
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-assembling amphiphilic peptide nanoparticle for the efficient entrapment of DNA cargoes up to 100 nucleotides in length†
Shabnam
Tarvirdipour
ab,
Cora-Ann
Schoenenberger
a,
Yaakov
Benenson
*b and
Cornelia G.
Palivan
 *a
*a
aDepartment of Chemistry, University of Basel, Mattenstrasse 24a, 4058 Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch
bDepartment of Biosystems Science and Engineering, ETH Zurich, Mattenstrasse 26, 4058 Basel, Switzerland. E-mail: kobi.benenson@bsse.ethz.ch
First published on 13th January 2020
Abstract
To overcome the low efficiency and cytotoxicity associated with most non-viral DNA delivery systems we developed a purely peptidic self-assembling system that is able to entrap single- and double-stranded DNA of up to 100 nucleotides in length. (HR)3gT peptide design consists of a hydrophilic domain prone to undergo electrostatic interactions with DNA cargo, and a hydrophobic domain at a ratio that promotes the self-assembly into multi-compartment micellar nanoparticles (MCM-NPs). Self-assembled (HR)3gT MCM-NPs range between 100 to 180 nm which is conducive to a rapid and efficient uptake by cells. (HR)3gT MCM-NPs had no adverse effects on HeLa cell viability. In addition, they exhibit long-term structural stability at 4 °C but at 37 °C, the multi-micellar organization disassembles overtime which demonstrates their thermo-responsiveness. The comparison of (HR)3gT to a shorter, less charged H3gT peptide indicates that the additional arginine residues result in the incorporation of longer DNA segments, an improved DNA entrapment efficiency and an increase cellular uptake. Our unique non-viral system for DNA delivery sets the stage for developing amphiphilic peptide nanoparticles as candidates for future systemic gene delivery.
Introduction
Gene therapy depends on viral and non-viral delivery systems to ferry therapeutic nucleic acids into target cells.1,2 While there are a few viral-based gene therapy products approved by regulatory agencies,3,4 there are numerous disadvantages associated with a viral strategy such as high immunogenicity, mutagenicity, limitation in size of transgenic DNA, low availability, and high development and manufacturing cost.5 These drawbacks led to a boost of non-viral systems for DNA delivery including some that are currently at various stages of clinical trials.6–8 The advantages of non-viral DNA delivery systems include lower immunogenicity and toxicity, better cell specificity, extensive flexibility in design with the option of large-scale production at comparatively low cost.9,10 So far, several types of nanoparticles have been studied for DNA delivery, for example quantum dots, carbon nanotubes, gold nanoparticles, silica nanoparticles, and polymer-based and lipid-based nanoparticles.11 Peptides as non-viral gene delivery vectors are particularly attractive as they offer nearly limitless design options, higher biocompatibility and biodegradability, high binding affinities and increased cellular penetration.12,13 Consistently, the comprehensive study of DNA/peptide complexes and conjugated peptides with regard to cellular targeting and fusogenic function corroborate their exceptional properties compared to other non-viral gene delivery systems.14 Moreover, nanovesicles assembled from branched amphipathic peptides that served as cationic nucleation centers around which DNA winds were reported to deliver plasmid DNA in vivo and in vitro.15 However, studies involving the self-assembly of supramolecular nanoparticles from pure peptides for DNA delivery are rare16,17 and to best of our knowledge, parameters conductive to the entrapment and delivery of DNA molecules have so far not been systematically investigated.A large number of non-viral gene-therapy systems focus on the delivery of antisense oligonucleotides (ASOs).18–21 These 20 to 40 nucleotide single-stranded DNAs (ssDNAs) can be designed to mediate target RNA degradation, block the translation of a specific RNA, and modify RNA splicing.22 Nevertheless, pro-inflammatory effects, inadequate pharmacokinetic properties such as high degradation rates and short half-lives, off-target effects, and preclinical toxicological challenges are associated with these systems and represent hurdles that impede their clinical application.23 One option to sidestep drawbacks of ASOs is to entrap entire genes. On the other hand, many non-viral nanosystems that are able to deliver entire plasmid DNA encoding various genes have been reported.24–27 However, these delivery systems are largely based on conjugation or complex formation of the DNA with lipids,28,29 polymers,30,31 and peptides32–35 rather than on the directed self-assembly of supramolecular nanostructures. The ability to entrap DNA sequences larger than 50 nucleotides in length, either single- or double-stranded, by self-assembly of peptides alone has not been systematically explored so far. By taking advantage of the concept of self-assembly rather than depending on tedious chemical reactions for producing supramolecular assemblies, several aspects of a rational design are fulfilled. Self-assembly allows for the specific selection of starting components and a subsequent optimization by adapting the building blocks to produce tailored, well-defined structures that are easy to handle.36–40 Amphipathic peptides are emerging as suitable candidates for fabricating well-ordered nanostructures.41–47
To take advantage of the vector properties offered by peptides and at the same time start closing the gap in cargo size between ASOs and entire genes, we intended to establish a purely peptidic, self-assembling delivery system that lends itself to the incorporation of single- and double-stranded DNA segments that are larger than the average ASO. Here, we report a novel amphiphilic peptide henceforth called (HR)3gT that is designed to self-assemble into multi-compartment micellar nanoparticles (MCM-NPs). Qualitative and quantitative analysis of amphiphilic peptide self-assembly leading to multi-compartment micellar nanoparticles with detailed statistical data of their inner structure was reported by de Bruyn Ouboter.48 One of the parameters that principally governs the self-assembly of amphiphilic peptides is hydrophilic–hydrophobic balance.35,49 Therefore, our sequence modifications to the H3gT peptide were designed to keep the hydrophobic to hydrophilic weight ratio relatively constant. We significantly modified the design of a previously published amphiphilic H3gT peptide that forms MCM-NPs50 by the addition of charged amino acid residues to allow for more electrostatic interactions between peptide and DNA. The extension of the hydrophilic peptide domain was accompanied by an increase of the hydrophobic domain to sustain the peptide's ability to self-assemble into the desired nanoparticle architecture. We address how changes in length and type (single-stranded versus double-stranded) of DNA sequences affect the self-assembly process of peptides. First and foremost, a detailed understanding of the physicochemical properties of the peptidic nanoparticles that are conducive to the incorporation of longer DNA sequences is required.
The impact of the aforementioned modifications on (HR)3gT self-assembly was systematically analysed in the absence and presence of single- and double-stranded DNA (ssDNA/dsDNA) of 22 and 100 nucleotide length. In particular, we examined how compared to H3gT, the modified physicochemical properties of (HR)3gT affected the loading efficiency of a 22-nucleotide ssDNA. By dissecting the relationship between the type and length of the DNA sequence and the MCM-NP formation, we demonstrate that (HR)3gT nanoparticles can accommodate longer DNA segments as payloads. While emphasizing the importance of the physicochemical properties of peptide nanoparticles in generating safe and efficient DNA-delivery systems, our results pave the way to incorporate protein-coding sequences in peptide assemblies as an efficient approach to non-viral gene-based therapy.
