
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solubility behaviour of random and gradient copolymers of di- and oligo(ethylene oxide) methacrylate in water: effect of various additives†
Maryam
Bozorg
a,
Birgit
Hankiewicz
a and
Volker
Abetz
 *ab
*ab
aInstitute of Physical Chemistry, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany
bInstitute of Polymer Research, Helmholtz-Zentrum Geesthacht, Max-Planck-Straße 1, 21502 Geesthacht, Germany. E-mail: volker.abetz@hzg.de
First published on 3rd December 2019
Abstract
Poly[oligo(ethylene oxide)] based gradient and random copolymers with different compositions are synthesized via Cu-based atom transfer radical polymerization. The solubility behavior of these copolymers in pure water and in the presence of different salts, surfactants and ethanol is investigated. According to dynamic light scattering results, the lower critical solution temperature (LCST) depends on the structure of the copolymer and changes slightly in the presence of additives. Good cosolvents like ethanol can increase the LCST through dissolving the collapsed copolymer chains to some extent. The same effect is observed for surfactants that make the copolymer solution more stable by preventing aggregation. Above a certain concentration of surfactant, depending on the copolymer structure, the solution is stable at all temperatures (no LCST). The effect of salts on the solubility of the copolymers follows the Hofmeister series and it is related linearly to the salt concentration. Based on their affinity to the copolymer, the salts can increase or decrease the LCST. There is a considerable difference in phase transition changes for gradient or random copolymers after salt addition. While both copolymers show a two-step phase transition in the presence of different salts, the changes in the hydrodynamic radius and normalized scattering intensity are rather broad for random compared to gradient copolymers. Contrary to what was expected, varying the cations has no distinguishable effect on the LCST for both copolymers. All chlorides decrease the LCST. This decrease is almost the same for gradient copolymers and fluctuates for random copolymers.
Introduction
POEOMAs are nonlinear poly(ethylene oxide) (PEO) or poly(ethylene glycol) (PEG) analogues. They can be up to 85 wt% composed of ethylene oxide units (EO), and therefore are water soluble and biocompatible in most cases. POEOMAs combine both the properties of PEG and poly(N-isopropylacrylamide) (PNIPAM) in a single macromolecule and therefore are considered as ideal structures for use in biomedicine. The LCST of these polymers does not depend much on the solution concentration in water (above approx. 1 g L−1), which is an important factor for application in biotechnology. Moreover, being produced from a commercially available monomer, POEOMA containing polymers are favorable to be used as smart biomaterials in biosensors, artificial tissues, smart gels for chromatography and hyperthermia-induced drug delivery. For POEOMAs, the phase transition temperature depends slightly on the molecular weight, main-chain end-groups, tacticity and ionic strength. However, the changes in LCST are generally rather small.1–5For a long time, PNIPAM has held the title of “gold standard” for thermoresponsive polymers in bio-application due to its LCST of 32 °C, which is close to the physiological temperature. However, POEOMAs can exhibit an adjustable LCST between 26 and 90 °C by simple copolymerization of OEOMAs with different amounts of EO in the side chain. The LCST can be precisely adjusted by the copolymer composition considering that the comonomers have a similar structure containing a methacrylate moiety and ethylene oxide units.1,6 Moreover, the phase transition of these copolymers is reversible, in comparison to PNIPAM which shows an irreversible phase transition.4,7 The solubility behavior of PNIPAM also shows a significant dependency to its end group.8 Furthermore, the presence of the amide group at the side chain of PNIPAM might cause hydrogen bonding with other polyamides like proteins and result in bio-adhesion.9 PNIPAM also produces low molecular weight amines during hydrolysis which complicates its use in biotechnological applications.10
The outstanding solubility behavior of POEOMA is due to its hydrogen bonding with water and lack of strong polymer–polymer interactions in the collapsed state. Like PNIPAM, the phase transition of POEOMA is attributed to the competition between hydrophilic polymer–water interactions and hydrophobic polymer–polymer interactions. At temperatures below the LCST, polymer–water interactions are thermodynamically favorable which makes the polymer soluble in water. Above LCST, the polymer–polymer interactions become more favored which results in the self-aggregation of the polymers and phase transition in the form of globules or micelles depending on the structure of the POEOMA copolymer.1,3,11 Maeda et al. discovered that both C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H groups are hydrated in poly di(ethylene oxide) methyl ether methacrylate (PMEO2MA) aqueous solution, but due to the crowded position of the carbonyl groups near the backbone, only about half of these moieties are hydrated. By increasing the temperature above LCST, the fraction of hydrogen-bonded carbonyl groups decreases. Moreover, the complete breakage of the H-bond between the ether oxygens with water is reported. The fraction of hydrogen-bonding methoxy oxygens changes from one below LCST to zero after LCST. These results indicate that the hydrogen bond breakage is the main reason for phase separation of POEOMAs.12,13
O and C–H groups are hydrated in poly di(ethylene oxide) methyl ether methacrylate (PMEO2MA) aqueous solution, but due to the crowded position of the carbonyl groups near the backbone, only about half of these moieties are hydrated. By increasing the temperature above LCST, the fraction of hydrogen-bonded carbonyl groups decreases. Moreover, the complete breakage of the H-bond between the ether oxygens with water is reported. The fraction of hydrogen-bonding methoxy oxygens changes from one below LCST to zero after LCST. These results indicate that the hydrogen bond breakage is the main reason for phase separation of POEOMAs.12,13
A similar behavior is observed for the solubility of P(MEO2MA-stat-OEGMA475) copolymers in D2O which shows a sharp change in the hydrodynamic radius at the LCST and a gradual change above LCST. The phase transition of P(MEO2MA-stat-OEGMA475) occurs due to multiple chain aggregation without pre-connection of individual polymer chains. Self-aggregation of P(MEO2MA-stat-OEGMA475) is mainly based on the conformation change of ethylene oxide side chains. They first collapse to be near the hydrophobic backbone and then distort to bring hydrophilic ether oxygen groups to the “outer shell” of polymer chains as far as possible. As a result of disturbance in the balance between hydrophobic and hydrophilic interactions, the single dehydrated polymer chains aggregate into more stable micelles and cause a sharp change below LCST.3
The P(MEO2MA-stat-PEGMA2080) copolymer in aqueous dilute solution undergoes a similar phase separation mechanism. However, compared to other random copolymers of MEO2MA and OEOMA, this copolymer shows a weird two stage thermally induced phase separation. Instead of loose aggregates formed at the phase transition, the copolymer chains associate at the first thermal transition, followed by a rearrangement process at the second thermal transition to form stable micellar structures consisting of a methacrylate core stabilized by the longer ethylene oxide chains at the shell.14 A more detailed study shows four conformation changes: “unimers–clusters–micelles–aggregates” during the two-step phase transition. Like other POEOMAs, the dehydration of the long hydrophilic ethylene oxide side chains takes place before the dehydration of carbonyl groups and backbones during the whole phase transition process. Therefore, the driving force of the phase transition of P(MEO2MA-stat-PEGMA2080) should be the hydration changes of the side chains. However, detailed FT-IR analysis reveals that the peculiar behavior in the phase transition process could be attributed to the complex transition between hydrated CO, semi-dehydrated CO and dehydrated CO.15
A two-step phase transition is also observed by Yao and Tam16 for the behavior of PMEO2MA-block-(PMEO2MA-stat-POEGMA300) block copolymers. They observed that by changing the ratio of MEO2MA/OEGMA300 from 80/20 to 70/30 the copolymer's solution behavior changes from showing one thermal transition to two. A similar phenomenon has also been observed by Gibson et al.17 for the mixture of two POEGMA chains with different molecular weights. The independent phase transitions are described by the weak molecular weight dependence of the polymers’ cloud point.
By increasing the number of blocks, Kudo et al.18 observed a multi-step phase separation for the hexa-block copolymer P[MEO2MA-block-(MEO2MA-stat-OEOMA)]. They synthesized this hexa-block by semi-batch RAFT polymerization with the addition of more OEOMA every two hours and therefore, the concentration of OEOMA in the reaction flask increased in batches, not gradually. Upon sequential dehydration, each block of the hexa-block copolymer showed its own temperature responsive behavior. The thermoresponsivity in each step was reversible with 2 °C hysteresis. Overall, the behavior of the block copolymer showed slight similarity to gradient copolymers. To the best of our knowledge, this is the only study mentioning the solubility behavior of gradient POEOMA copolymers with a similar comonomer structure.
Studies on the solubility behavior of gradient copolymers consisting of a hydrophilic and a hydrophobic monomer show that the solubility behavior changes drastically, depending more on the interaction of the comonomers with water and their hydrophobicity rather than their sequential order in the copolymer structure.19,20 On the other hand, the thermal phase transition of other temperature responsive gradient copolymers consisting of monomers with similar chemical structure and therefore, similar hydrophobicity has shown considerable differences from the respective random and block copolymers and dependent on their sequential order in the gradient structure. This characteristic makes such gradient copolymers a great potential in biomimetic applications.21–29
Various studies are done on the effect of additives on the solubility behavior of thermoresponsive polymers30–39 including POEOMAs,40–44 but so far there is no comprehensive study to compare the effect of different additives on the phase transition of thermoresponsive copolymers with different structure.
In this study, the synthesis of gradient copolymers of MEO2MA and OEOMA via semi-batch ATRP is investigated. The injection procedure of the second monomer (OEOMA) is optimized to reach the best sequence control and gradient structure. Moreover, the solubility behavior of gradient and random copolymers with different compositions as well as their behaviors in the presence of various additives are compared. As additives, different anions and cations, as well as ethanol as an organic polar solvent and sodium dodecyl sulfate SDS (anionic surfactant) and cetyltrimethylammonium bromide CTAB (cationic surfactant) are used and the effects on the solubility behavior of random and gradient copolymers of MEO2MA and OEOMA are investigated. Knowledge of the copolymer's responsivity towards different additives is essential for in vivo applications, as well as separation and membrane technology.37,45 While adjusting the phase transition behavior of POEOMAs by changing the structure seems to be expensive, time consuming and complicated, an easier way to alter the dynamic thermal transition is by addition of small amounts of additives to the system.
Experimental
Materials
The monomers, di(ethylene oxide) methyl ether methacrylate (MEO2MA) (Aldrich, 95%) and oligo(ethylene oxide) methyl ether methacrylate (OEOMA) (Aldrich Mn = 500), were purified by passing them through a basic alumina column to remove the inhibitors. The catalyst copper(I) bromide (CuBr) (Sigma-Aldrich > 98%) was purified by mixing with glacial acetic acid, followed by filtration. It was further washed three times with absolute ethanol and two times with diethyl ether. The colorless solid product was dried overnight under vacuum at room temperature and was stored under nitrogen. The initiator, ethyl α-bromoisobutyrate (EBiB) (Sigma-Aldrich 98%), the ligands, 4,4′-dinonyl-2,2′-dipyridyl (dNbpy) (Alfa Aesor 97%) and N,N,N′,N′′,N′′-pentamethyl diethylene triamine 99% (PMDETA) (TCI 98%), the reaction solvents, anisole (Merck 99%) and ethanol (Acros 99.5%) as well as other chemicals were used as received without further purification.Homopolymerization and random copolymerization of MEO2MA and OEOMA via ATRP
The polymerization was performed using [M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]0:[CuI]0:[L] ([Monomer]0:[Initiator]0:[CuBr]0:[Ligand]) at a ratio of 100:1:0.5:1. For a general ATRP reaction, CuBr and a magnetic stirring bar were added to a 50 mL two necked flask connected to a condenser and Schlenk line. The reaction flask was then evacuated and backfilled with nitrogen at least three times and kept for each repetition at least 15 min under vacuum. The monomer and the initiator were added to a 50 mL Schlenk flask. The solvent and the ligand were added to another 50 mL Schlenk flask. The liquids were degassed via three freeze–pump–thaw cycles and kept under nitrogen. The solvent and the ligand were afterwards added to the reaction flask containing CuBr via a degassed airtight syringe and mixed for half an hour until the CuBr/ligand complex was formed. The monomer and the initiator were added afterwards with a degassed airtight syringe and the reaction started by placing the reaction flask in a preheated oil bath at 60 °C. The reaction proceeded under nitrogen flow. Samples were withdrawn with a degassed syringe at defined time intervals for kinetic studies. The reaction was stopped by opening the flask to air and cooling it in an ice bath. The reaction solution was diluted with tetrahydrofuran and passed through a neutral aluminum oxide column to remove the catalyst. The extra amount of solvent was removed via a rotary evaporator. The polymer was purified by precipitating the concentrated polymer solution in cyclohexane and dried under vacuum overnight.
:[I]0:[CuI]0:[L] ([Monomer]0:[Initiator]0:[CuBr]0:[Ligand]) at a ratio of 100:1:0.5:1. For a general ATRP reaction, CuBr and a magnetic stirring bar were added to a 50 mL two necked flask connected to a condenser and Schlenk line. The reaction flask was then evacuated and backfilled with nitrogen at least three times and kept for each repetition at least 15 min under vacuum. The monomer and the initiator were added to a 50 mL Schlenk flask. The solvent and the ligand were added to another 50 mL Schlenk flask. The liquids were degassed via three freeze–pump–thaw cycles and kept under nitrogen. The solvent and the ligand were afterwards added to the reaction flask containing CuBr via a degassed airtight syringe and mixed for half an hour until the CuBr/ligand complex was formed. The monomer and the initiator were added afterwards with a degassed airtight syringe and the reaction started by placing the reaction flask in a preheated oil bath at 60 °C. The reaction proceeded under nitrogen flow. Samples were withdrawn with a degassed syringe at defined time intervals for kinetic studies. The reaction was stopped by opening the flask to air and cooling it in an ice bath. The reaction solution was diluted with tetrahydrofuran and passed through a neutral aluminum oxide column to remove the catalyst. The extra amount of solvent was removed via a rotary evaporator. The polymer was purified by precipitating the concentrated polymer solution in cyclohexane and dried under vacuum overnight.
