unless stated otherwise). For standard measurements at room temperature, the same concentrations were used as for the temperature-dependent measurements. Analysis of all DLS measurements was conducted with a self-written program based on a cumulant approach up to the third order cumulant. For specific samples an additional CONTIN analysis was made.
Author contributions
F. L. contributed to the experimental setup, data acquisition and interpretation, analysis as well as validation, and the writing of the manuscript. V. A. was responsible for funding acquisition, project administration, as well as interpretation of the experimental data and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. L. and V. A. would like to thank Dr Birgit Fischer for prolific discussions about the analysis of DLS data. F. L. furthermore acknowledges the work of Katharina Nieswandt, Vanessa Meyen, and Sascha B. Lemich on preceding studies regarding the present polymer system.References
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS PubMed.
- W. D. Harkins, J. Polym. Sci., 1950, 5, 217–251 CrossRef CAS.
- R. W. Lines and B. V. Miller, Powder Technol., 1979, 24, 91–96 CrossRef CAS.
- E. Gombocz, D. Tietz, S. S. Hurtt and A. Chrambach, Electrophoresis, 1987, 8, 261–271 CrossRef CAS.
- R. T. Fisk, US Pat., US3088875A, 1959.
- S. Kasempimolporn, N. Chaiyabutr and V. Sitprija, Current Laboratory Techniques in Rabies Diagnosis, Research and Prevention, Academic Press, 2014, pp. 141–145 Search PubMed.
- E. D. Korn and R. A. Weisman, J. Cell Biol., 1967, 34, 219–227 CrossRef CAS PubMed.
- M. Monzon, M. Lock and J. Galloway, EP0364629A1, 1988.
- J. Dejeu, M. Bechelany, L. Philippe, P. Rougeot, J. Michler and M. Gauthier, ACS Appl. Mater. Interfaces, 2010, 2, 1630–1636 CrossRef CAS.
- Y. Li, R. Liu, J. Wang and X. Tang, Prog. Org. Coat., 1992, 21, 101–107 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Macromolecules, 2002, 35, 5417–5425 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811 CrossRef CAS PubMed.
- M. F. Cunningham, J. D. Campbell, Z. Fu, J. Bohling, J. G. Leroux, W. Mabee and T. Robert, Green Chem., 2019, 21, 4919–4926 RSC.
- J. Ho, B. Mudraboyina, C. Spence-Elder, R. Resendes, M. F. Cunningham and P. G. Jessop, Green Chem., 2018, 20, 1899–1905 RSC.
- J. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572–7573 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- P. G. Griffiths, E. Rizzardo and D. H. Solomon, Tetrahedron Lett., 1982, 23, 1309–1312 CrossRef CAS.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, WO1998001478A1, 1997.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- T. Otsu and M. Yoshida, Die Makromol. Chemie, Rapid. Commun., 1982, 3, 127–132 CrossRef CAS.
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Metal-catalyzed living radical polymerization, 2001, vol. 101 Search PubMed.
- A. Reyhani, T. G. McKenzie, H. Ranji-Burachaloo, Q. Fu and G. G. Qiao, Chem. – Eur. J., 2017, 23, 7221–7226 CrossRef CAS PubMed.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- S. Kumagai, K. Nagai, K. Satoh and M. Kamigaito, Macromolecules, 2010, 43, 7523–7531 CrossRef CAS.
- Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu and K. Matyjaszewski, Macromolecules, 2017, 50, 7872–7879 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- C. N. Urbani and M. J. Monteiro, Macromolecules, 2009, 42, 3884–3886 CrossRef CAS.
- S. Eggers and V. Abetz, Polymers, 2017, 9, 668–691 CrossRef PubMed.
- N. Lucht, S. Eggers and V. Abetz, Polym. Chem., 2017, 8, 1196–1205 RSC.
- S. Eggers, T. Eckert and V. Abetz, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 399–411 CrossRef CAS.
- M. Wang, S. Harrisson, M. Destarac and J.-D. Marty, J. Supercrit. Fluids, 2019, 152, 104572 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed.