Materials and methods
Solvents and reagents
Solvents and reagents were purchased from Sigma-Aldrich unless otherwise specified. Triisopropylsilane, piperidine and N,N′-diisopropylcarbodiimide (DIC) were of synthesis grade. Rink Amide AM resin (0.71 mmol g−1) and Fmoc-Trp(Boc)-OH were purchased from IRIS Biotech GmbH. All other Fmoc-protected amino acids and ethyl cyano(hydroxyimino)acetate (Oxyma Pure) were purchased from Novabiochem. Dimethylformamide (DMF) was purchased from J.T. Baker. Dichloromethane (DCM) and acetonitrile (ACN) were purchased from VWR chemical. Solvent exchange was performed in dialysis tubes from Spectrum Laboratories (cellulose ester, MWCO 500–1000 Da, 3.2 cm mL−1). Atto550 was purchased from ATTO-TEC GmbH. Atto550-labeled, 22 nucleotide (nt) and 100 nt DNA and their unlabelled complementary stands were purchased from Microsynth. Fetal calf serum (FCS) and phosphate buffer saline (PBS) was purchased from BioConcept. Live cell imaging solution was obtained from Thermo Fisher Scientific. Dulbecco's modified Eagle's medium (DMEM), Opti-MEM, and pen/strep were obtained from Gibco life technologies. CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) was purchased from Promega.Peptide synthesis and purification
(HR)3gT peptide consists of nineteen amino acids (AA), H2N-[H-R]3-[W-DL]6-W-NH2, (with DL = D-leucine) and H3gT of 10 AA, H2N-[H]3-[W-DL]3-W-NH2, as previously reported.50 The (HR)3gT and H3gT peptides were synthesized using Liberty Blue™ automated microwave peptide synthesizer (CEM, Kamp-Lintfort, Germany). The synthesis of both peptides was performed on a rink amide resin using standard fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesizer chemistry and DIC/OXYMA coupling protocols. After completing synthesis in the Liberty Blue™ synthesizer, the peptidyl resin was collected and washed alternatingly with DMF and dichloromethane. Peptides were manually cleaved from the resin by 5 mL trifluoroacetic acid (TFA) under gentle agitation over a period of 2 h at room temperature in the presence of scavengers (standard cleavage solution: TFA/triisopropylsilane (TIPS)/water 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5). The cleavage cocktail was filtered and the resin was washed with 1 mL cleavage solution. The peptides were precipitated from the combined cleavage solution by the addition of 40 mL cold diethyl ether and pelleted by centrifugation at 4500 rpm for 3 min at 4 °C. The pellet together with supernatant were incubated at −20 °C for 1 h and again centrifuged under the same conditions and supernatant was discarded. The pellet was dissolved in ACN/H2O (1:1) and TFA (0.05%, v/v) solution. The crude peptide was filtered and lyophilized.
:2.5:2.5). The cleavage cocktail was filtered and the resin was washed with 1 mL cleavage solution. The peptides were precipitated from the combined cleavage solution by the addition of 40 mL cold diethyl ether and pelleted by centrifugation at 4500 rpm for 3 min at 4 °C. The pellet together with supernatant were incubated at −20 °C for 1 h and again centrifuged under the same conditions and supernatant was discarded. The pellet was dissolved in ACN/H2O (1:1) and TFA (0.05%, v/v) solution. The crude peptide was filtered and lyophilized.
Purification of the crude (HR)3gT and H3gT peptide was carried out by reversed phase high performance liquid chromatography (RP-HPLC) (Prominence 20A, Shimadzu, Japan) on a C18-TSE (VDSpher OptiBio PUR 300 C18-TSE, 20 × 250 mm, VDS Optilab, Germany) and a C18 (RP18e, 100 mm × 10 mm, Merck Chromolith, Germany) column, respectively. A mobile phase of water and acetonitrile containing 0.1% TFA with a gradient of 25–60% ACN over 30 min for (HR)3gT and 20–65% ACN over 30 min for H3gT was used to separate peptides, and purification was monitored at 280 nm. Purified peptides were lyophilized and stored at −20 °C. The molecular mass of each peptide was determined by PerSeptive Biosystems Voyager-DE-PRO time-of-flight mass spectrometer (MALDI-TOF-MS) in positive mode. Peptides were characterized by analytical high performance liquid chromatography (HPLC) and (MALDI-TOF) mass spectroscopy before and after purification.
Self-assembly of peptide MCM-NPs
For self-assembly of empty and DNA-loaded peptide nanoparticles, a 1 mg mL−1 peptide stock solution was prepared in 50/50 ethanol/water from the respective lyophilized peptide and filtered through a 0.2 μm hydrophilic syringe filter. Self-assembly was achieved via the solvent exchange method by dialysing the organic solvent (ethanol) against Milli-Q H2O (Merck Millipore, Milli-Q Direct 8 water purification system). For loading nanoparticles with DNA, the following synthetic DNA cargoes were prepared at a concentration of 100 μM in water: 5′Atto550-labeled 22 nt and 100 nt single-stranded DNA (22 nt ssDNA/100 nt ssDNA), and 22 and 100 base pair double-stranded DNA (22 bp dsDNA/100 bp dsDNA) with one 5′Atto550-labeled strand (sequence data are provided in Table S1, ESI†). In order to form DNA-loaded peptide nanoparticles, 100 μL of peptide stock solution were mixed with 3 μg of DNA (100 μM solution). The DNA–peptide mixture was then adjusted to a final volume of 500 μL in a final concentration of 20%, 35%, or 50% ethanol, and then filled in a prewashed 500–1000 MWCO dialysis tube. The self-assembly was induced by dialysis at 4 °C for approximately 20 h against three changes of 1 L water. To prepare the control nanoparticles without DNA, peptide solutions were diluted to a concentration of 0.2 mg mL−1 in corresponding final ethanol concentrations (20%, 35% or 50%) and subsequently dialyzed under the same conditions.Characterization of DNA-free and DNA-loaded peptide MCM-NPs