The procedure for random copolymerization was similar to homo-polymerization; instead of a single monomer, two different monomers (MEO2MA and OEOMA) with a defined molar ratio were added depending on the planned copolymer composition.
Gradient copolymerization of MEO2MA and OEOMA via ATRP
Gradient copolymerization was performed with a [M]0:[I]0:[CuI]0:[L]: of 100:1:0.5:1 (where [M]0 is a mixture of MEO2MA and OEOMA with a defined molar ratio) using PMDETA as the ligand and anisole as the solvent (Fig. 1). In the procedure of optimization of the injection program, the aspired ratio of two monomers was chosen to be [MEO2MA]:[OEOMA]:80:20. It was subsequently changed according to the desired composition.
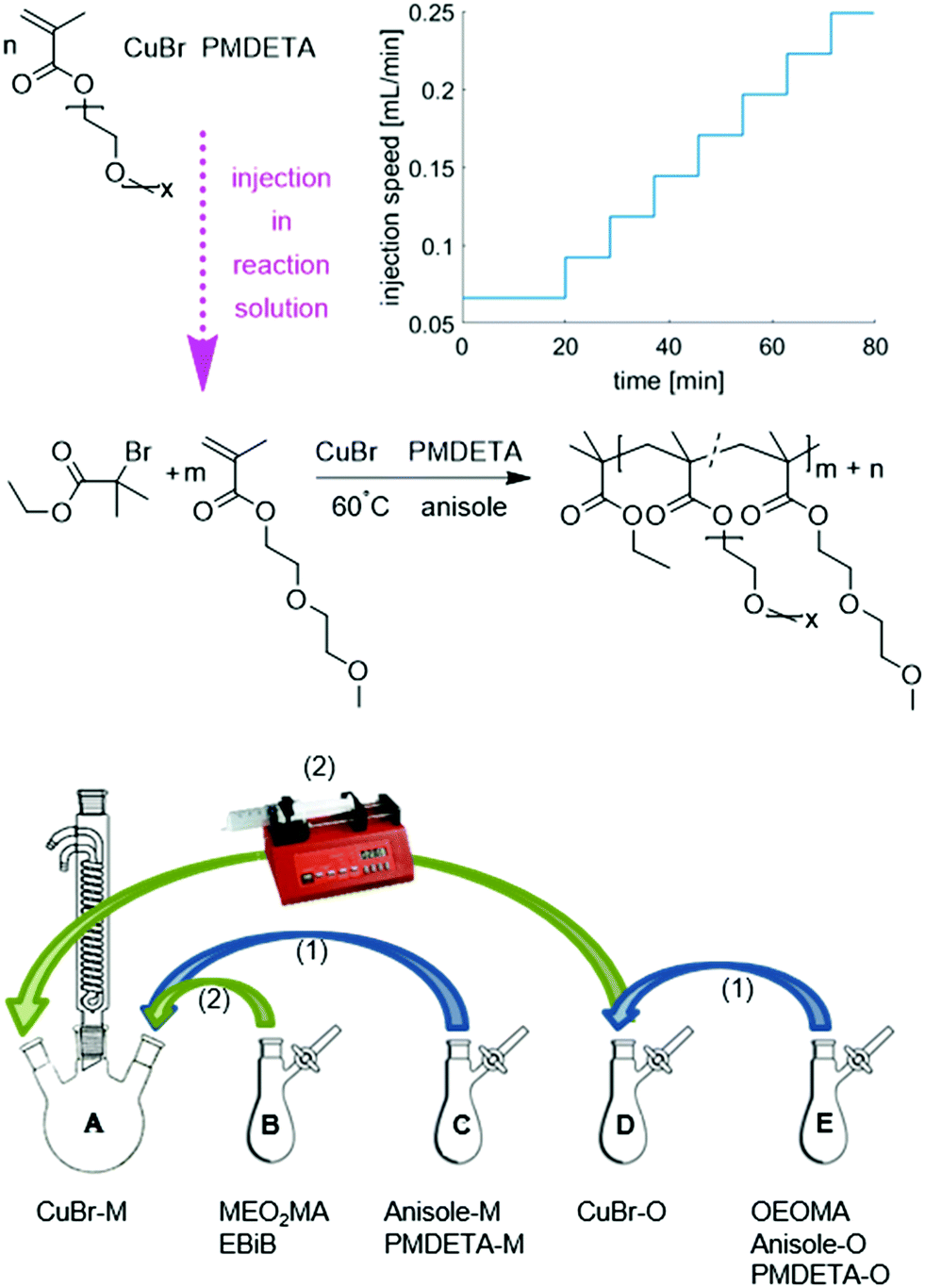 | ||
| Fig. 1 The reaction scheme of gradient copolymerization of MEO2MA and OEOMA via ATRP at 60 °C with CuBr as catalyst, PMDETA as ligand and anisole as solvent and the addition procedure of different reagents for gradient polymerization; M assigns for equivalent of MEO2MA and O for OEOMA. The graph of injection speed versus time belongs to the injection program (I). Other injection programs tried in this study are represented in ESI.† | ||
To achieve the gradual composition of the product, a programmable single syringe pump was used. The injection procedures differed in the overall injection time as well as the number of steps and the injection volume of each step of gradual addition of OEOMA to the reaction solution. The injection programs are shown in Fig. S1 (ESI†). They were calculated for the given injection volume and injection time with an increasing injection speed in order to compensate the increasing viscosity of the reaction solution and the decreasing reaction rate. Based on the results of the first syntheses, the injection program was adjusted to achieve the gradient copolymer with desired composition and good gradient quality. The effect of the injection procedure on the copolymer structure will be explained later. In order to avoid the very fast reaction at the beginning due to the high concentration of the catalyst, the CuBr/L complex and the reaction solvent are divided into two different monomer flasks with the same equivalent ([M]0:[CuI]0:[L] of 100:0.5:1). This also helps to keep the ratio between catalyst/ligand and monomer constant during the reaction. To prevent a reaction in the syringe, the whole amount of initiator is added at the beginning to the reaction flask. Fig. 1 also shows the addition procedure of the different reagents. In general, the reaction procedure is as mentioned before for homopolymerization, with an extra step of monomer solution preparation. The reaction flask was prepared and afterwards charged with MEO2MA, while another flask of OEOMA was prepared in a similar way (without initiator). MEO2MA and the initiator were not added to the CuBr solution before the reaction started but degassed separately.
Both flasks containing CuBr were degassed via evacuation and backfilling with nitrogen as mentioned before. The mixtures of liquids were degassed via three freeze–pump–thaw cycles. To dissolve the solids and build up the copper complex, solutions C (anisole and PMDETA, the equivalent amount for MEO2MA) and E (anisole and PMDETA, the equivalent amount for OEOMA and the monomer OEOMA) were added to flask A (containing CuBr, the equivalent amount for MEO2MA) and D (containing CuBr, the equivalent amount for OEOMA) respectively and mixed for 30 minutes. The reaction was started by adding MEO2MA and EBriB from flask B to the reaction solution A and immersing the flask into an oil bath (T = 60 °C). The injection program of the OEOMA solution was started simultaneously. The reaction was quenched as described for homopolymerization before and the final product was purified by column chromatography and precipitation in cyclohexane.
Analytics
A 0.1 M solution of lithium bromide (LiBr) (Acros Organics) in N,N-dimethylacetamide (DMAc) (HPLC Optigrade, Promo-chem) with a flow rate of 1.0 mL min−1 at a temperature of 50 °C was utilized as the eluent. Methyl benzoate was added as the internal standard to the analyzed polymer solutions which had a polymer concentration of 2 mg mL−1. The injection volume was 100 μL. For determination of the relative MWs and Đ-values, the system was calibrated with narrowly distributed PMMA standards with molecular weights between 2.2 and 1190 kDa.
The polymer solutions were prepared with a concentration of 1 mg mL−1 and were shaken overnight to ensure complete dissolution. Each solution was filled in a dust-free glass tube through a microporous regenerated cellulose filter with an average pore diameter of 200 nm. The DLS samples were let to rest at least 1 h prior to measurement in order to ensure that the possible dust particles present in the system settle and not interfere with the measurement. To investigate the effect of different additives, the solution of a certain additive with an exact concentration was prepared beforehand and added to the polymer instead of pure solvent.
Temperature-dependent DLS measurements were performed at temperature steps of 2 °C with 3 runs per temperature, except for more detailed measurements which were done with 1 °C temperature steps. A Julabo F25 thermostat functioning with a mixture of water and ethylene glycol with a temperature accuracy of 0.01 °C was used as a heating system. There was a 3 min time interval for the stabilization of the temperature prior to each measurement.
A MATLAB program was used to analyze the electric field autocorrelation functions g1(q,t) by means of a cumulant fit up to the second order for a monomodal distribution, eqn (1),
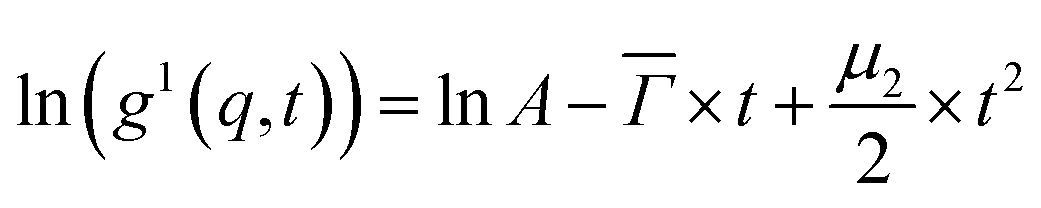 | (1) |
![[capital Gamma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ba.gif) is the average decay rate, t is the time, μ2 is the second moment, and
is the average decay rate, t is the time, μ2 is the second moment, and 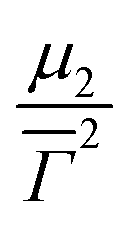 presents a measure of the relative width of the size distribution (particle size dispersity, PSD):
presents a measure of the relative width of the size distribution (particle size dispersity, PSD):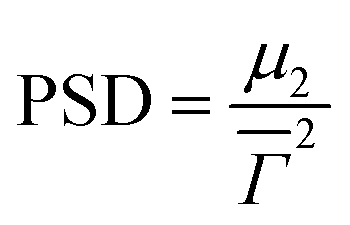 | (2) |
The translational diffusion coefficient D was determined from = Dq2 with
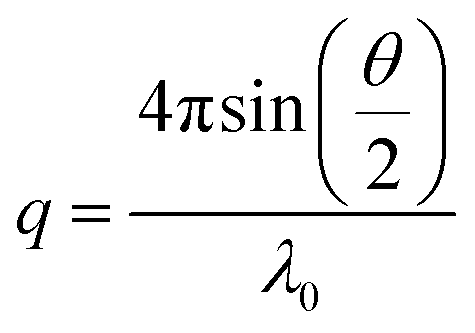 | (3) |
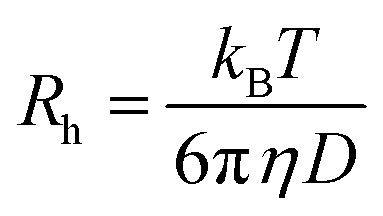 | (4) |
Results and discussion
Controlled radical polymerization (CRP) techniques like ATRP provide versatile routes for macromolecular engineering such as synthesis of tailor-made polymers with controlled chain-length, dispersity, functionality, composition and architecture. Moreover, the polymers synthesized via CRP have a reactive end group which could be used in further end functionalization or even as a macroinitiator for new polymerization.1The first polymerization of an OEOMA via ATRP was carried out by Armes et al. in aqueous medium. But the high polarity of water makes it very complicated to control the reaction.51 In early studies, Ishizone et al. also synthesized various poly[oligo(ethylene glycol) methacrylate]s using living anionic polymerization. They studied the effect of methyl, ethyl, vinyl and hydroxyl end groups, as well as the number of ethylene oxide moieties in the side chain, on the polymers’ solubility in water and organic solvents.5,52–54 ATRP as a synthesis route is advantageous compared to anionic polymerization due to its flexibility and simpler reaction conditions. Moreover, the prospects of obtaining new structures like gradient, random or star copolymers are higher via ATRP. It is worth mentioning that the precise dependency of LCST on the copolymer composition could only be observed for copolymers prepared by controlled polymerization techniques. In a conventional free radical polymerization, the chains can present a strong chain-to-chain deviation of composition, which results in different phase transitions.1,37
The synthesis route of this research is an optimized work based on the research of Lutz et al.55 and Matyjaszewski et al.6 Lutz et al. reported a well-controlled ATRP of OEOMAs in pure ethanol. The polarity of ethanol establishes fast polymerization kinetics,55 but since OEOMA monomers have a slight polarity, their polymerization can also be successfully done in apolar solvents like anisole as reported by Matyjaszewski's group.6 For selecting the best reaction conditions for ATRP gradient polymerization of OEOMAs, first the homopolymerization of MEO2MA was optimized. Several test reactions with different solvents, ligands and reaction times were conducted. Samples were taken at defined time intervals and analyzed via NMR and SEC.
