
Suppression of hypersynchronous network activity in cultured cortical neurons using an ultrasoft silicone scaffold
 *ab
and
Ayumi
Hirano-Iwata
ab
*ab
and
Ayumi
Hirano-Iwata
ab
Abstract
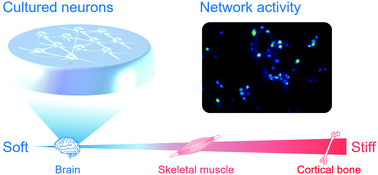
The spontaneous activity pattern of cortical neurons in dissociated culture is characterized by burst firing that is highly synchronized among a wide population of cells. The degree of synchrony, however, is excessively higher than that in cortical tissues. Here, we employed polydimethylsiloxane (PDMS) elastomers to establish a novel system for culturing neurons on a scaffold with an elastic modulus resembling brain tissue, and investigated the effect of the scaffold's elasticity on network activity patterns in cultured rat cortical neurons. Using whole-cell patch clamp to assess the scaffold effect on the development of synaptic connections, we found that the amplitude of excitatory postsynaptic current, as well as the frequency of spontaneous transmissions, was reduced in neuronal networks grown on an ultrasoft PDMS with an elastic modulus of 0.5 kPa. Furthermore, the ultrasoft scaffold was found to suppress neural correlations in the spontaneous activity of the cultured neuronal network. The dose of GsMTx-4, an antagonist of stretch-activated cation channels (SACs), required to reduce the generation of the events below 1.0 event per min on PDMS substrates was lower than that for neurons on a glass substrate. This suggests that the difference in the baseline level of SAC activation is a molecular mechanism underlying the alteration in neuronal network activity depending on scaffold stiffness. Our results demonstrate the potential application of PDMS with biomimetic elasticity as cell-culture scaffold for bridging the in vivo–in vitro gap in neuronal systems.
 Please wait while we load your content...
Please wait while we load your content...