Fluorescence signals were measured in a real time (5 s with 30 repetitions) and autocorrelation function was obtained by a QuickFit 3.0 software calculator. For determining the DNA entrapment in peptide MCM-NPs, experimental auto correlation curves were fitted to a two-component model including triplet state with a structural parameter of 5 (eqn (1)):51–53
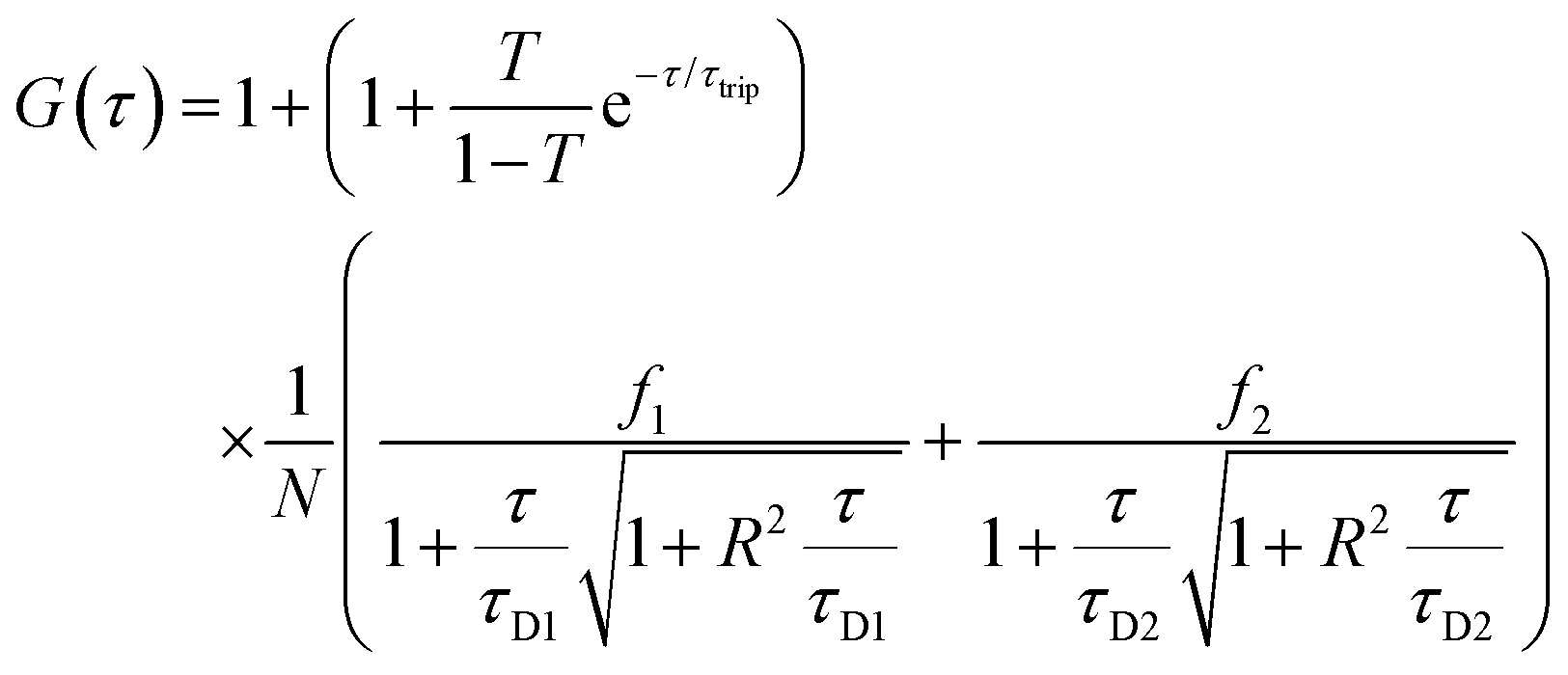 | (1) |
For the three-dimensional (3-D) Gaussian-shaped observation volume (Vobs), identical fluorescent species with the volume much smaller than Vobs moving in and out of it and, autocorrelation function (G(τ)) determine with eqn (2):51–53
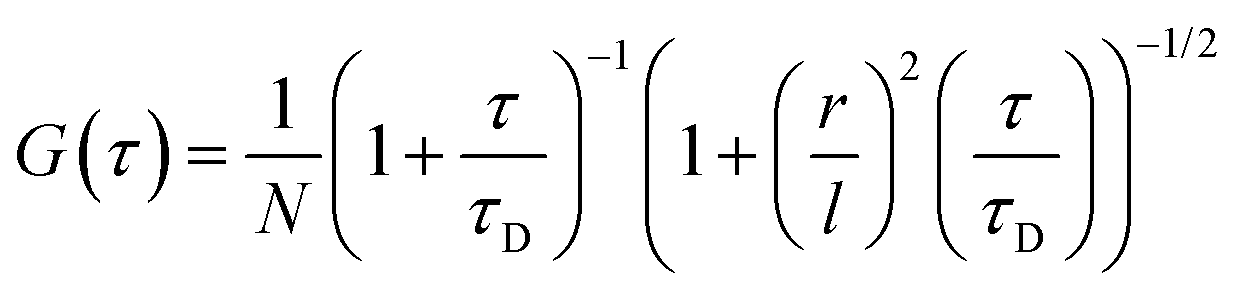 | (2) |
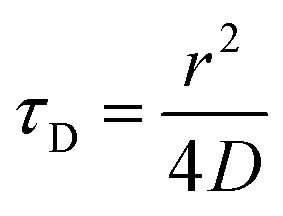 | (3) |
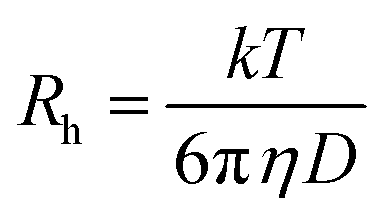 | (4) |
490 nm using a Spectramax plate reader (Molecular Devices LLC, USA). Background absorbance of wells without cells was subtracted from all test wells. The viability in treated wells was determined by normalizing the absorbance to that of untreated control cells.
Results and discussion
Characterization of DNA-free and DNA-loaded peptide MCM-NPs
The urgent need for a versatile non-viral delivery system with advanced physicochemical characteristics that allows for the efficient incorporation of DNA other than single-stranded short antisense oligonucleotides motivated us to develop a purely peptidic DNA delivery system that self-assembles. To systematically explore the molecular parameters that are associated with producing such a DNA-delivery system, we designed a 19 amino acid amphiphilic peptide henceforth termed (HR)3gT (Fig. 1A). The core of the peptide is based on a 10 amino acid amphiphilic H3gT peptide, which has previously been shown to self-assemble into multi-compartment micellar nanoparticles (MCM-NPs) when employing the solvent exchange method.50 To accommodate the negative charge of DNA segments during self-assembly, the positive charge of the hydrophilic domain was increased by adding an arginine residue after each histidine. The extension of the hydrophilic domain was compensated by increasing the hydrophobic domain of H3gT from three to six repetitive L-tryptophan-D-leucine [LW-DL] motifs in (HR)3gT (Fig. 1A) as the ratio of hydrophilic to hydrophobic domain is a critical determinant for the assembly of micelles.54,55 The newly designed (HR)3gT peptide and the 10 amino acid H3gT (for comparison) were synthesised using standard Fmoc-based solid phase peptide synthesis and purified by RP-HPLC. MALDI-TOF mass spectrometry confirmed the expected molecular mass of 2878 g mol−1 for purified (HR)3gT and 1512.7 g mol−1 for purified H3gT (Fig. S2, ESI†). The addition of the hydrophilic and hydrophobic residues in (HR)3gT might interfere with the ability to form nanoparticles although the weight ratio between hydrophilic and hydrophobic domains was kept in the range that typically leads to micelle formation. Thus, we tested the self-assembly of (HR)3gT under the same solvent exchange conditions as reported for H3gT. An overview transmission electron microscopy (TEM) image (Fig. S3A, ESI†) showed that (HR)3gT NPs had a similar appearance. (HR)3gT assembled MCM-NPs with a morphology similar to that observed for H3gT (Fig. 1B). Intriguingly, (HR)3gT MCM-NPs appeared slightly smaller than H3gT MCM-NPs in negatively stained specimens. Consistent with this notion, analysis of particle size by DLS showed a shift from DH = 211 ± 46 nm for H3gT (Table S2, ESI†) to DH = 112 ± 21 nm for (HR)3gT (Fig. 1B, right panel). Further evidence for the assembly of several micelles into a multicompartment structure was provided by high resolution TEM of an individual nanoparticle (Fig. S3B, ESI†).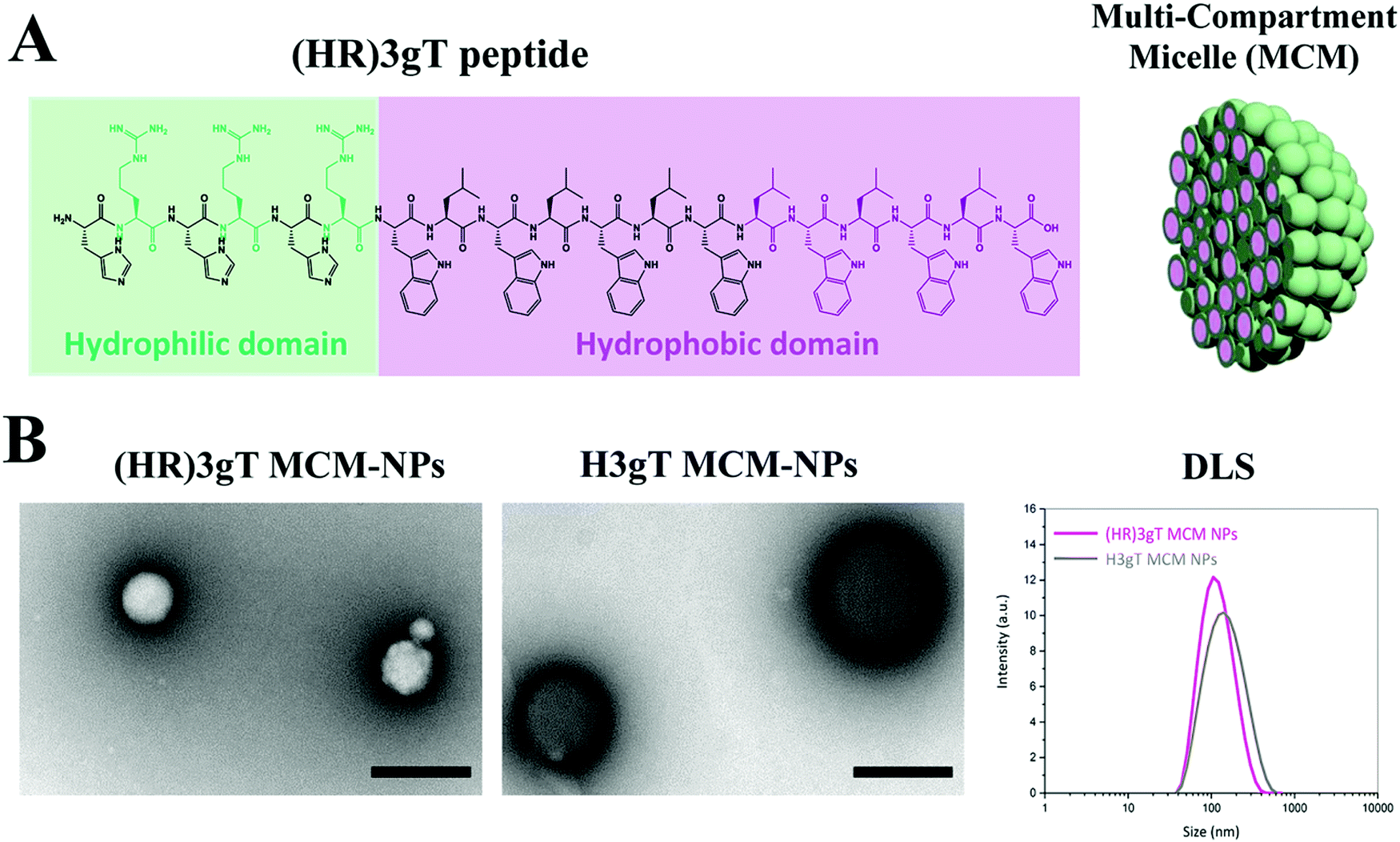 | ||
| Fig. 1 Self-assembling amphiphilic (HR)3gT peptide. (A) The (HR)3gT peptide consists of a hydrophilic (green) and hydrophobic (purple) domain based on modifications of the amphiphilic H3gT peptide (represented in black). The panel on the right is a schematic representation of the self-assembled multi-compartment micelle nanoparticle. (B) TEM micrographs and DLS of (HR)3gT MCM-NPs and H3gT MCM-NPs, scale bars = 200 nm. | ||
To optimize the yield of (HR)3gT MCM-NP formation and simultaneously entrap 22 nt ssDNA during their self-assembly, we performed the solvent exchange at 4 °C to different final ethanol concentrations (Fig. S4, ESI†). Formation of MCM-NPs was observed at final concentrations of 35% and 50% ethanol, while at 20% ethanol, predominantly individual micelles and smaller MCM-NPs were detected with few contaminants of un-assembled peptide. However, at 50% ethanol concentration, the 22 nt single stranded DNA precipitated rather than being entrapped into the MCM-NPs as revealed by precipitation of fluorescently labelled DNA to the bottom of dialysis tube. Hence, 35% ethanol was chosen as a final solvent concentration to allow for DNA incorporation into self-assembled MCM-NPs.