The NMR spectra of the MEO2MA monomer and its changes during polymerization are plotted in Fig. S2 (ESI†) where the calculation of monomer conversion is explained as well. To analyze and compare the control over different reaction systems, in Fig. 2(a) the semi-logarithmic plot of monomer conversion i.e. ln([M]0/[M]) is plotted versus reaction time. The reaction time is 100 min for all reactions, except for the solvent ethanol and the ligand PMDETA, which is stopped after 80 min due to its higher rate. The semi-logarithmic plots are linear for all of the polymerizations indicating that the polymerization rate is proportional to the monomer concentration (first order polymerization with respect to monomer) and moreover, the radical concentration is constant during the polymerization, according to.5,51,56
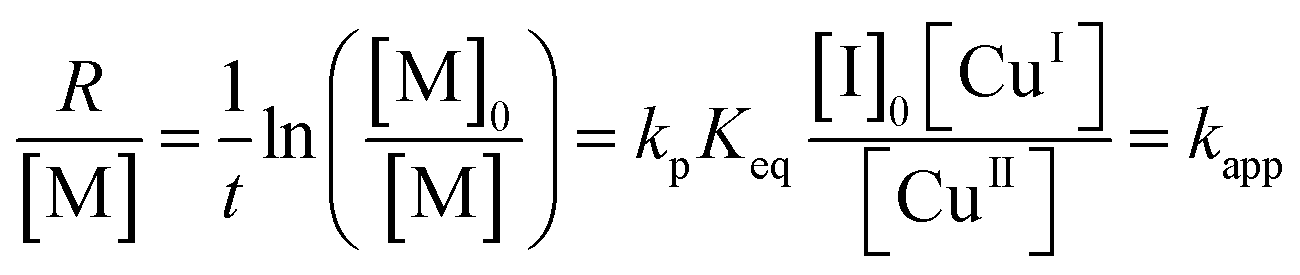 | (5) |
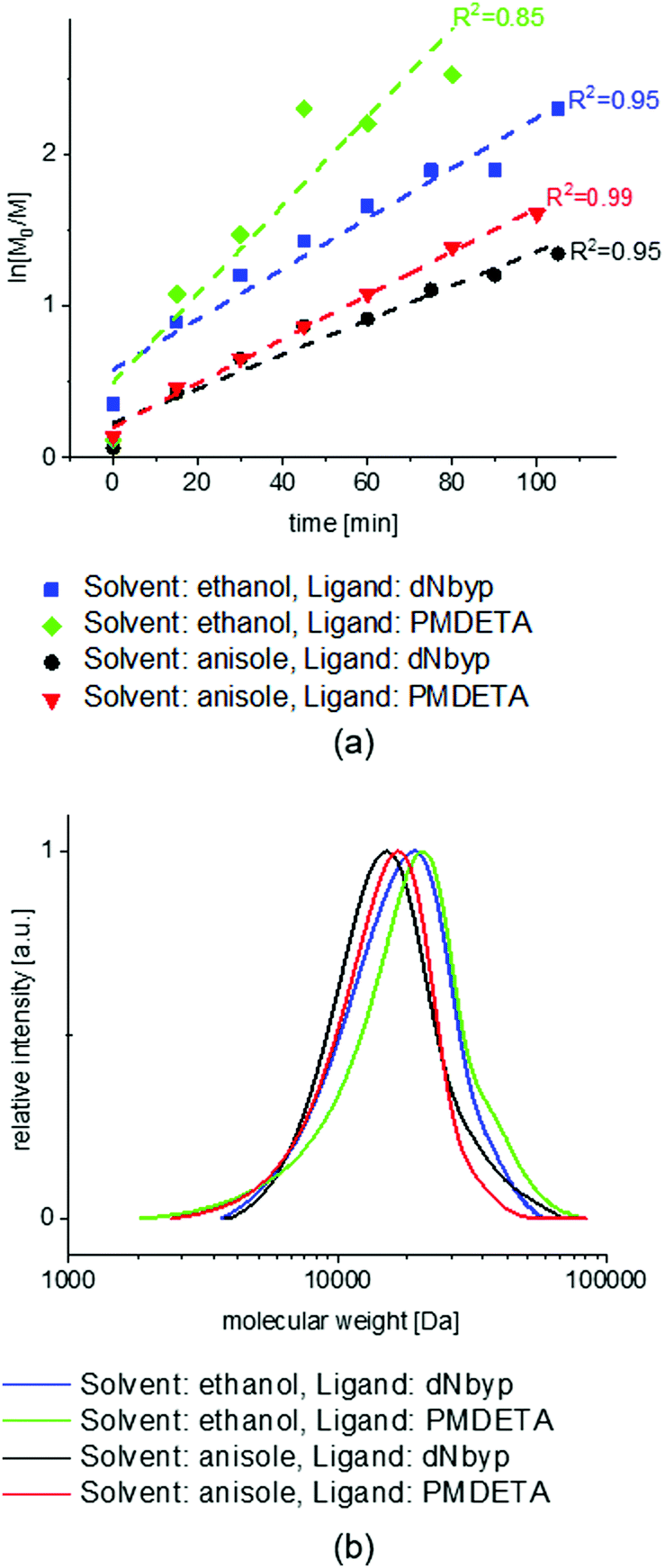 | ||
| Fig. 2 (a) Comparison of semi-logarithmic plot of monomer conversion vs. time in ATRP homopolymerization of MEO2MA with different solvents and ligands. (b) The SEC graphs of MEO2MA homopolymerization with different solvents and ligands. The best control is obsereved for anisole as solvent and PMDETA as ligand. The worst control as well as the fastest polymerization is observed for ethanol as solvent and PMDETA as ligand. [M]0:[I]0:[CuI]0:[L] = 100:1:0.5:1 for all reactions. | ||
The highest slope in the semi-logarithmic plots is observed for the ethanol and PMDETA system showing the highest apparent reaction rate with lower control over polymerization compared to the other systems.
The best control over polymerization is observed when using anisole as a solvent and PMDETA as a ligand. In comparison to anisole, the polymerization in ethanol is faster and less controlled. This could be explained by the relative polarity of ethanol (0.654) which is higher than anisole (0.198); (relative polarity, normalized from measurements of solvent shifts in absorption spectra).57
Among different solvents used to conduct ATRP, good control mostly resulted from bulk polymerization or using nonpolar solvents, while using polar solvents especially water, often leads to limited control over polymerization and poor livingness and the reaction is incredibly fast.58,59 Wang and Armes suggested that, by increasing the solvent polarity, a higher concentration of mononuclear copper catalyst is produced which increases the radical concentration and the polymerization rate. As a result, the molecular weight distribution is broadened distinctly.51 According to other studies, increasing the solvents’ polarity increases the kact while decreasing kdeact with approximately the same ratio: Δlogkact ≈ −Δlogkdeact. Therefore, changing to a more polar solvent increases KATRP since KATRP = kact/kdeact and results in overall faster and less controlled polymerization.59–63
Furthermore, the solvent effect on the Cu ions’ redox potential appears to be higher for complexes of copper with ligands having high degrees of freedom, compared to more rigid structures.59 Moreover, by increasing the number of coordination sites of a ligand, the redox potential of its copper complex increases.62 Comparing the ligands that are used in this research, PMDETA has the highest redox potential considering the fastest polymerization in both ethanol and anisole. Incidentally, the reaction with PMDETA in anisole is still well controlled based on its first order kinetics and SEC results.
The SEC results of different reaction conditions (Fig. 2) and summarized information (in Table 1) prove the good control over polymerization and low Đ for all systems excluding the polymerization using ethanol as a solvent and PMDETA as a ligand. The above-mentioned system shows a small shoulder at higher molecular weights which is related to faster activation than deactivation reaction in ATRP equilibrium and therefore a small portion of bimolecular termination close to the end of the reaction. The addition of CuIIBr or decreasing the CuIBr concentration does not help to decrease the propagating radicals’ concentration and improving the control over polymerization noticeably.
| Solvent | Ligand | [M]0:[I]0:[CuI]0:[L]a |
Conv.b | Time [min] |
M
theorync [kDa] |
M SECn [kDa] | M w/Mn | |
|---|---|---|---|---|---|---|---|---|
a [M]0:[I]0:[CuI]0:[L]:[MEO2MA]0:[EBiB]0:[CuIBr]0:[Ligand]: the ratio of the ingredients at the start of the reaction.
b Monomer conversion is determined by 1H NMR.
c
M
theoryn: theoretical number-average molecular weight is calculated via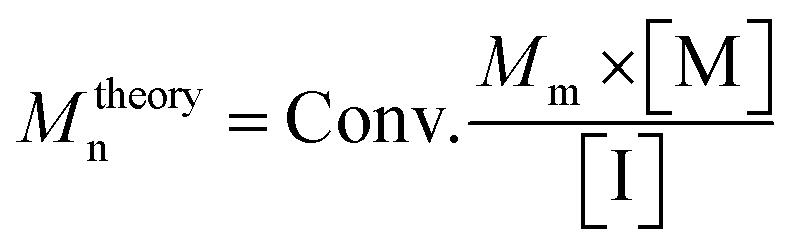 , Conv.: conversion, Mm: monomer's molecular weight, [M]: molar amount of monomer, [I]: molar amount of initiator. , Conv.: conversion, Mm: monomer's molecular weight, [M]: molar amount of monomer, [I]: molar amount of initiator.
|
||||||||
| 1 | Ethanol | dNbyp | 100:1:0.5:2 |
0.85 | 100 | 15.99 | 12.03 | 1.36 |
| 2 | Ethanol | PMDETA | 100:1:0.5:1 |
0.92 | 80 | 17.32 | 14.95 | 1.43 |
| 3 | Anisole | dNbyp | 100:1:0.5:2 |
0.65 | 100 | 12.23 | 12.86 | 1.22 |
| 4 | Anisole | PMDETA | 100:1:0.5:1 |
0.75 | 100 | 14.12 | 13.66 | 1.23 |
Table 1 shows that the apparent molecular weights of the polymers synthesized in anisole are in good agreement with the theoretical calculation, while for the polymers synthesized in ethanol the molecular weights achieved are slightly lower than from theoretical calculations which is in agreement with previous studies.6 A possible explanation for such behavior is the chain transfer reaction to solvent due to the higher reactivity of ethanol than anisole. Such a chain transfer reaction to solvent was also indicated in previous ATRP studies.64–66 Among the test polymerizations conducted, the one in ansiole using PMDETA as a ligand shows a very good control over the reaction while being fast. Considering the fact that less molar amount of PMDETA is needed to provide a good control compared to dNbyp and the easier way of handling such small amounts (because PMDETA is liquid while dNbyp is solid) the final choice was to carry out ATRP polymerization of MEO2MA and further copolymerizations in anisole using PMDETA as a ligand.
The shift of SEC peaks over time as well as the growth of molecular weight vs. conversion for the anisole and PMDETA system are plotted in Fig. S4 (ESI†). Above a conversion around 0.3, the molecular weight increases linearly with conversion and it's in agreement with the theoretical prediction.
The fast increase of molecular weight at the beginning is explained due to the low deactivator concentration which results in rapid early growth of a small amount of polymer chains. The polydispersity decreases at the beginning and then remains constant during the reaction, indicating a fast exchange between active and dormant species. According to the SEC results in Fig. S3 (ESI†), the narrow SEC peaks are shifted gradually to higher molecular weights which indicates that termination and chain transfer reactions are not happening since neither a high molecular tail nor a shoulder is observable. Thus, the PMDETA–anisole system provides the desired characteristics of high initiation efficiency, low dispersity, and a moderate apparent polymerization rate which is in accordance with the literature.56 This system should permit fast initiation, fast deactivation of the propagating radicals by CuII species, and reduced side reactions of the Pn–Br growing chains formed by halogen exchange, thereby providing a good control during ATRP.56
Random and gradient copolymerization of MEO2MA and OEOEMA
Since MEO2MA and OEOMA differ only in the side chain, therefore, their reactivity ratios are very similar. This is also proven by very similar ATRP kinetics of these monomers under similar reaction conditions as can be seen in Fig. S4 and S5 in the ESI.† Therefore, forced gradient copolymerization could be an optimal synthesis route to prepare gradient copolymers of MEO2MA and OEOMA. This technique involves a semi-batch reaction, in which one of the monomers is added continuously from an external reservoir, via a syringe pump into the polymerization mixture. The forced gradient method with living polymerization allows precise synthesis of gradient compositions with high sequence control.21To follow the change of the copolymer structure during the reaction, the copolymers’ compositions were studied by NMR spectroscopy. The NMR spectra as well as the calculation of the copolymers’ compositions are provided in the ESI† (Fig. S6 and S7). The calculated copolymer composition for a random copolymer of MEO2MA and OEOMA with a designed composition of EO2MA:OEOMA is plotted in Fig. 3. The composition remains constant during the reaction which shows a perfect random copolymerization of the two monomers.38
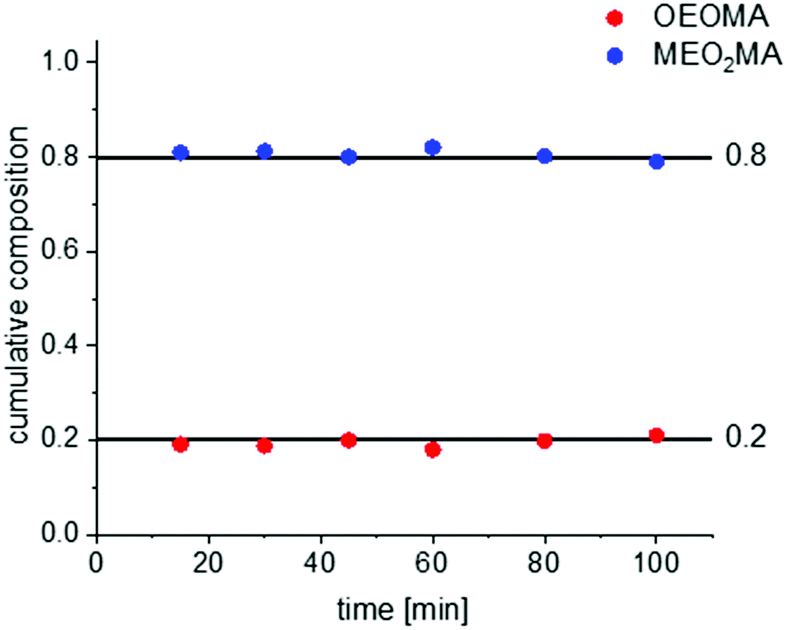 | ||
| Fig. 3 The composition change for random copolymerization of two monomers with the ratio of MEO2MA:OEOMA = 80:20. | ||
Since the reactivity ratios of the two monomers are very similar, a gradient copolymerization in a batch system is impossible,67 as also proven by the perfect random copolymerization of the two monomers in a batch system (Fig. S4, ESI†). Therefore, gradient copolymerization was done via ATRP semi-batch polymerization. Three different injection programs (P1–P3) were used in this work to inject the OEOMA solution to the reaction system. In all programs, the injection speed was increased stepwise during the reaction leading to the parabolic curve of the injected volume against time. The reason for injecting OEOMA to the system is its lower ratio in the copolymer's desired composition compared to MEO2MA and its higher viscosity. Since the two monomers have almost the same structure and they differ only at the side chain, their reactivity ratios are the same and almost equal to one. The similar reactivity ratios of MEO2MA and OEOMA make it easier to adjust the injection program according to the desired composition.