- S. Eggers, B. Fischer and V. Abetz, Macromol. Chem. Phys., 2016, 217, 735–747 CrossRef CAS.
- R. G. Gilbert, Emulsion polymerization: a mechanistic approach, Academic Press, London, 1995 Search PubMed.
- A. van Herk, Chemistry and Technology of Emulsion Polymerisation, 2007 Search PubMed.
- M. Semsarilar, E. R. Jones and S. P. Armes, Polym. Chem., 2014, 5, 195–203 RSC.
- T. Boursier, I. Chaduc, J. Rieger, F. D’Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- Y. Luo, X. Wang, B. G. Li and S. Zhu, Macromolecules, 2011, 44, 221–229 CrossRef CAS.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- K. Jung, J. Xu, P. B. Zetterlund and C. Boyer, ACS Macro Lett., 2015, 4, 1139–1143 CrossRef CAS.
- A. Jung, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Macromol. Mater. Eng., 2012, 297, 790–798 CrossRef CAS.
- R. A. Mulvenna, J. L. Weidman, B. Jing, J. A. Pople, Y. Zhu, B. W. Boudouris and W. A. Phillip, J. Membr. Sci., 2014, 470, 246–256 CrossRef CAS.
- V. Abetz, Macromol. Rapid Commun., 2015, 36, 10–22 CrossRef CAS.
- A.-C. Hellgren, P. Weissenborn and K. Holmberg, Prog. Org. Coat., 1999, 35, 79–87 CrossRef CAS.
- M. J. Monteiro, M. Sjöberg, J. van der Vlist and C. M. Göttgens, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4206–4217 CrossRef CAS.
- J. I. Amalvy, M. J. Unzué, H. A. S. Schoonbrood and J. M. Asua, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2994–3000 CrossRef CAS.
- N. Shirakbari, M. Ebrahimi, H. Salehi-Mobarakeh and M. Khorasani, J. Macromol. Sci., Part B: Phys., 2014, 53, 1286–1292 CrossRef CAS.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D’Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- H. Vihola, A.-K. Marttila, J. S. Pakkanen, M. Andersson, A. Laukkanen, A. M. Kaukonen, H. Tenhu and J. Hirvonen, Int. J. Pharm., 2007, 343, 238–246 CrossRef CAS PubMed.
- L. M. Johnson, Z. Li, A. J. LaBelle, F. S. Bates, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2017, 50, 1102–1112 CrossRef CAS.
- L. Esser, N. P. Truong, B. Karagoz, B. A. Moffat, C. Boyer, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 7325–7337 RSC.
- J. Lesage de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D’Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS PubMed.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- S. M. Lepizzera and A. E. Hamielec, Macromol. Chem. Phys., 1994, 195, 3103–3115 CrossRef CAS.
- B. R. Morrison and R. G. Gilbert, Macromol. Symp., 1995, 92, 13–30 CrossRef CAS.
- E. M. Coen, R. G. Gilbert, B. R. Morrison, H. Leube and S. Peach, Polymer, 1998, 39, 7099–7112 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- A. Pich, A. Tessier, V. Boyko, Y. Lu and H.-J. P. Adler, Macromolecules, 2006, 39, 7701–7707 CrossRef CAS.
- A. Pich, S. Bhattacharya, Y. Lu, V. Boyko and H.-J. P. Adler, Langmuir, 2004, 20, 10706–10711 CrossRef CAS PubMed.
- S. Eggers, F. Lauterbach and V. Abetz, Polymer, 2016, 107, 357–367 CrossRef CAS.
- Y. S. Jo, A. J. van der Vlies, J. Gantz, S. Antonijevic, D. Demurtas, D. Velluto and J. A. Hubbell, Macromolecules, 2008, 41, 1140–1150 CrossRef CAS.
- X. Xu, A. E. Smith, S. E. Kirkland and C. L. McCormick, Macromolecules, 2008, 41, 8429–8435 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm02483b |
| ‡ For the use of TTC1 and TTC2 endgroups the solution pH was in all cases around 5 while for TTC3 the solution pH remained at 7. |
| This journal is © The Royal Society of Chemistry 2020 |