Ultrastructural analysis by TEM showed that in the presence of 22 nt ssDNA, the newly designed (HR)3gT peptide assembled NPs with a multi-compartment micellar architecture similar to that of H3gT MCM-NPs (Fig. 2A, upper panel). To test the ability of (HR)3gT to entrap dsDNA, we examined (HR)3gT self-assembly in the presence of 22 bp dsDNA and compared it to H3gT (Fig. 2A, lower panel).
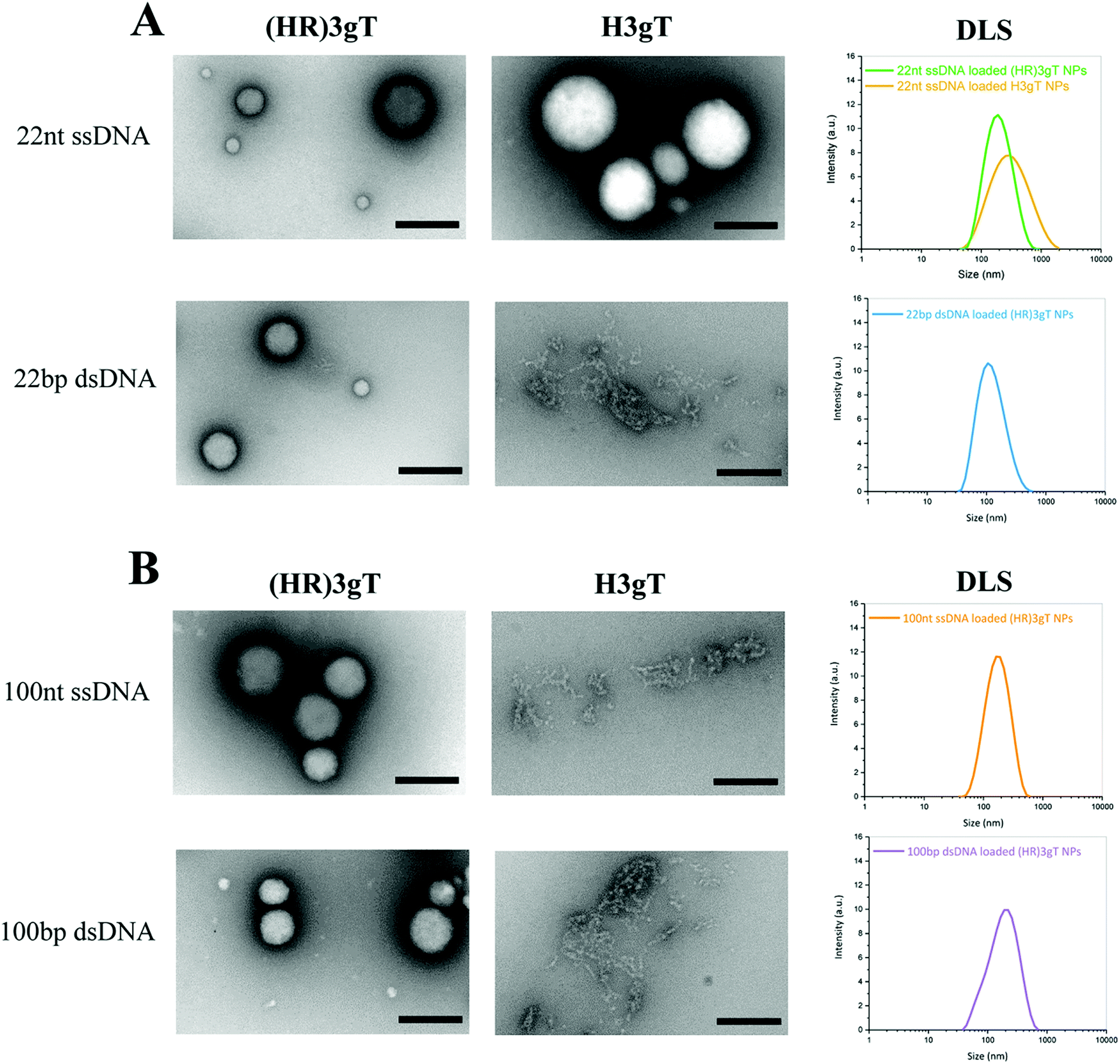 | ||
| Fig. 2 TEM micrographs and DLS of (HR)3gT and H3gT MCM-NPs. (A) 22 nt ssDNA and 22 bp dsDNA loaded MCM-NPs, and (B) 100 nt ssDNA and 100 bp dsDNA loaded MCM-NPs. Scale bars = 200 nm. | ||
In contrast to H3gT peptide, the newly designed (HR)3gT peptide was able to form spherical MCM-NPs also in the presence of 22 bp dsDNA. Conceivably, the higher positive charge of (HR)3gT in comparison to H3gT results in an increased electrostatic interaction between the negatively charged DNA and positively charged peptide. In addition, because dsDNA is more rigid than ssDNA and has double the amount of negatively charged phosphate in its backbone, it is likely to require a higher driving force, i.e. more positive charges that are available for the electrostatic interactions needed to condense the stiffer dsDNA. To bridge the gap in DNA loading between existing non-viral systems that deliver short single-stranded ASO and those that are based on complex formation to transfer entire genes, we assessed the self-assembly process of (HR)3gT in the presence of single- and double-stranded DNA sequences of 100 nucleotide length (Fig. 2B). While both 100 nt ssDNA and 100 bp dsDNA were entrapped during the self-assembly process of (HR)3gT, the shorter H3gT peptide was not able to self-assemble into MCM-NPs in the presence of the longer DNA segments. In the self-assembly solution at pH 7.1, the net charge of the (HR)3gT peptide is +3 while H3gT has a net charge of +0.3 with regard to their isoelectric point. Therefore, H3gT peptide does not provide adequate positive charge to enable electrostatic forces that condense the longer ss/dsDNA. Thus, increasing the positive charge of hydrophilic domain of H3gT by arginine is conducive to the entrapment of the longer DNA segment in the (HR)3gT MCM-NPs. Although the hydrophilic to hydrophobic weight ratio in (HR)3gT was slightly higher (44.6% compared to 38.4%), it was still within the range where micelles formed. Our data suggest that optimizing the charge and tuning the ratio of hydrophilic to hydrophobic domains are key parameters in the development of DNA delivery systems based on peptidic MCM-NPs.