In the first synthesis (P1), the injection was carried out for the first 80 min of the total reaction time of 100 min. In the second synthesis (P2), the starting injection speed was decreased to 30% while the injection time was reduced to 70 min of the overall reaction time of 100 min leading to a steeper injection volume curve. The third reaction (P3) was done like the first one (injecting for 80 min of 100 min reaction time) but with 30% higher starting speed and less injection steps which changes the injection volume curve toward a linear graph.
The composition development during the three polymerizations is shown in Fig. 4. The plots of all syntheses show a gradual, almost linear increase of OEOMA and decrease of MEO2MA in the copolymer composition which indicates a gradient structure for all systems. For syntheses of P1 and P2, the change in composition at the beginning of the reaction is very small which led us to increase the starting injection speed for the last reaction. It should also be noted that for both P1 and P2, the aspired copolymer composition (OEOMA:MEO2MA of 80:20) was not achieved. The final composition is 87:13 for P1 and 85:15 for P2. In the last reaction P3, a perfect linear graph of composition vs. time is observed and the copolymer reached the desired composition.
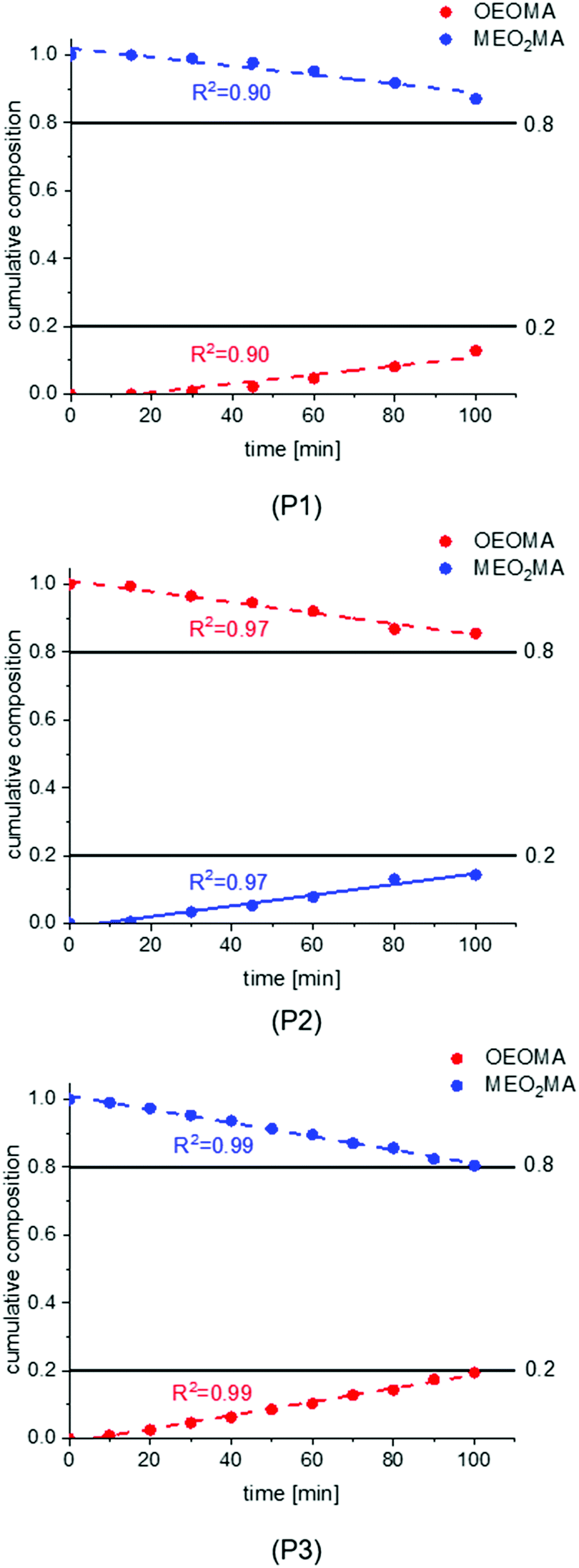 | ||
| Fig. 4 The composition development during the first ATRP gradient copolymerization (injection program P1–P3). | ||
The developments of the composition qualitatively follow the injection program. Since the chain growth is directional with only one active end, the increasing incorporation of OEOMA during the reaction indicates an increasing OEOMA concentration from one chain end to the other. Hence, the synthesized polymers exhibit the aspired gradient structure.
A direct conclusion about the kinetics or the control over the reaction is difficult because of the continuous injection of the monomer. A kinetic study of polymerization control via conversion calculation is hence not possible. However, the implied high control over the reaction is reflected in the SEC analysis and the molecular weight development (see Fig. S2, ESI†). Furthermore, the molecular weight change for gradient and random copolymerization versus time is linear showing a good controlled polymerization. The lower molecular weight for gradient copolymer is due to the lower concentration of OEOMA during reaction, which results in a slightly slower growth of molecular weight as explained in SI. The molar amount of OEOMA incorporated in the gradient and random copolymer chains are proven to be the same by NMR.
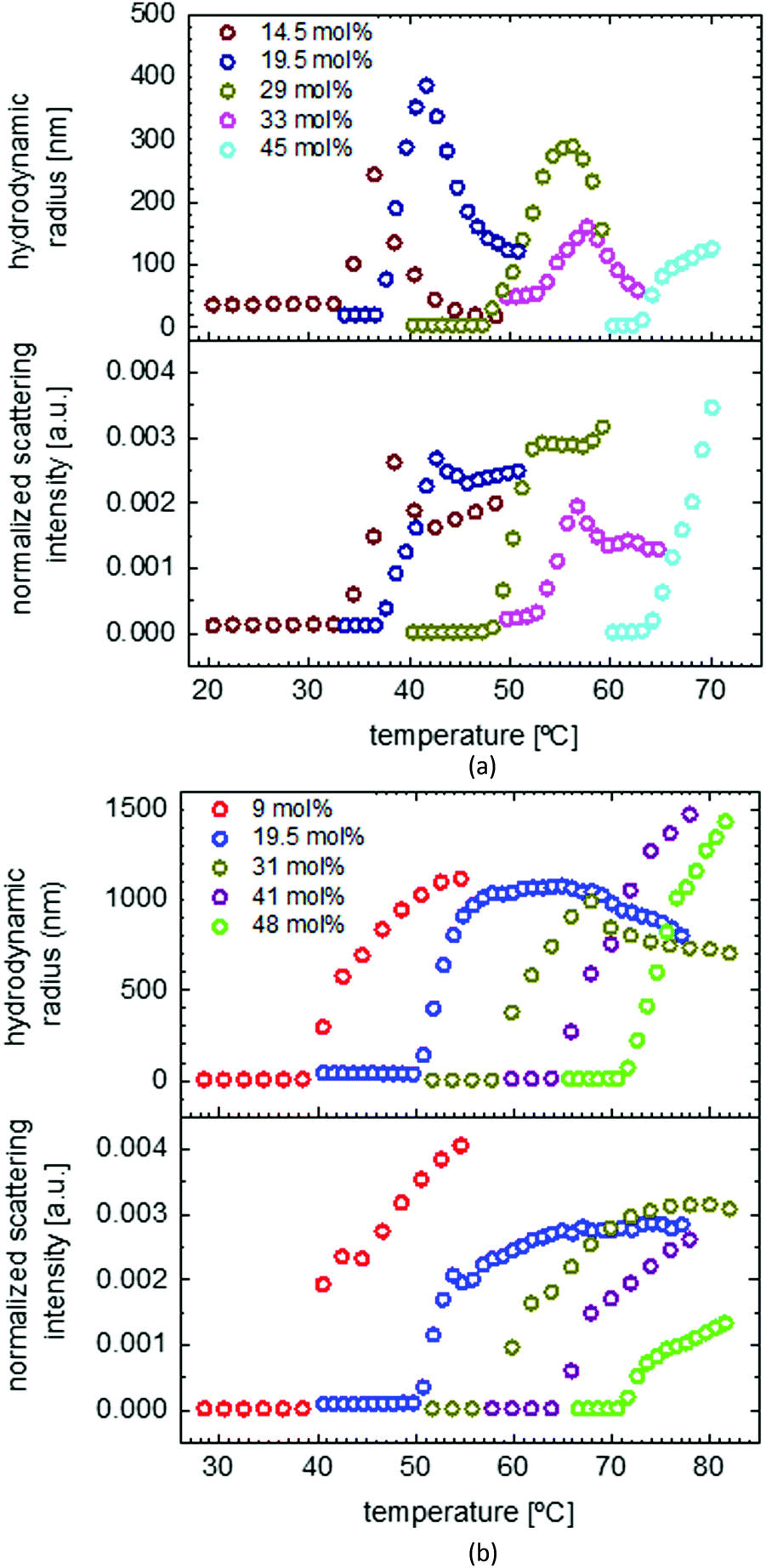
Solubility study of PMEO2MA homopolymer and random and gradient copolymers of MEO2MA and OEOMA in water
To investigate the solubility behavior of a temperature responsive polymer, mostly turbidimetry is used, which gives good information about the cloud point; but it cannot explain the transition procedure well. By using DLS, we were able to follow the changes in the particle size, size distribution and scattering intensity, and therefore, study the solution behavior in more detail. Fig. 5 shows the solubility behavior of PMEO2MA as well as the solubility behavior of a random and a gradient copolymer of MEO2MA and OEOMA with approximately 20 mol% OEOMA in a dilute water solution. As seen in the graph, both normalized scattering intensity (I) and hydrodynamic radius (Rh) increase sharply above LCST, then they increase gradually to reach a maximum and then decrease very slowly (the red graph). The amphiphilic structure of PMEO2MA is the reason for its thermoresponsive behavior. Below LCST, the ether oxygens of the side chains form hydrogen bonds with water. On the other hand, the apolar carbon–carbon backbone causes a competitive hydrophobic effect. This results in a balance between favorable polymer–water interactions and unfavorable polymer–polymer interactions which grants solubilization. By increasing the temperature above LCST, this balance is disrupted as the hydrogen bonds break and hydrophobic polymer–polymer interactions become thermodynamically favored as compared to polymer–water interactions.1 Therefore, the hydrophobicity of the polymer increases as the temperature increases and this results in aggregation of the polymer chains and phase separation.6 This also explains why the LCST increases upon increasing the amount of ethylene oxide in the side chain of POEOMA, as more EO units result in an increase in the amount of hydrogen bonds and therefore stronger polymer–water interaction.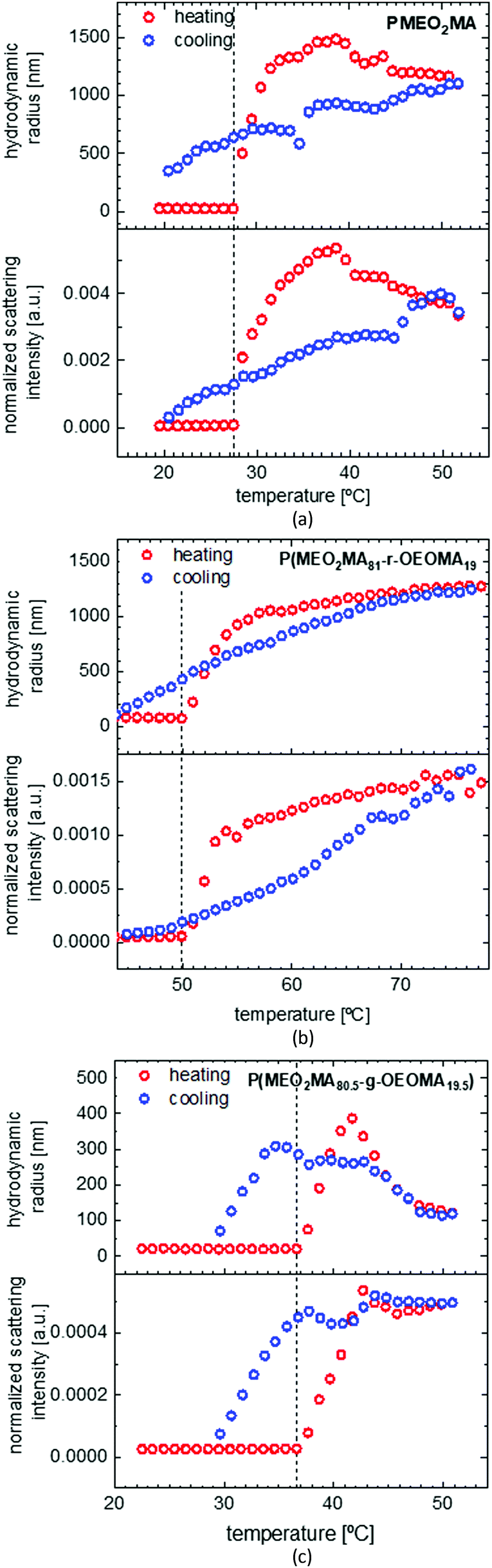 | ||
| Fig. 5 DLS measurement of PMEO2MA, Mn = 13.66 kDa, Đ = 1.23 (a), the random copolymer P(MEO2MA81-r-OEOMA19), Mn = 15.96 kDa, Đ = 1.17 (b) and gradient copolymer P(MEO2MA80.5-g-OEOMA19.5), Mn = 15.21 kDa, Đ = 1.24 (c) in water. The red points show the heating cycle and blue points the cooling cycle. The vertical dashed line points out the abrupt change in hydrodynamic radius and normalized scattering intensity and indicates the LCST at 27.5 °C. | ||
At temperatures lower than LCST, the polymer is dissolved in the form of unimers with an Rh of around 20 nm. When the temperature reaches LCST, as was explained before, the polymer chains form aggregates and Rh suddenly increases to 1300 nm at 32 °C. The size growth continues until Rh reaches 1500 nm, due to binding of more PMEO2MA chains to the aggregate. The aggregate size then starts to decrease while more and more water molecules are expelled and PMEO2MA chains get dehydrated and as a result, the aggregates slightly shrink. It should also be noted that there is no size change observed for PMEO2MA before LCST, proving that the coil of PMEO2MA does not collapse (mainly by the distortion of backbones) into a crumpled structure at low temperatures. This is totally different from PNIPAM which shows a precontraction of individual polymer chains before the phase transition.3,68
It is also obvious from Fig. 5(a) that there is a hysteresis in the heating and cooling cycle of the solubility behavior of PMEO2MA, which is due to the small delay in the dissolution of large precipitated globular particles. This is attributed to the procedure of DLS measurements which excludes any type of stirring while measuring and makes it more difficult for the large globules to dissolve fast. However, the hysteresis according to the literature is still much less than for PNIPAM that is assigned as the golden standard of temperature responsive polymers.1,3
Wu and coworkers explain the large hysteresis in the solution behavior of PNIPAM as a result of the intramolecular and intermolecular NH⋯OC hydrogen bonding interactions that are formed in the collapsed state. These strong hydrogen bonds hinder the rehydration of PNIPAM during the cooling process and result in a pronounced hysteresis.69 In contrast, a reversible dehydration is observed for POEOMAs due to the lack of strong H-bond donors in the molecular structure of these polymers and as a result, there is no possibility of stabilizing H-bond formation in the collapsed state.1