Consistent with TEM analysis, the scattered intensities measured by DLS demonstrated a narrow size distribution in the range of 100–180 nm for all (HR)3gT assemblies (Fig. 2). It is noteworthy that the average size for 22 nt ssDNA-loaded (HR)3gT NPs was smaller than the corresponding H3gT NPs (160 ± 18 nm versus 241 ± 73 nm in diameter). This can be attributed to the higher charge interaction between the new peptide and DNA resulting in more compacted NPs.
Furthermore, increasing the length of the entrapped DNA sequence from 22 nt/bp to 100 nt/bp did not significantly affect the average size of (HR)3gT NPs. It is widely accepted that particle size plays a critical role in developing successful therapeutic delivery systems.56,57 For example, size is one of the most crucial determinants of nanoparticle half-life. In order to decrease the likelihood of liver or splenic trapping and increase systemic circulation time, a particle size below 200 nm is desired for in vivo applications of non-viral delivery systems.58 Thus, maintaining a particle size below 200 nm independent of whether 22 nt/bp or 100 nt/bp were entrapped represents an important advance of the (HR)3gT peptide towards an efficient DNA delivery system. Moreover, as the upper size limit for a clathrin-mediated endocytosis is 200 nm,59,60 the characteristic size of our newly generated DNA-loaded (HR)3gT MCM nanoparticles make them well-suited for cellular uptake. DLS and TEM demonstrated that (HR)3gT NPs with and without DNA do not tend to aggregate over time. This is a further advantage over H3gT peptide nanoparticles which tend to aggregate.50
To determine the size and polydispersity index (PDI) of (HR)3gT MCM-NPs that had entrapped either single- or double-stranded DNA sequences of different lengths, we employed DLS, NTA and FCS measurements and compared the corresponding data (Table 1). From the FCS measurements, we determined the hydrodynamic radius of NPs from eqn (4) (see Methods) and extracted the diffusion time by solving eqn (2), using the hydrodynamic diameter and the known diffusion coefficients of ss/dsDNA of different size.61
| (HR)3gT MCM-NPs | PDI | D H (nm) DLS | D H (nm) NTA | D H (nm) FCS | Zeta potential (mV) |
|---|---|---|---|---|---|
| a D H is a hydrodynamic diameter. | |||||
| (HR)3gT NPs | 0.19 ± 0.024 | 112 ± 21 | 102 ± 6 | N/A | +8.2 ± 2.1 |
| 22 nt ssDNA loaded (HR)3gT NPs | 0.23 ± 0.026 | 160 ± 18 | 151 ± 12 | 144 ± 58 | +2.8 ± 3.35 |
| 22 bp dsDNA loaded (HR)3gT NPs | 0.22 ± 0.023 | 115 ± 19 | 129 ± 10 | 124 ± 62 | +3.67 ± 3.11 |
| 100 nt ssDNA loaded (HR)3gT NPs | 0.28 ± 0.021 | 176 ± 11 | 164 ± 4 | 186 ± 78 | +3.46 ± 3.87 |
| 100 bp dsDNA loaded (HR)3gT NPs | 0.27 ± 0.02 | 165 ± 12 | 150 ± 11 | 174 ± 99 | +4.18 ± 3.36 |
In addition, the surface charge of nanoparticles is known to play a key role in cellular uptake. In many instances, positively charged particles were shown to be efficiently endocytosed due to their interactions with the negatively charged cell membrane.62,63 Accordingly, the zeta potentials of empty and DNA-loaded (HR)3gT MCM-NPs were measured (Table 1).
A PDI ≤ 0.3, which for polymer-based nanosystems implies a relatively monodisperse system, was obtained by DLS measurements of DNA-free and DNA-loaded (HR)3gT MCM-NPs.64,65 It is noteworthy that (HR)3gT MCM-NPs (DNA-free and DNA-loaded) had a lower PDI than corresponding H3gT MCM-NPs (Table S2, ESI†). The mean diameter obtained by NTA was similar that determined by DLS for each kind of (HR)3gT MCM-NPs (Table 1). The respective sizes were further corroborated by average sizes calculated from FCS measurements (Table 1). Indeed, FCS is a suitable method for characterizing the size of fluorescent NPs if the number of fluorescent species is ≤3 and the particle size <500 nm.53 In contrast to the hydrodynamic diameters of (HR)3gT MCM-NPs loaded with different DNAs, which were all below 200 nm, H3gT MCM-NPs loaded with 22 nt ssDNA were above 200 nm in diameter (Table S2, ESI†), which also limited their cellular uptake (see below, Fig. 7). It has been reported that nanoparticles above 200 nm may be excluded from cellular internalization altogether.66,67
With the entrapment of negatively charged DNA in self-assembling (HR)3gT the surface charge of the resulting MCM-NPs only slightly decreased compared to DNA-free NPs. These data support the presence of corresponding DNA payload between individual micelles of the multi-compartment rather than its accumulation on the NP surface (Table 1) which would lead to larger decrease of the zeta potential. As expected, in the absence of DNA, (HR)3gT MCM-NPs displayed a higher zeta potential than H3gT MCM-NPs (Table 1 and Table S2, ESI†) due to the extra arginines included in the hydrophilic domain of (HR)3gT. With a surface charge of 2.8 ± 3.35 mV, 22 nt ssDNA-loaded (HR)3gT MCM-NPs experience repulsion forces between each other that hinder their aggregation. On the other hand, 22 nt ssDNA-loaded H3gT MCM-NPs are neutral (0.6 ± 2.3 mV) and thus, tend to aggregate after formation. A positive surface charge has been shown to be advantageous for delivering DNA in a number of NP systems.68 For example, PEGylated DNA/transferrin–PEI complexes bearing a positive charge of only +4 mV, were found to be the most efficient gene delivery system among other positively charged complexes.69 In another study on 140 distinct polymer compositions, DNA-loaded NPs bearing a charge of +1.8 mV demonstrated the third highest uptake and best overall transfection of a luciferase plasmid in NIH 3T3 cells.70 Accordingly, the overall positive charge of +2mV to +4 mV of (HR)3gT MCM-NPs loaded with different DNA segments is expected to be conducive to an efficient DNA delivery. Thus, the modifications of our newly designed (HR)3gT peptide, which led to MCM-NPs below 200 nm with a slightly positive surface charge, represent a crucial advance in fulfilling the physiochemical criteria required for generating successful therapeutic nanosystems.