For comparing the solubility behavior of random and gradient copolymers MEO2MA and OEOMA both copolymers are chosen to have similar molecular weight and dispersity index, as the LCST shows a slight decrease upon increasing the molecular weight.6 As shown in Fig. 5(b), the LCST for the random copolymer (50.7 °C) is almost 15 °C higher than that for the gradient copolymer (36.6 °C). Around 8 °C hysteresis is observable for the gradient copolymer like in the case of the PMEO2MA homopolymer. But it is less (5 °C) for the random copolymer. The behaviors of gradient and random copolymers also differ distinguishably above LCST. Despite the random copolymer which shows a large broad transition at normalized scattering intensities (4.7 × 10−5 to 1.6 × 10−3 a.u.) and hydrodynamic radii (20–1280 nm), the change in gradient copolymers (I: 2.7 × 10−5 to 5.2 × 10−4 a.u., Rh: 24–292 nm) is rather sharp but with less increase in the amount. This shows the formation of micelles rather than globular aggregates in terms of gradient copolymers which are held in solution by the hydrophilic OEOMA segments that are not yet aggregated.6
As mentioned before, there are no strong intermolecular hydrogen bonding interactions between polymer chains. Therefore, the phase transition occurs mainly because of the multiple chain aggregation without a precontraction process of individual polymer chains. Moreover, the self-aggregation process of P(MEO2MA-co-OEGMA) is mainly dominated or driven by the conformation changes of oxyethylene side chains, which collapse first to get close to the hydrophobic backbones and then distort to expose hydrophilic ether oxygen groups to the “outer shell” of polymer chains as much as possible.2,3,11,12 According to Sun and Wu,3 P(MEO2MA-co-OEGMA) random copolymers exhibit “hydrated chains, dehydrated chains, loosely aggregated agglomerates and finally densely aggregated agglomerates” conformations during the phase transition. As there is no precontraction process before phase transition, the conformation of hydrophobic backbones with the slowest response does not change much and the micelle size remains constant. It should be noted that the cores in the micelles are only physically or loosely cross-linked by hydrogen bond bridges between ether oxygen groups and water molecules. By increasing the temperature above the LCST, due to the increased molecular motion and decreased density, the amount of water molecules which participate in hydrogen bonding decrease or in other words, more water molecules are expelled from micelles. Therefore, the micelles get more densely aggregated resulting in a gradual change as seen in DLS.3
The case is significantly different for the gradient copolymer based on its phase behavior in water (Fig. 5(c)). The change of Rh and I are rather sharp than broad and, in terms of the amount, less pronounced than the random copolymer. Above LCST, upon further temperature increase, first Rh decreases and then stays constant. This could be attributed to the formation of micelles rather than big agglomerates. Peng et al. reported a similar behavior in the case of poly(MEO2MA-co-PEGMA2080) due to the association of the polymer chains as a result of the dehydration while increasing the temperature, followed by a rearrangement process and micelle formation.14 In the system of P(MEO2MA-grad-OEOMA), there are no long PEGMA2080 side chains to stabilize the micelles in the system. Whereas the dense OEOMA at one end of the polymer chain can help the micelle stabilization to some extent. The decrease in Rh is explained by further dehydration and water expelling from the system which results in shrinking of the micelles and decreasing the particle size while the scattering intensity remains constant. This is also to a certain degree similar to the behavior of block copolymers,16,18 but without showing multiple transition temperatures, while micelles cannot stay in the system long enough to reach a second LCST.
The solubility behaviors of several gradient and random copolymers with various comonomer compositions were studied via DLS and the change of hydrodynamic radii and normalized scattering intensities are plotted in Fig. 6. For simplicity, only the heating cycles are shown. The solubility behavior depends strongly on the copolymer structure and therefore is similar for all gradient copolymers or all random copolymers, in a similar manner to that shown in Fig. 5. For both gradient and random structures, the LCST increases at a higher portion of OEOMA in the comonomer composition. This change is plotted in Fig. 7. By increasing the mol% of OEOMA, the LCSTs of gradient and random copolymers were found to be closer to each other. The higher slope of LCST versus temperature for random copolymers shows the higher dependency of LCST to OEOMA's ratio for random copolymers rather than gradient copolymers.
 | ||
| Fig. 6 The change of LCST versus the copolymer composition for gradient (a) and random (b) copolymers with different molar ratio of OEOMA:MEO2MA. The molar percentages on the graph show the amount of OEOMA in the copolymer. | ||
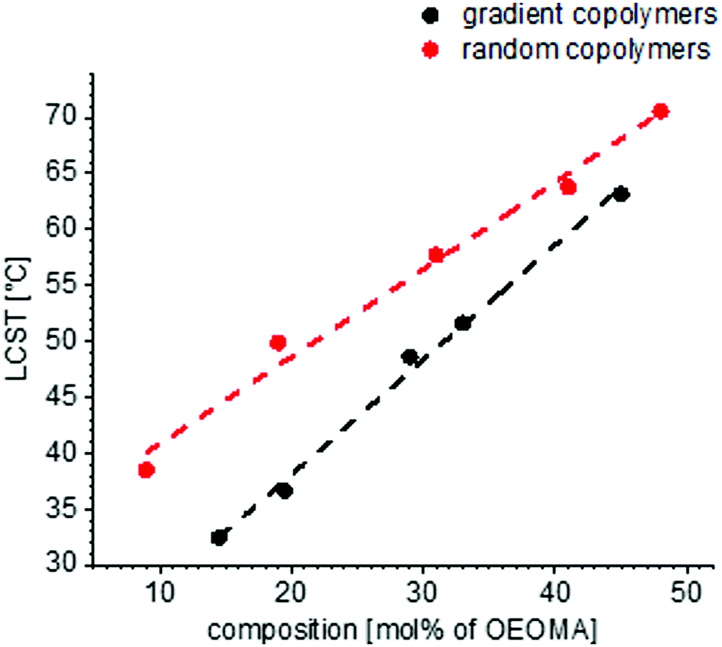 | ||
| Fig. 7 The change of LCST versus the copolymer composition for gradient and random copolymer. As the amount of OEOMA in the copolymer increases, the LCSTs of gradient and random copolymer get closer to each other. | ||
Effect of additives
| CO32− > SO42− > H2PO4− > F− > Cl− > Br− ≈ NO3− > I− > ClO4− > SCN− |
Fig. 8 shows the change of LCST for gradient and random copolymers of MEO2MA and OEOMA with 20% OEOMA in their chains in the presence of different salts. The results of DLS measurements (normalized scattering intensity and hydrodynamic radius versus temperature) in the presence of various salts, are plotted in Fig. S8–S24 (ESI†). To make the comparison easier, the concentration of all the salts was set to 0.5 M. Although, generally less amount of salt is present in most of the biological systems such as blood plasma, this higher concentration was used as for some of the salts at concentrations lower than 0.5 M, no change was observed in the LCST of the copolymers in water. Surprisingly, there is no distinguishable trend in the behavior of different cations on the LCST for both random and gradient copolymers. All chlorides studied in this work decrease the LCST. The decrease of LCST is almost similar for the various chlorides to the solution of gradient copolymers and is fluctuating for random copolymers.
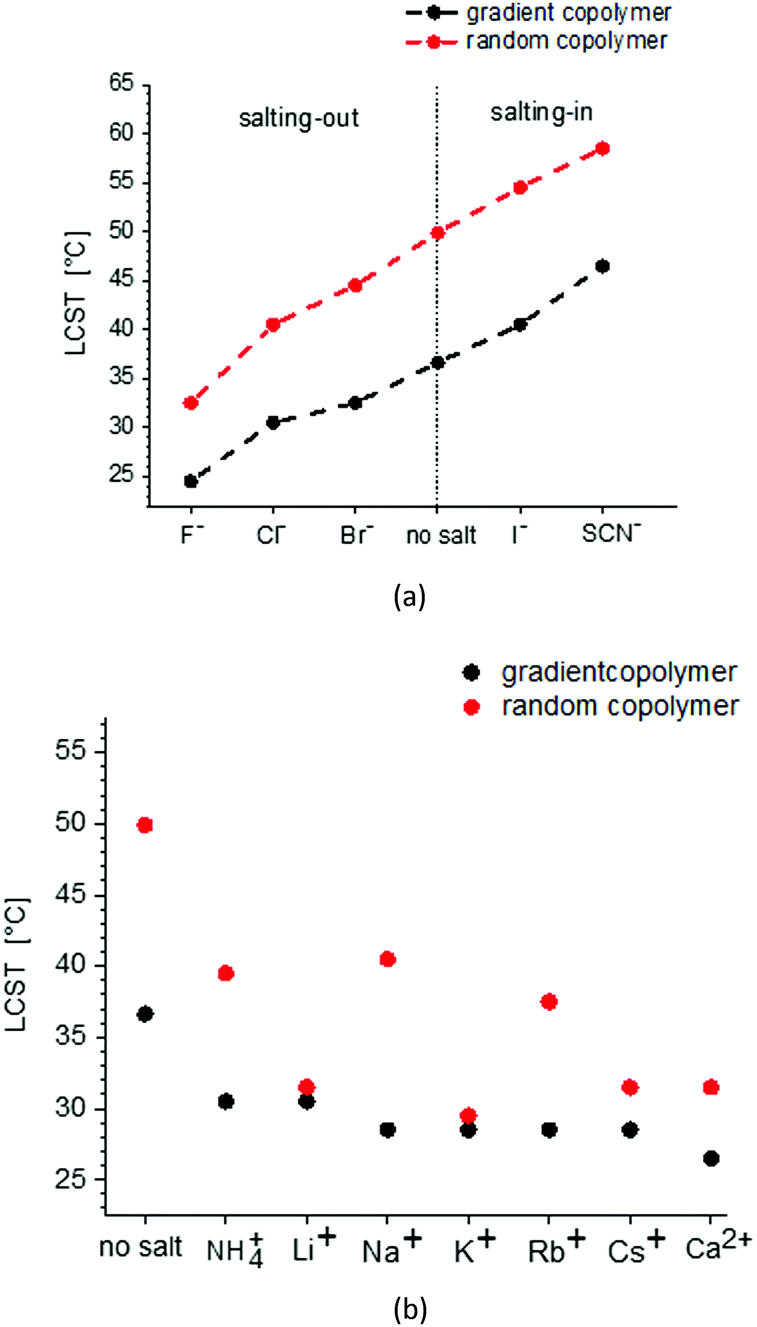 | ||
| Fig. 8 The effect of anions (a) and cations (b) on the LSCT of random and gradient copolymers. The salt concentration in all measurements is 0.5 M. The anions are studied as sodium salts and the anions as chloride salts. | ||
The effect of anions on the solubility behavior of gradient and random copolymers is in agreement with the Hofmeister series and similar to their effect on the solubility of PNIPAM.33 F−, Cl− and Br− decrease the LCST and therefore, show a salting-out effect while I− and SCN− increase the LCST and show a salting-in effect. The biggest change in solubility is observed for SO42− which makes the copolymer precipitate immediately after it is added to the solution and therefore, it is not plotted in Fig. 8. The next maximum change is observed for F− with the most salting-out effect that decreases the LCST around 17.5 °C for the random copolymer and 12.2 °C for the gradient copolymer. The biggest salting-in effect is observed for SCN− which causes an 8.5 °C increase in LCST for the random copolymer and 10 °C increase for the gradient copolymer.