Quantification of DNA entrapped in (HR)3gT MCM-NPs
The incorporation of different sizes and types of DNA into (HR)3gT MCM-NPs was examined by FCS using Atto550-labelled DNAs. Diffusion time (τD) and count per molecule (CPM) of free Atto550 dye, Atto550-DNA in solution, and of Atto550-DNA-loaded (HR)3gT MCM-NPs were calculated by fitting the experimental autocorrelation curves (Fig. 3) to a two-component fit (eqn (1)). Subsequently, the number of DNA molecules per particle was calculated by dividing the CPM of DNA-loaded (HR)3gT MCM-NPs by the corresponding Atto550-DNA in solution (Table 2). The significant increase in the diffusion time of all Atto550-DNA-loaded NPs compared to Atto550-labelled DNA indicates that the DNA is entrapped in (HR)3gT MCM-NPs. For (HR)3gT MCM-NPs, we calculated 19.9 ± 13.7 22 nt ssDNA molecules per particle while the H3gT peptide was able to entrap 3.95 ± 2.39 22 nt ssDNA per particle. Although 22 nt ssDNA-loaded (HR)3gT MCM-NPs had entrapped five times more 22mer ssDNA than H3gT MCM-NPs, their average size is significantly smaller (160 ± 18 versus 241 ± 73 nm, Table 1, p ≤ 0.05) (Table 2 and Table S3, ESI†). Hence, the enhanced positive charge of (HR)3gT peptide compared to H3gT peptide results in increased electrostatic interactions between DNA and peptide and more compact assemblies. Consequently, the nanoparticle size remains smaller than the corresponding H3gT MCM-NPs despite more DNA and a longer peptide (19 versus 10 amino acids). For (HR)3gT MCM-NPs loaded with double-stranded 22 bp DNA, the average diffusion time of 3235 ± 1607 μs indicated that the number of DNA molecules per particle is approximately half the number of corresponding ssDNA per particle (Fig. 3B and Table 2). Considering that electrostatic interactions between DNA and peptide are responsible for the DNA entrapment during the self-assembly process, doubling the amount of negatively charged phosphates in the backbone of the double-stranded DNA may explain why the entrapment is reduced by half. In addition, the higher rigidity of the 22 bp dsDNA may interfere with entrapment.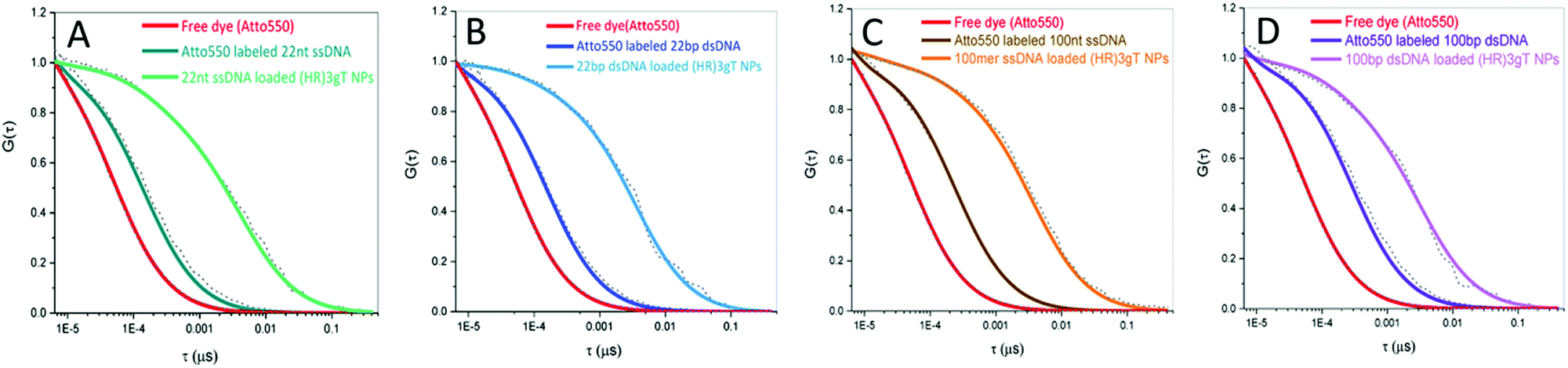 | ||
| Fig. 3 Normalised FCS autocorrelation curves for (A) Atto550-labelled 22 nt ssDNA (dark green), 22 nt ssDNA-loaded (HR)3gT MCM-NPs (green), (B) Atto550-labelled 22 bp dsDNA (dark blue), 22 bp dsDNA-loaded (HR)3gT MCM-NPs (blue), (C) Atto550-labelled 100 nt ssDNA (brown), 100 nt ssDNA-loaded (HR)3gT MCM-NPs (orange), (D) Atto550-labelled 100 bp dsDNA (dark purple), 100 bp dsDNA-loaded (HR)3gT MCM-NPs (purple). Atto550 free dye is shown in red in all panels. | ||
| Fluorescent specious | Diffusion time (μs) | DNA/particle |
|---|---|---|
| a Not applicable. | ||
| Atto550 fluorescent dye | 48 ± 3 | N/Aa |
| Atto550 labelled 22 nt ssDNA | 139 ± 5 | N/A |
| Atto550 labelled 22 bp dsDNA | 157 ± 9 | N/A |
| Atto550 labelled 100 nt ssDNA | 237 ± 13 | N/A |
| Atto550 labelled 100 bp dsDNA | 283 ± 12 | N/A |
| 22 nt ssDNA loaded (HR)3gT MCM-NPs | 3894 ± 1337 | 19.9 ± 13.7 |
| 22 bp dsDNA loaded (HR)3gT MCM-NPs | 3235 ± 1607 | 11.7 ± 7.2 |
| 100 nt ssDNA loaded (HR)3gT MCM-NPs | 3875 ± 1778 | 2.12 ± 2.43 |
| 100 bp dsDNA loaded (HR)3gT MCM-NPs | 3553 ± 1900 | 1.47 ± 1.12 |
Likewise, for the longer DNA segments (100 nt/bp ss/dsDNA), the number of DNA molecules incorporated into (HR)3gT MCM-NPs decreased compared to both, single- and double-stranded 22 nucleotide-long DNA (Fig. 3C, D and Table 2). (HR)3gT with an isoelectric point of 12.4 (versus 7.61 for H3gT) has a charge of +3 compared to 0 for H3gT at neutral pH. Because more electrostatic interactions that drive the DNA incorporation can occur, (HR)3gT is able to condense longer DNA segments (100 nt and 100 bp). Thus, the modifications of the (HR)3gT peptide, in particular the enhanced positive charge over H3gT, conveys not only the ability to entrap longer single- and double-stranded DNA upon nanoparticles but also the potential to more efficiently incorporate higher amounts of 22 nt ssDNA. Generating (HR)3gT MCM-NPs with improved DNA loading and smaller particle size compared to H3gT represents a significant progress towards developing an efficient and functional DNA delivery system.