The salting-out effect is assumptively related to the high surface charge density of the kosmotropic anions; this increases the surface tension in the inner hydration shell of the polymer, leading also to a relatively rigid and well-ordered anion hydration shell. As a result, the kosmotropic salts show a highly negative hydration entropy.76 Therefore, in the presence of kosmotropic anions, less water molecules are available to hydrate the polymer and the LSCT decreases. Furthermore, in the presence of salt, the solvent polarity increases which enforces the hydrophobic–hydrophobic interactions. On the other hand, the salting-in effect is related to the high polarizability of the chaotropic salts which results in less negative hydration entropy. This can partially distribute the rigid cage-like water structure and as a result, more water molecules can hydrate the polymer. Moreover, chaotropic anions can bind directly to the polymer and increase its surface charge and thus its solubility.37
According to Zhang et al.,33 the change of LCST after adding salt to a polymeric solution could be modeled based on three facts:
1. If the concentration of an inorganic salt is not too high, the surface tension of water at the hydrophobic/aqueous interface changes linearly with salt concentration.
2. At the first hydration shell of a macromolecular solute the polarization of water molecules is also dependent linearly on the salt concentration.
Each of these two effects can be the cause of the polymer precipitation depending on whether the anion is a kosmpotrope or a chaotrope.
3. For the most weakly hydrated anions, enthalpically favorable anion–polymer interactions can result in a salting-in effect.
As a result, the change of LCST by addition of salt could be described by:
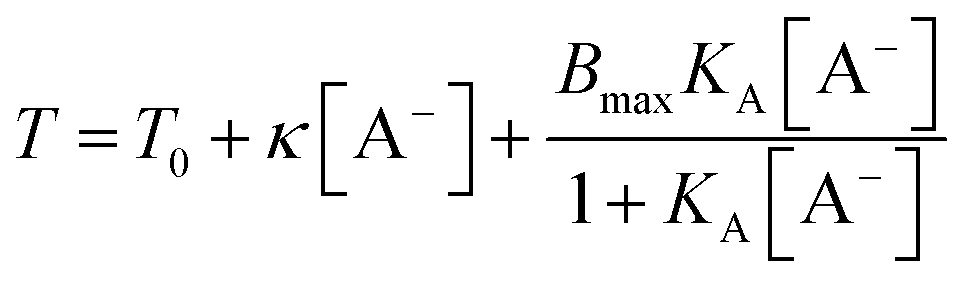
The binding isotherm 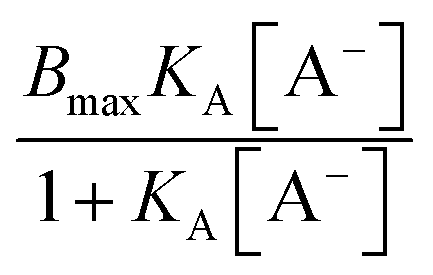 is attributed to the direct ion binding to the polymer (third fact) which is considered to be a saturation phenomenon and is relevant just for the chaotropic salts. For kosmotropic salts, the LCST is related linearly to the salt concentration as:
is attributed to the direct ion binding to the polymer (third fact) which is considered to be a saturation phenomenon and is relevant just for the chaotropic salts. For kosmotropic salts, the LCST is related linearly to the salt concentration as:
| T = T0 + κ[A−] |
Fig. 9 shows the effect of salt concentration of different salts on the LCST of gradient and random copolymers. From these graphs it is visible that the effect of salt concentration on the LCST of gradient copolymers is sharper than for random copolymers. This is especially distinguishable in NaSCN's salting-in effect. The calculated amount of κ as well as Bmax and KA are presented in Table 2. As also obvious from Fig. 9, the increase of κ for both gradient and random copolymers is in agreement with the order in the Hofmeister series. Moreover, except for SO42− which shows a considerably lower value of κ for the random copolymer as compared to the gradient copolymer, for the rest of the anions, κ and as a result the effect of anion on the LCST is similar for gradient and random copolymers.
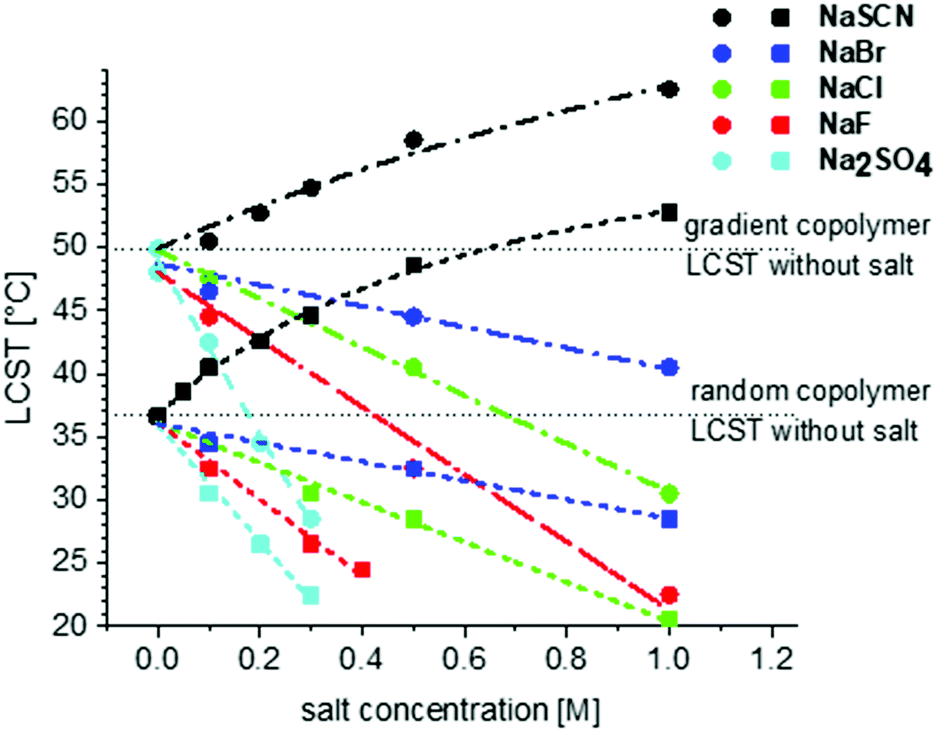 | ||
| Fig. 9 The effect of salt concentration of different sodium salts on the LCST of random (●) and gradient (■) copolymers containing 20 mol% OEOMA measured via DLS in water. All the salts in this study show a salting-out effect except for NaSCN which shows salting-in effect. | ||
| Anion | κ [M−1] | B max [°C] | K A [M−1] | |||
|---|---|---|---|---|---|---|
| Random copolymer | Gradient copolymer | Random copolymer | Gradient copolymer | Random copolymer | Gradient copolymer | |
| SO42− | −69 | −46 | — | — | — | — |
| F− | −27 | −30 | — | — | — | — |
| Cl− | −19 | −16 | — | — | — | — |
| Br− | −8 | −7 | — | — | — | — |
| SCN− | −2.5 | −12 | 34.5 | 69.2 | 0.65 | 0.68 |
Effect of surfactant
Fig. 10 shows the change of LCST for gradient and random copolymer after addition of SDS (anionic surfactant) and CTAB (cationic surfactant). The surfactant concentration is chosen to be lower than or close to the critical micelle concentration (CMC) (∼8.1 mM for SDS77 and ∼1 mM for CTAB78,79). In general, addition of a surfactant increases the LCST, due to its effect on stabilizing the formed polymeric micelles or globules in the solution. The surfactant molecules interact with the polymer and anchor on the surface of the formed micelles or globules in solution and increase the repulsion between adjacent polymer-bound micelles.32 This can also explain the lesser increase of particle size above the LCST when increasing the surfactant concentration (Fig. S25–S28, ESI†). As shown in Fig. 10, LCST increases linearly with increasing surfactant concentration until it reaches the boiling point. This result is completely different from PNIPAM, which shows the abnormal behavior of not precipitating in the presence of SDS until the concentration of SDS reaches the critical aggregation concentration (CAC).80 The increase in LCST is larger for CTAB than for SDS, which is different from the general trend observed for PNIPAM.81–83 For PNIPAM in general, the LCST increases in the order of nonionic < cationic < anionic which is the general surfactant adsorption on the polymer.32 The reason could be the longer alkyl chain for CTAB which according to the literature can also influence the increase of LCST after addition of surfactant.82,84 Although it was mentioned that the effect of ionic structure is more relevant than the length of alkyl chain, this is not in agreement with our observation for POEOMAs.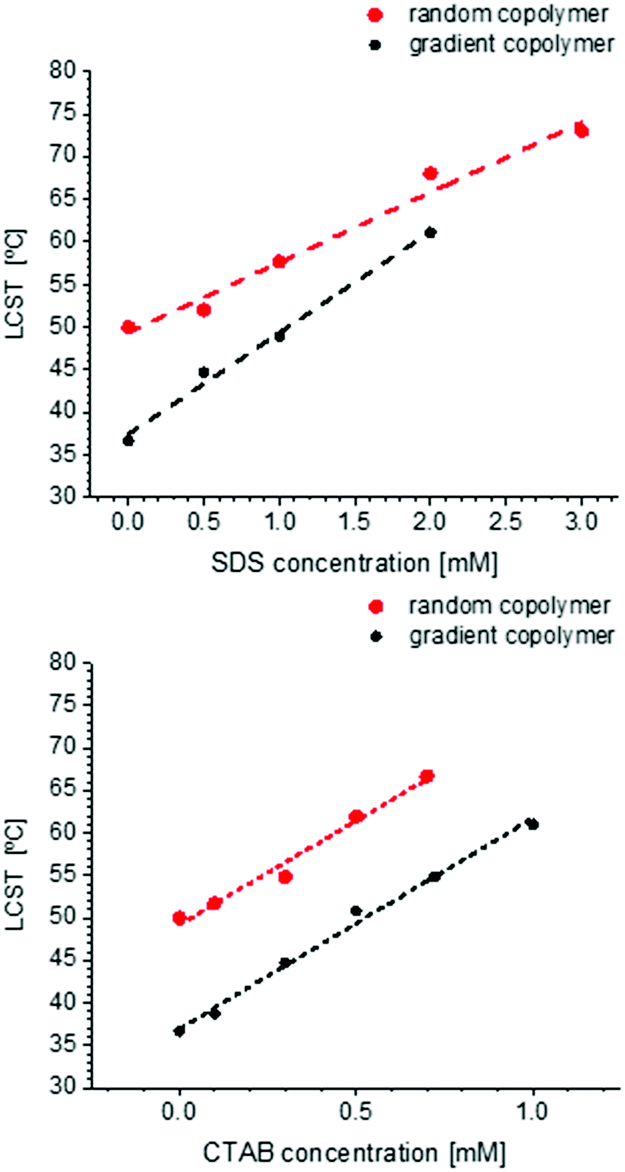 | ||
| Fig. 10 The change of LCST for gradient and random copolymers containing 20 mol% OEOMA versus surfactant concentration. The upper graph shows the effect of SDS and the lower graph of CTAB. The LCST is increasing drastically as the surfactant concentration increases. | ||
Effect of ethanol
The change of LCST of the gradient and random copolymers of MEO2MA and OEOMA with 20 mol% OEOMA in the presence of ethanol and a cosolvent is plotted in Fig. 11. The results of DLS measurements (normalized scattering intensity and hydrodynamic radius versus temperature) for gradient and random copolymers in the presence of various amounts of ethanol in aqueous solution, are plotted in Fig. S29 and S30 (ESI†). The LCST increases with increasing amount of ethanol until the polymer is completely soluble and does not show any LCST below the solvents’ boiling point. The change of LCST is exponential for the random copolymer and linear for the gradient copolymer. This behavior is in agreement with copolymers of similar structure in the literature.85 Addition of ethanol as a good solvent for POEOMAs increases their solubility. Moreover, the competitive interaction with polymer between water and alcohol enhances the solubility, thus increasing the LCST.No cononsolvency effect (i.e. lower compatibility of the polymer with the solvent at a certain range of solvent composition) is observed for these copolymers in the water–ethanol system in contrast to other temperature responsive polymers like PNIPAM32,36,86,87 or other thermoresponsive polymers with nitrogen atoms as the source of hydrogen bonding.38
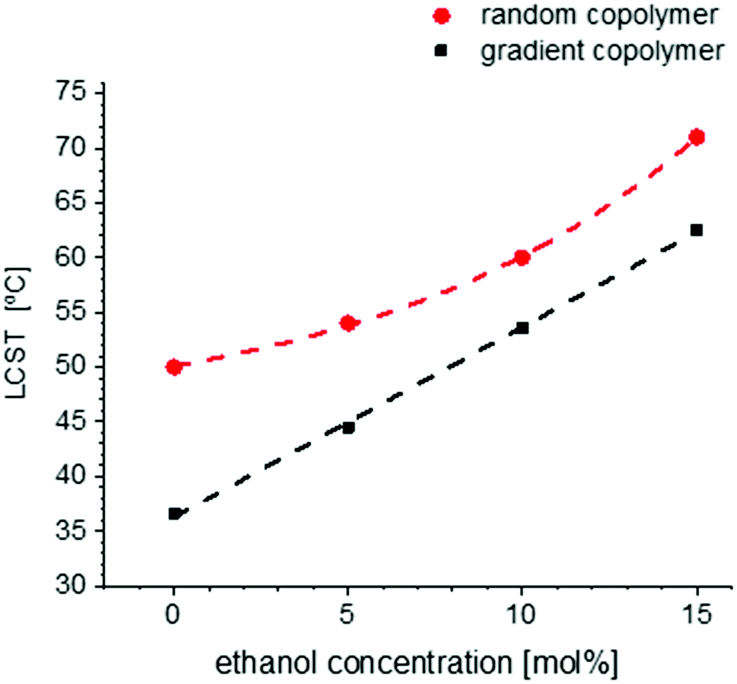 | ||
| Fig. 11 The change of LCST for gradient and random copolymers containing 20 mol% OEOMA by adding absolute ethanol to the copolymer's aqueous solution. LCST increases as the amount of ethanol in the solution increases till the copolymer gets fully soluble at all temperatures and shows no LCST anymore. | ||
This can be a result of no preference to form water–ethanol interactions rather than water–polymer or polymer–polymer interactions in this system. It is assumed that due to high interaction of both water and ethanol with the polymer rather than water-ethanol interaction, no hydrophobic hydration of ethanol molecules occurs in this system. According to literature, hydrophobic hydration happens at low fraction of ethanol, when water molecules form a hydration shell around ethanol molecules as a result of strong hydrogen bonds between them and therefore, there is not enough water molecules to hydrate the polymer. By increasing the ethanol fraction, there are no more sufficient water molecules to hydrate all ethanol molecules. As a result, the mobility of ethanol molecules increases and destroys the water network built by hydrogen bonds. At very high concentrations of ethanol, the water molecules form clusters which are surrounded by ethanol molecules.88,89 Cononsolvency is also a reason for lower polymer solubility of this type of copolymer at high alcohol concentration and the appearance of a UCST as reported by Roth et al.40
Conclusion
Poly[oligo(ethylene glycol)] (POEOMA) based gradient and random copolymers with various compositions were synthesized via semi-batch Cu-based ATRP. The continuous injection of OEOMA to the system is a very straightforward method of force gradient copolymerization to provide optimized sequence control. The solubility behaviors of gradient and random copolymers of MEO2MA and OEOMA with various amounts of OEOMA were investigated by DLS. Both copolymers show reversible thermoresponsivity with a small hysteresis that is slightly higher for gradient copolymers compared to random copolymers. Both copolymers undergo the phase transition in one step, but with different mechanisms. While the random copolymer shows a simple coil to globule transition, the gradient copolymer undergoes micelle formation followed by micelle shrinkage as the temperature increases. By increasing the amount of OEOMA in the copolymer composition, the LCST of both gradient and random copolymers increases linearly, and their values tend to converge.The effect of different additives including various salts, ethanol and surfactants on the solubility behavior of a gradient and a random copolymer was investigated by DLS. The copolymers show different phase transition behavior in the presence of various additives. While the random copolymer shows a broad transition with a vast change in hydrodynamic radius and normalized scattering intensity, the gradient copolymer displays a rather sharp transition but with less changes in hydrodynamic radius and normalized scattering intensity. The effect of anions on the solubility of both copolymers follows the Hofmeister series. Among the anions studied in this work, SO42−, F−, Cl− and Br− show kosmotropic effects while I− and SCN− show chaotropic effects on the solubility of copolymers in pure water. However, there is no distinguishable trend observed for the decrease of LCST in the presence of various cations. The phase transition behavior of both gradient and random copolymers changes from one-step to two-step phase transition in the presence of salts.
Addition of a good solvent as well as an anionic or cationic surfactant increases the LCST of both gradient and random copolymers. While the gradient copolymer shows a linear increase in LCST vs. the amount of ethanol added to the solution, the LCST of the random copolymer changes exponentially. The increase in LCST in the presence of ethanol is attributed to the improvement of polymer's solubility with ethanol as a good solvent for POEMAs which itself is a result of the competitive interaction with polymer between water and ethanol. There is no cononsolvency observed despite the cononsolvency behavior found for other temperature responsive polymers in the presence of ethanol.38 On the other hand, the addition of surfactants to the aqueous solution of gradient and random copolymers makes the solution more stable by stabilizing the formed micelles and prevention of the aggregation. The latter results in a stable solution regardless of temperature over a certain concentration (depending on the copolymer's structural architecture) of surfactant in the system.
Conflicts of interest
There are no conflicts to declare.References
- J. F. Lutz, Polymerization of oligo(ethylene glycol) methacrylates: Toward new generations of smart biocompatible materials, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(11), 3459–3470 CrossRef CAS.
- J. F. Lutz, Thermo-switchable materials prepared using the OEGMA-platform, Adv. Mater., 2011, 23(19), 2237–2243 CrossRef CAS.
- S. Sun and P. Wu, On the thermally reversible dynamic hydration behavior of oligo(ethylene glycol) methacrylate-based polymers in water, Macromolecules, 2013, 46(1), 236–246 CrossRef CAS.
- J. F. Lutz, Ö. Akdemir and A. Hoth, Point by point comparison of two thermosensitive polymers exhibiting a similar LCST: Is the age of poly(NIPAM) over?, J. Am. Chem. Soc., 2006, 128(40), 13046–13047 CrossRef CAS PubMed.
- S. Han, M. Hagiwara and T. Ishizone, Synthesis of Thermally Sensitive Water-Soluble Polymethacrylates by Living Anionic Polymerizations of Oligo(ethylene glycol) Methyl Ether Methacrylates, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- S.-I. Yamamoto, J. Pietrasik and K. Matyjaszewski, The Effect of Structure on the Thermoresponsive Nature of Well-Defined Poly(oligo(ethylene oxide) methacrylates) synthesized by ATRP, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 194–202 CrossRef CAS.
- X. Wang, X. Qiu and C. Wu, Comparison of the Coil-to-Globule and the Globule-to-Coil Transitions of a Single Poly(N-isopropylacrylamide) Homopolymer Chain in Water, Macromolecules, 1998, 31(9), 2972–2976 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada and F. Tanaka, et al., Impact of End-Group Association and Main-Chain Hydration on the Thermosensitive Properties of Hydrophobically Modified Telechelic Poly(N-isopropylacrylamides) in Water, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- B.-P. Havazelet and S. Gryc, Binding of Amino Acids to “Smart” Sorbents: Where Does Hydrophobicity Come into Play?, Langmuir, 2004, 20(1), 169–174 CrossRef PubMed.
- J.-Y. Wu, S.-Q. Liu, P. W.-S. Heng and Y.-Y. Yang, Evaluating proteins release from, and their interactions with, thermosensitive poly (N-isopropylacrylamide) hydrogels, J. Controllled Release, 2005, 10(2), 361–372 CrossRef PubMed.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, About the phase transitions in aqueous solutions of thermoresponsive copolymers and hydrogels based on 2-(2-methoxyethoxy) ethyl methacrylate and oligo (ethylene glycol) methacrylate, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- Y. Maeda, T. Kubota, H. Yamauchi, T. Nakaji and H. Kitano, Hydration changes of poly(2-(2-methoxyethoxy)ethyl methacrylate) during thermosensitive phase separation in water, Langmuir, 2007, 23(22), 11259–11265 CrossRef CAS PubMed.
- Y. Maeda, H. Yamauchi and T. Kubota, Confocal micro-Raman and infrared spectroscopic study on the phase separation of aqueous poly(2-(2-methoxyethoxy)ethyl (meth)acrylate) solutions, Langmuir, 2009, 25(1), 479–482 CrossRef CAS PubMed.
- B. Peng, N. Grishkewich, Z. Yao, X. Han, H. Liu and K. C. Tam, Self-assembly behavior of thermoresponsive oligo(ethylene glycol) methacrylates random copolymer, ACS Macro Lett., 2012, 1(5), 632–635 CrossRef CAS.
- B. Zhang, H. Tang and P. Wu, In depth analysis on the unusual multistep aggregation process of oligo(ethylene glycol) methacrylate-based polymers in water, Macromolecules, 2014, 47(14), 4728–4737 CrossRef CAS.
- Z. L. Yao and K. C. Tam, Temperature induced micellization and aggregation of biocompatible poly (oligo(ethylene glycol)methyl ether methacrylate) block copolymer analogs in aqueous solutions, Polymer, 2012, 53(16), 3446–3453 CrossRef CAS.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O’Reilly and M. I. Gibson, Polymers with molecular weight dependent LCSTs are essential for cooperative behaviour, Polym. Chem., 2012, 3, 794–799 RSC.
- Y. Kudo, H. Mori and Y. Kotsuchibashi, Preparation of an ethylene glycol-based block copolymer consisting of six different temperature-responsive blocks, Polym. J., 2018, 50, 1013–1020 CrossRef CAS.
- Y. Ogura, T. Terashima and M. Sawamoto, Amphiphilic PEG-Functionalized Gradient Copolymers via Tandem Catalysis of Living Radical Polymerization and Transesterification, Macromolecules, 2017, 50(3), 822–831 CrossRef CAS.
- S. Medel, J. M. Garcia, L. Garrido, I. Quijada-Garrido and R. Paris, Thermo- and pH-responsive gradient and block copolymers based on 2-(2-methoxyethoxy)ethyl methacrylate synthesized via atom transfer radical polymerization and the formation of thermoresponsive surfaces, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 690–700 CrossRef CAS.
- K.-I. Seno, I. Tsujimoto, S. Kanaoka and S. Aoshima, Synthesis of Various Stimuli-Responsive Gradient Copolymers by Living Cationic Polymerization and Their Thermally or Solvent Induced Association Behavior, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6444–6454 CrossRef CAS.
- K.-I. Seno, I. Tsujimoto, T. Kikuchi, S. Kanaoka and S. Aoshima, Thermosensitive Gradient Copolymers by Living Cationic Polymerization: Semibatch Precision Synthesis and Stepwise Dehydration-Induced Micellization and Physical Gelation, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6151–6164 CrossRef CAS.
- K.-I. Seno, K. Shokoyoku and A. Sadahito, Thermosensitive Diblock Copolymers with Designed Molecular Weight Distribution: Synthesis by Continuous Living Cationic Polymerization and Micellization Behavior, J. Polym. Sci., Part A: Polym. Chem., 2007, 46, 2212–2221 CrossRef.
- R. Konefal, J. Spevacek and P. Cernoch, Thermoresponsive poly(2-oxazoline) homopolymers and copolymers in aqueous solutions studied by NMR spectroscopy and dynamic light scattering, Eur. Polym. J., 2018, 100, 241–252 CrossRef CAS.
- S. Matsumoto, A. Kanazawa, S. Kanaoka and S. Aoshima, Dual stimuli-responsive copolymers with precisely arranged degradable units: synthesis by controlled alternating copolymerization of oxyethylene-containing vinyl ethers and conjugated aldehydes, Polym. Chem., 2019, 10, 4134–4141 RSC.
- S. Eggers, T. Eckert and V. Abetz, Double thermoresponsive block-random copolymers with adjustable phase transition temperatures: From block-like to gradient-like behavior, J. Polym. Sci., Part A: Polym. Chem., 2017, 399–411 Search PubMed.
- N. Oleszko-Torbus, A. Utrata-Wesolek, W. Walach and A. Dworak, Solution behavior of thermoresponsive random and gradient copolymers of 2-n-propyl-2-oxazoline, Eur. Polym. J., 2017, 88, 613–622 CrossRef CAS.
- S. Jaksch, A. Schulz, K. Kyriakos, J. Zhang, I. Grillo and V. Pipich, et al., The collapse and aggregation of thermoresponsive poly(2-oxazoline) gradient copolymers: a time-resolved SANS study, Colloid Polym. Sci., 2014, 292(10), 2413–3425 CrossRef CAS.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Block and gradient copolymers of 2-hydroxyethyl acrylate and 2-methoxyethyl acrylate via RAFT: Polymerization kinetics, thermoresponsive properties, and micellization, Macromolecules, 2013, 46(4), 1447–1460 CrossRef CAS.
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Studies on thermoresponsive polymers: Phase behaviour, drug delivery and biomedical applications, Asian J. Pharm. Sci., 2015, 10(2), 99–107 CrossRef.
- M. C. M. Costa, S. M. C. Silva and F. E. Antunes, Adjusting the low critical solution temperature of poly(N-isopropyl acrylamide) solutions by salts, ionic surfactants and solvents: A rheological study, J. Mol. Liq., 2015, 210, 113–118 CrossRef CAS.
- D. Dhara and P. Chatterji, Phase Transition in Linear and Cross-Linked Poly(N-Isopropylacrylamide) in Water: Effect of Various Types of Additives, Polym. Rev., 2000, 40(1), 51–68 Search PubMed.