Determination of MCM-NP concentrations by NTA
The total concentration of particles in solution and the percentage of loaded particles were determined by nanoparticle tracking analysis (Table 3). The concentration of MCM-NPs formed in the presence of 100 nt long sequences was similar for single- and double-stranded DNA (3.29 × 108 ± 7.65 × 107 mL−1 and 3.34 × 108 ± 8.64 × 107 mL−1, respectively), and comparable to that of NPs assembled in the presence of 22 bp dsDNA (3.02 × 108 ± 3.06 × 107 mL−1). Significantly higher numbers of MCM-NPs (5.57 × 108 ± 7.03 × 107) were obtained when (HR)3gT was assembled with 22 nt ssDNA. For all types of DNA, more than 60% of the MCM-NPs were loaded with DNA whereby the percentage of loaded particles for dsDNA (22 bp and 100 bp) was clearly higher than for ssDNA (22 nt and 100 nt). Considering the FCS data presented above, which show that (HR)3gT MCM-NP entrap approximately twice as many ssDNA as dsDNA molecules, the overall amount of loaded DNA is higher for ssDNA compared to dsDNA MCM-NPs. Thus, while the total concentration of particles is similar for 100 nt ssDNA and 100 bp dsDNA MCM-NPs, the overall amount of entrapped DNA is higher with single-stranded than with double-stranded DNA despite the lower percentage of loaded particles (68.5% versus 82%). Similarly, the percentage of loaded particles for the 22 nt long DNA was 68.5% for ssDNA versus 95% for dsDNA (3.82 × 108 ± 2.29 × 107versus 2.87 × 108 ± 3.10 × 107). Due to the significantly higher concentration of MCM-NPs that self-assemble in the presence of 22 nt ssDNA, the difference in the amount of entrapped DNA between single- and double stranded is higher for 22 nucleotide long DNA sequences than for 100 nucleotide sequences.| MCM peptide NPs | Total concentration (particles per mL) | Concentration of labelled particles (particles per mL) | Loaded particles (%) |
|---|---|---|---|
| (HR)3gT NPs | 4.29 × 108 ± 9.17 × 107 | N/A | N/A |
| 22 nt ssDNA loaded (HR)3gT NPs | 5.57 × 108 ± 7.03 × 107 | 3.82 × 108 ± 2.29 × 107 | 68.5 |
| 22 bp dsDNA loaded (HR)3gT NPs | 3.02 × 108 ± 3.06 × 107 | 2.87 × 108 ± 3.10 × 107 | 95 |
| 100 nt ssDNA loaded (HR)3gT NPs | 3.29 × 108 ± 7.65 × 107 | 2.25 × 108 ± 1.10 × 107 | 68.3 |
| 100 bp dsDNA loaded (HR)3gT NPs | 3.34 × 108 ± 8.64 × 107 | 2.74 × 108 ± 1.52 × 107 | 82 |
The stability of DNA-free and DNA-loaded (HR)3gT MCM-NPs
To obtain insight on the stability of DNA-free and DNA-loaded (HR)3gT peptide NPs, we analyzed the size of NPs stored at 4 °C by DLS over time (Fig. 4A). Nanoparticle size measurements recorded every 30 days indicated that the average diameter of DNA-free and DNA-loaded (HR)3gT MCM-NPs did not change over five months. TEM analyses of empty and DNA-loaded (HR)3gT MCM-NPs after 5 months (Fig. 4B) showed that nanoparticles retained their initial multi-compartment micellar structure. In contrast, H3gtT MCM-NPs, especially when loaded with 22 nt ssDNA, aggregated due to their neutral surface charge.50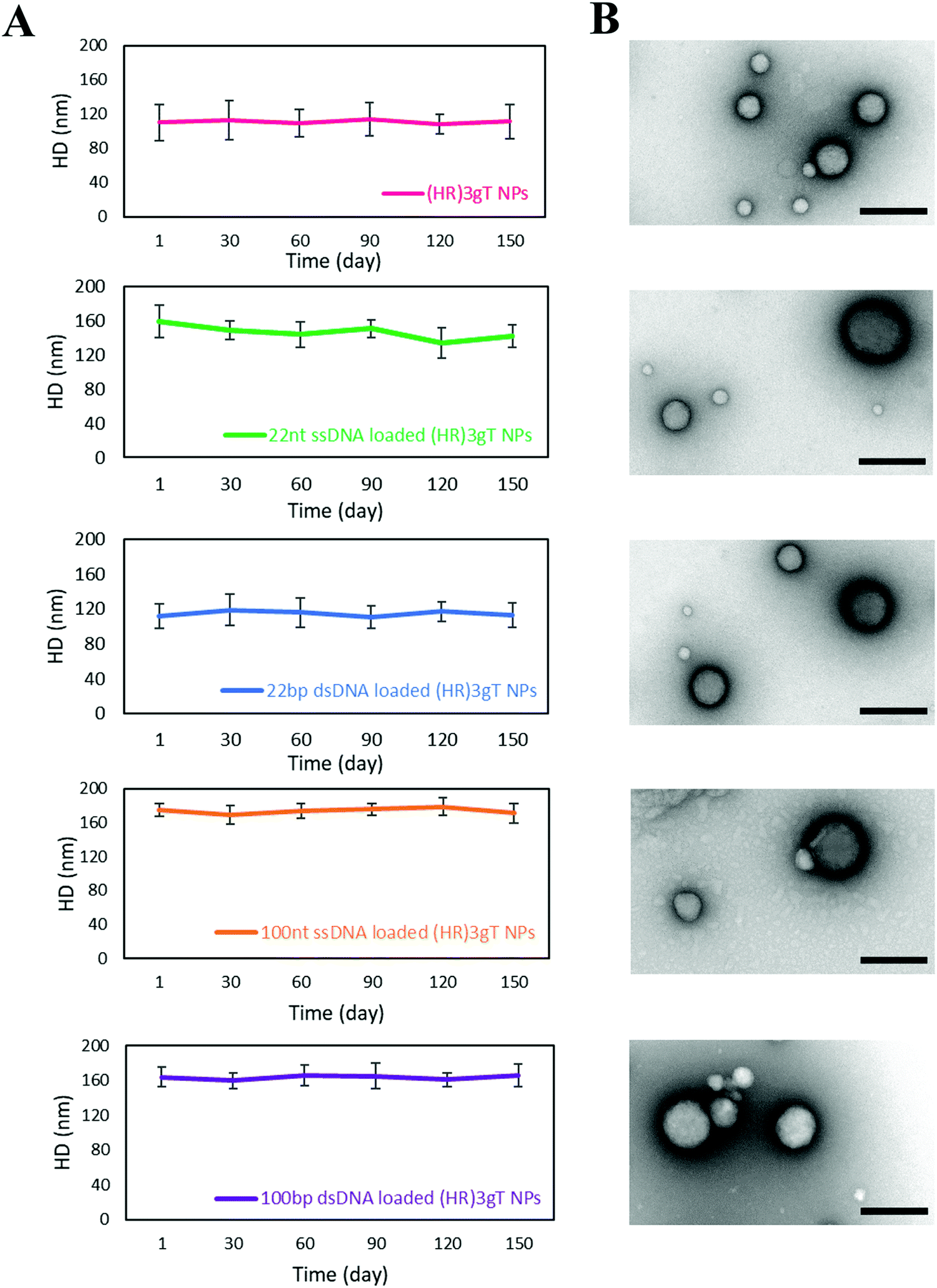 | ||
| Fig. 4 Stability of (HR)3gT MCM-NPs. From top to bottom: (A) DLS over 150 days, and (B) TEM after 150 days for (HR)3gT MCM-NPs, 22 nt ssDNA-loaded (HR)3gT MCM-NPs, 22 bp dsDNA-loaded (HR)3gT MCM-NPs, 100 nt ssDNA-loaded (HR)3gT MCM-NPs, and 100 bp dsDNA-loaded (HR)3gT MCM-NPs. Scale bars = 200 nm. | ||