- Y. J. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, Specific Ion effectis on the water solubility of macromolecules PNIPAM and the Hofmeister series, J. Am. Chem. Soc., 2005, 127(23), 14505–14510 CrossRef CAS PubMed.
- E. A. Algaer and N. F. A. Van Der Vegt, Hofmeister ion interactions with model amide compounds, J. Phys. Chem. B, 2011, 115(46), 13781–13787 CrossRef CAS PubMed.
- L. Pérez-Fuentes, D. Bastos-González, J. Faraudo and C. Drummond, Effect of organic and inorganic ions on the lower critical solution transition and aggregation of PNIPAM, Soft Matter, 2018, 14(38), 7818–7828 RSC.
- V. I. Michailova, D. B. Momekova, H. A. Velichkova, E. H. Ivanov, R. K. Kotsilkova and D. B. Karashanova, et al., Self-Assembly of a Thermally Responsive Double-Hydrophilic Copolymer in Ethanol-Water Mixtures: The Effect of Preferential Adsorption and Co-Nonsolvency, J. Phys. Chem. B, 2018, 122(22), 6072–6078 CrossRef CAS PubMed.
- S. Eggers, B. Fischer and V. Abetz, Aqueous Solutions of Poly[2-(N-morpholino)ethyl methacrylate]: Learning about Macromolecular Aggregation Processes from a Peculiar Three-Step Thermoresponsive Behavior, Macromol. Chem. Phys., 2015, 217(6), 735–747 CrossRef.
- N. Lucht, S. Eggers and V. Abetz, Cononsolvency in the ‘drunken’ state: the thermoresponsiveness of a new acrylamide copolymer in water–alcohol mixtures, Polym. Chem., 2017, 8(7), 1196–1205 RSC.
- M. Dilip, N. J. Bridges, H. Rodriguez, J. F. B. Pereira and R. D. Rogers, Effect of Temperature on Salt-Salt Aqueous Biphasic Systems: Manifestations of Upper Critical Solution Temperature, J. Solution Chem., 2014, 44, 454–468 CrossRef.
- P. J. Roth, F. D. Jochum and P. Theato, UCST-type behavior of poly[oligo(ethylene glycol) methyl ether methacrylate] (POEGMA) in aliphatic alcohols: solvent, co-solvent, molecular weight, and end group dependences, Soft Matter, 2011, 7(6), 2484 RSC.
- N. Guang, Li X Shou-xin Liu, L. Tian and M. Hongguang, Micellization and Gelation of the Double Thermoresponsive ABC-type Triblock Copolymer by One-pot RAFT Synthesis, Chin. J. Polym. Sci., 2016, 34(8), 956–980 CrossRef.
- W. Yuan and J. Wang, Oligo(ethylene glycol) and quaternary ammonium-based block copolymer micelles: from tunable thermoresponse to dual salt response, RSC Adv., 2014, 4, 38855 RSC.
- Q. Fang, T. Chen, Q. Zhong and J. Wang, Thermoresponsive polymers based on oligo(ethylene glycol) methyl ether methacrylate and modified substrates with thermosensitivity, Macromol. Res., 2017, 25(3), 206–213 CrossRef CAS.
- T. J. Murdoch, B. A. Humphreys, J. D. Willott, S. W. Prescott, A. Nelson and G. B. Webber, et al., Enhanced specific ion effects in ethylene glycol-based thermoresponsive polymer brushes, J. Colloid Interface Sci., 2017, 490, 869–878 CrossRef CAS PubMed.
- N. Badi and J. F. Lutz, Sequence control in polymer synthesis, Chem. Soc. Rev., 2009, 38(12), 3383–3390 RSC.
- A. Troy and J. Scott, Refractive Index of Ethanol-Water Mixtures and Density and Refractive Index of Ethanol-Water-Ethyl ether Mixtures, J. Phys. Chem., 1946, 50, 406–412 CrossRef PubMed.
- J. V. Herraez and R. Belda, Viscous Synergy of Pure Monoalcohol Mixtures in Water and its Relation to Concentration, J. Solution Chem., 2004, 33(2), 117–129 CrossRef CAS.
- Y. Tanaka, Y. Matsuda, H. Fujiwara, H. Kubota and T. Makita, Viscosity of (water + alcohol) mixtures under high pressure, Int. J. Thermophys., 1987, 8(2), 147–163 CrossRef CAS.
- Y. Tanaka, T. Yamamoto, Y. Satomi, H. Kubota and T. Makita, Specific Volume and Viscosity of Ethanol-Water Mixtures under High Pressure, Rev. Phys. Chem. Jpn., 1977, 47(1), 12–24 CAS.
- M. Radjabian, C. Abetz, B. Fischer, A. Meyer and V. Abetz, Influence of Solvent on the Structure of an Amphiphilic Block Copolymer in Solution and in Formation of an Integral Asymmetric Membrane, ACS Appl. Mater. Interfaces, 2017, 9(37), 31224–31234 CrossRef CAS PubMed.
- X. Wang and S. P. Armes, Facile Atom Transfer Radical Polymerization of Methoxy-Capped Oligo (ethylene glycol) Methacrylate in Aqueous Media at Ambient Temperature, Macrolecules, 2000, 33, 6640–6647 CrossRef CAS.
- J. Yamanaka, T. Kayasuga, M. Ito, H. Yokoyama and T. Ishizone, Synthesis of water-soluble poly[oligo(ethylene glycol) methacrylate]s by living anionic polymerization of oligo(ethylene glycol) vinyl ether methacrylates, Polym. Chem., 2011, 2(8), 1837 RSC.
- T. Ishizone, S. Han, M. Hagiwara and H. Yokoyama, Synthesis and surface characterization of well-defined amphiphilic block copolymers containing poly[oligo(ethylene glycol)] methacrylatel segments, Macromolecules, 2006, 39(3), 962–970 CrossRef CAS.
- T. Ishizone, S. Han, S. Okuyama and S. Nakahama, Synthesis of Water-soluble Polymethacrylates by Living Anionic Polymerizations of Trialkylsilyl-Protected Oligo (ethylene glycol) Methacrylates, Macromolecules, 2003, 36, 42–49 CrossRef CAS.
- J. F. Lutz and A. Hoth, Preparation of Ideal Analoues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- L. Xue, U. S. Agarwal and P. J. Lemstra, High molecular weight PMMA by ATRP, Macromolecules, 2002, 35(22), 8650–8652 CrossRef CAS.
- C. Reichardt and T. Welston, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, 4th edn, 2011 Search PubMed.
- G. Coullerez, A. Carlmark, E. Malmström and M. Jonsson, Understanding copper-based Atom Transfer Radical Polymerization in aqueous media, J. Phys. Chem., 2004, 108(35), 1–4 Search PubMed.
- H. Bergenudd, G. Coullerez, M. Jonsson and E. Malmström, Solvent effects on ATRP of oligo(ethylene glycol) methacrylate. exploring the limits of control, Macromolecules, 2009, 42(9), 3302–3308 CrossRef CAS.
- M. Horn and K. Matyjaszewski, Solvent effects on the activation rate constant in atom transfer radical polymerization, Macromolecules, 2013, 46(9), 3350–3357 CrossRef CAS.
- K. Matyjaszewski, B. Go1belt, H. Paik and C. P. Horwitz, Tridentate Nitrogen-Based Ligands in Cu-Based ATRP: A Structure - Activity Study, Macrolecules, 2001, 34, 430–440 CrossRef CAS.
- J. Qiu, K. Matyjaszewski, L. Thouin and C. Amatore, Cyclic voltammetric studies of copper complexes catalyzing atom transfer radical polymerization, Macromol. Chem. Phys., 2000, 201(14), 1625 CrossRef CAS.
- K. Matyjaszewski, Structure-reactivity correlation in Atom Transfer Radical Polymerization, Macromol. Symp., 2002, 182, 209–224 CrossRef CAS.
- B. Göbelt and K. Matyjaszewski, Diimino- and diaminopyridine complexes of CuBr and FeBr2 as catalysts in atom transfer radical polymerization (ATRP), Macromol. Chem. Phys., 2000, 201(14), 1619–1624 CrossRef.
- J. Huang, T. Pintauer and K. Matyjaszewski, Effect of variation of [PMDETA]0/[Cu(I)Br]0 ratio on atom transfer radical polymerization of n-butyl acrylate, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(13), 3285–3292 CrossRef CAS.
- H. Bergenudd, G. Coullerez, M. Jonsson and E. Malmstro, Solvent Effects on ATRP of Oligo (ethylene glycol) Methacrylate. Exploring the Limits of Control-SI, Macromolecules, 2009, 42, 3302–3308 CrossRef CAS.
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, Gradient copolymers by atom transfer radical copolymerization, J. Phys. Org. Chem., 2000, 13, 775–786 CrossRef CAS.
- X. Wang, X. Qiu and C. Wu, Comparison of the Coil-to-Globule and the Globule-to-Coil Transitions of a Single Poly(N-isopropylacrylamide) Homopolymer Chain in Water, Macromolecules, 1998, 31(9), 2972–2976 CrossRef CAS.
- X. Wang, X. Qiu and C. Wu, Comparison of the Coil-to-Globule and the Globule-to-Coil Transitions of a Single Poly(N-isopropylacrylamide) Homopolymer Chain in Water, Macromolecules, 1998, 31(9), 2972–2976 CrossRef CAS.
- F. Hofmeister, Zur Lehre von der Wirkung der Salze – Dritte Mittheilung, Arch. Exp. Pathol. Pharmakol., 1889, 25(1), 1–30 Search PubMed.
- W. Kunz, J. Henle and B. W. Ninham, “Zur Lehre von der Wirkung der Salze” (about the science of the effect of salts): Franz Hofmeister's historical papers., Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS.
- J. Heyda and J. Dzubiella, Thermodynamic Description of Hofmeister Effects on the LCST of Thermosensitive Polymers, J. Phys. Chem. B, 2014, 118, 10979–10988 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Chemistry of Hofmeister anions and osmolytes, Annu. Rev. Phys. Chem., 2010, 61, 63–83 CrossRef CAS PubMed.
- H. Peter and T. S. von Hippel, Ion effects on the Solution Structure of Biological macromolecules, Acc. Chem. Res., 1969, 2, 257–265 CrossRef.
- Y. Q. Gao, Simple Theory for Salt Effects on the Solubility of Amide, J. Phys. Chem. B, 2012, 116, 9934–9943 CrossRef CAS PubMed.
- A. Ciferri and A. Perico, Ionic interations in Natural and Synthetic Macromolecules, Wiley, 2012 Search PubMed.
- P. Mukerjee and K. J. Mysels, Critical Micelle Concentration of Aqueous Surfactant Systems, US Government Printing Office, Washington DC, vol. 36, 1971 Search PubMed.
- J. M. Neugebauer, Detergents: An overview, Methods Enyzmol., 1990, 182, 239–253 CAS.
- S. Roe, Protein Purification Application: A Practical Approach, Oxford University Press, 1990 Search PubMed.
- R. Walter, J. Ricka, Ch. Quellet and Ch. Nyffenegger T. B., Coil−Globule Transition of Poly(N-isopropylacrylamide), A Study of Polymer−Surfactant Association, Macromolecules, 1996, 29, 4019–4028 CrossRef CAS.
- C. Wu and S. Zhou, Effects of surfactants on the phase transition of poly(N-isopropylacrylamide) in water, J. Polym. Sci., Part B: Polym. Phys., 1996, 34(9), 1597–1604 CrossRef CAS.
- M. Sakai, N. Satoh, K. Tsujii, Y. Q. Zhang and T. Tanaka, Effects of Surfactants on the Phase Transition of a Hydrophobic Polymer Gel, Langmuir, 1995, 11(7), 2493–2495 CrossRef CAS.
- E. Kokufuta, Y.-Q. Zhang, T. Tanaka and A. Mamada, Effects of surfactants on the phase transition of poly(N-isopropylacrylamide) Gel, Macromolecules, 1996, 26, 1053–1059 CrossRef.
- H. G. Schild and D. A. Tirrell, Interaction of Poly(N-isopropylacrylamide) with Sodium n-Alkyl Sulfates in Aqueous Solution, Langmuir, 1991, 7(4), 665–671 CrossRef CAS.
- Q. Fang, T. Chen, Q. Zhong and J. Wang, Thermoresponsive polymers based on oligo(ethylene glycol) methyl ether methacrylate and modified substrates with thermosensitivity, Macromol. Res., 2017, 25(3), 206–213 CrossRef CAS.
- K. Kyriakos, M. Philipp, C. H. Lin, M. Dyakonova, N. Vishnevetskaya and I. Grillo, et al., Quantifying the Interactions in the Aggregation of Thermoresponsive Polymers: The Effect of Cononsolvency, Macromol. Rapid Commun., 2016, 37(5), 420–425 CrossRef CAS PubMed.
- R. O. Costa and R. F. Freitas, Phase behavior of poly(N-isopropylacrylamide) in binary aqueous solutions, Polymer, 2002, 43(22), 5879–5885 CrossRef CAS.
- S. Y. Noskov, G. Lamoureux and B. Roux, Molecular dynamics study of hydration in ethanol-water mixtures using a polarizable force field, J. Phys. Chem. B, 2005, 109(14), 6705–6713 CrossRef CAS PubMed.
- P. J. Roth, M. Collin and C. Boyer, Advancing the boundary of insolubility of non-linear PEG-analogues in alcohols: UCST transitions in ethanol-water mixtures, Soft Matter, 2013, 9(6), 1825–1834 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetics graphs, SEC traces, 1H NMR and DLS graphs. See DOI: 10.1039/c9sm02032b |
| This journal is © The Royal Society of Chemistry 2020 |