Thermo-responsiveness of DNA-free and DNA-loaded (HR)3gT MCM-NPs
To assess thermo-responsiveness, DNA-free and DNA-loaded (HR)3gT MCM-NPs were incubated at 37 °C for 5 h and 24 h. Ultrastructural analysis by TEM revealed that compared to the MCM structure of NPs at 4 °C, (HR)3gT MCM-NPs disassembled into smaller MCMs and/or individual micelles after 5 h at 37 °C (Fig. 5A). The tendency to disassembly increased over time and after 24 h at 37 °C (Fig. 5B), only few MCM-NPs were detected. When kept at 37 °C, all peptide MCM-NPs exhibited a similar trend in the change of their structure and size, independent of the length and type (single or double stranded) of the entrapped DNA, whereas at 4 °C, (HR)3gT MCM-NPs remained stable for as long as 5 months (Fig. 4). Likewise, H3gT MCM-NPs demonstrated a similar disassembly behaviour in response to temperature (Fig. S6, ESI†). Our data are consistent with the study by Schuster et al. who reported that the formation of MCM peptide NPs occurs as a function of temperature and solvent composition.71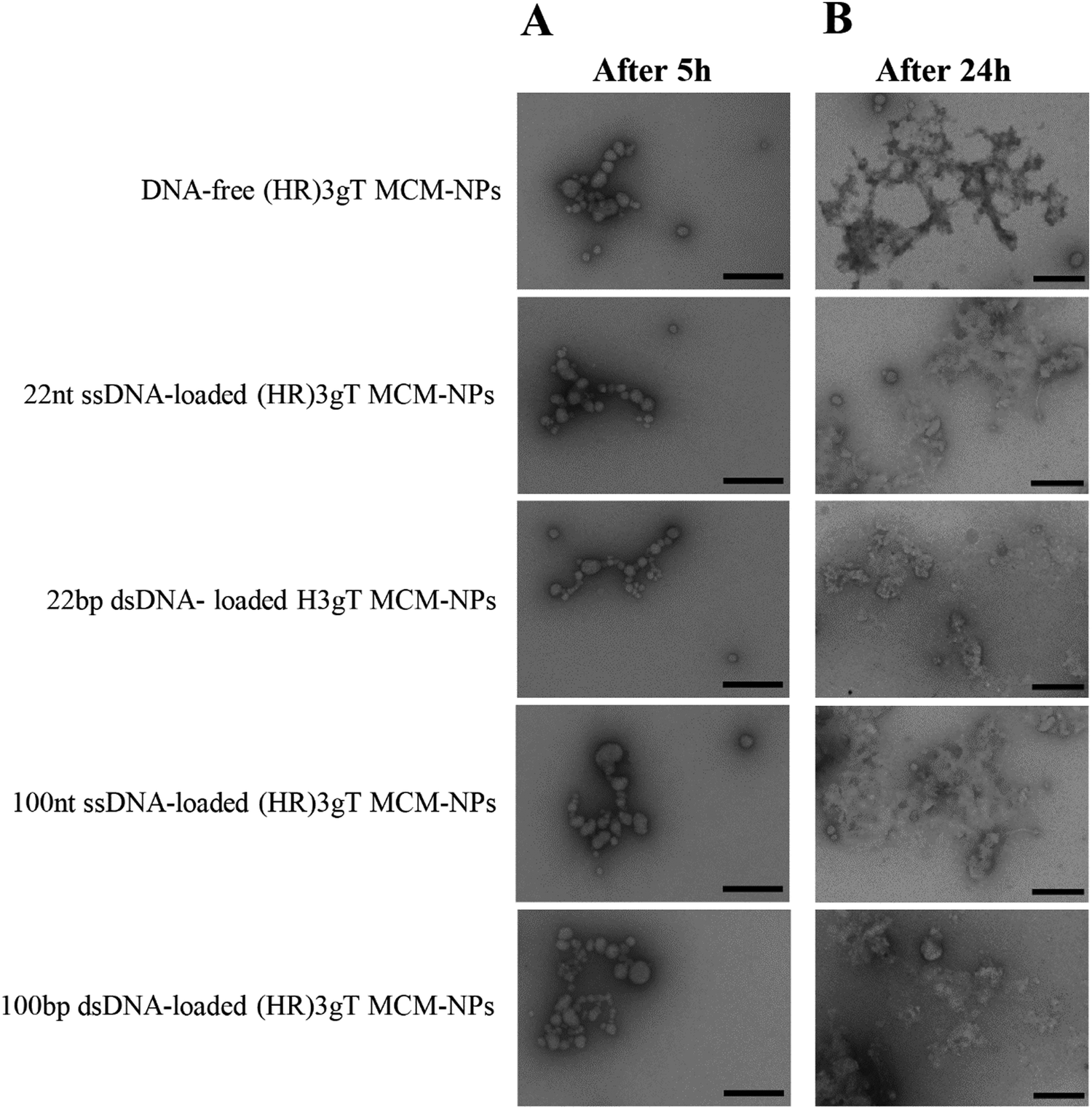 | ||
| Fig. 5 TEM micrographs of (HR)3gT MCM-NPs with different DNA loads after (A) 5 h incubation, and (B) 24 h incubation at 37 °C. Scale bars = 200 nm. | ||
Effect of (HR)3gT MCM-NPs on cell viability
In order to examine the cytotoxicity of our non-viral DNA delivery system, MTS cell proliferation assays with different concentrations of DNA-loaded and DNA-free (HR)3gT MCM-NPs (up to 1550 μg mL−1) were evaluated (Fig. 6). The data showed that after 24 h of incubation at 37 °C, DNA-loaded and DNA-free (HR)3gT MCM-NPs up to a concentration of 1550 μg mL−1 had no toxic effects on HeLa cells. Interestingly, 22 nt ssDNA-loaded (HR)3gT MCM-NPs had no significant inhibitory effect on cell proliferation even though they are loaded with a 5-times higher amount of 22 nt ssDNA compared to corresponding H3gT MCM-NPs (MTS assays for H3gT MCM-NPs in Fig. S7, ESI†). These findings suggest that the viability of treated cells remains largely unaffected by the peptide MCM-NP concentration and the amount of entrapped DNA. Based on the apparent biocompatibility, (HR)3gT MCM-NPs are a viable candidate for the future development of safe gene delivery systems.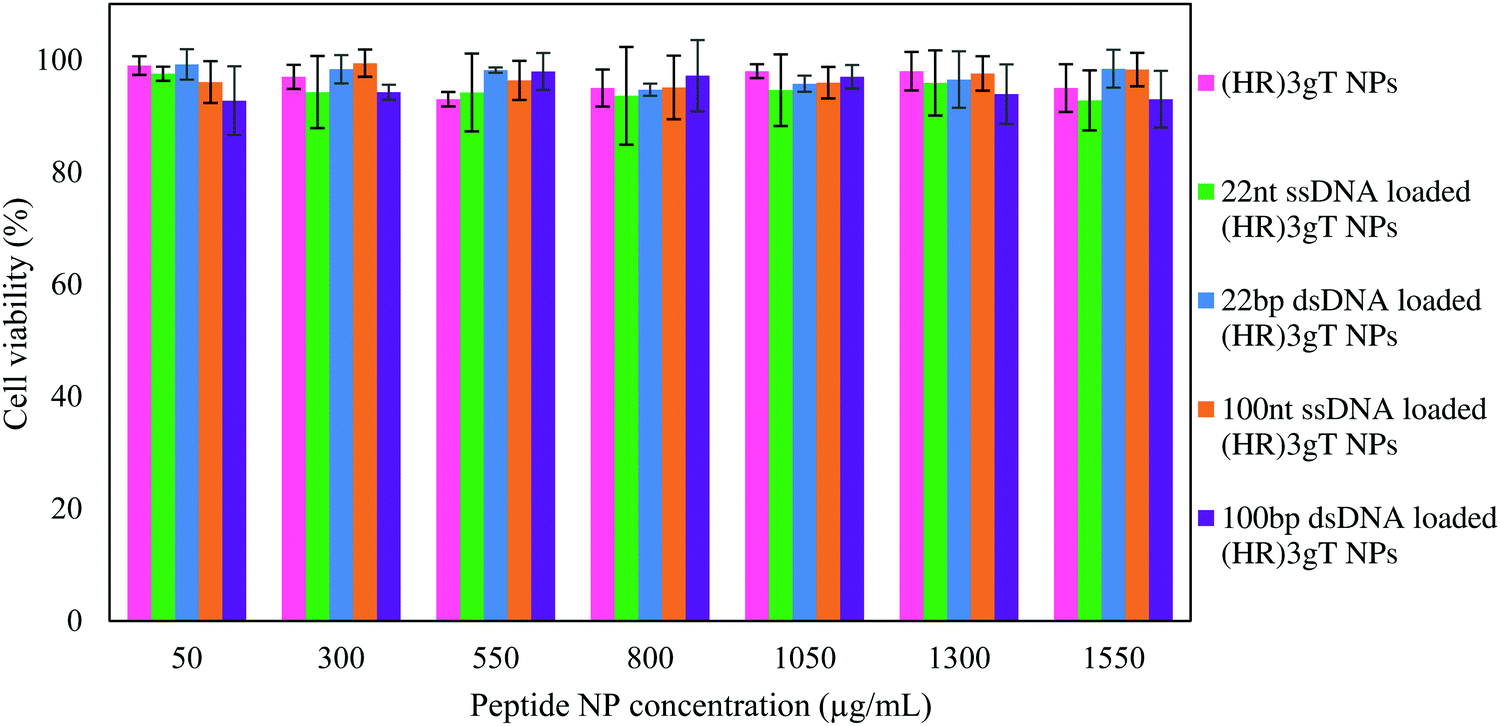 | ||
| Fig. 6 HeLa cell viability (MTS assay). Cells treated for 24 h with (HR)3gT MCM-NPs (pink), 22 nt ssDNA-loaded (HR)3gT MCM-NPs (green), 22 bp dsDNA-loaded H3gT MCM-NPs (blue), 100 nt ssDNA-loaded (HR)3gT MCM-NPs (orange), 100 bp dsDNA-loaded (HR)3gT MCM-NPs (purple). Cell viability was normalized to untreated HeLa cells (negative control, 100% viability). All data presented as the mean ± SD (n = 3). | ||
Cellular uptake
The cellular uptake of peptide MCM-NPs loaded with fluorescently labelled Atto550-DNA segments was examined in H2B-GFP expressing HeLa cells by confocal laser scanning microscopy (CLSM). We first compared cells that were treated with 22 nt ssDNA-loaded (HR)3gT or H3gT MCM-NPs (Fig. 7A). Confocal images of cells incubated with NPs for 24 h revealed that at a similar cell density, a significantly higher number of cells had taken up 22 nt ssDNA-loaded (HR)3gT MCM-NPs. Besides the higher rate of uptake, the fluorescence signal in these cells was considerably stronger than in cells containing H3gT MCM-NPs under identical imaging conditions. Also after 48 h of NP incubation, the uptake efficiency for 22 nt ssDNA-loaded (HR)3gT was higher than for H3gT MCM-NPs (Fig. S8, ESI†). Several properties of NPs assembled from the newly designed (HR)3gT peptide could account for the higher uptake efficiency of 22 nt ssDNA-loaded (HR)3gT MCM-NPs versus 22 nt ssDNA-loaded H3gT MCM-NPs. Nanoparticle size, shape, surface charge, and surface functionality are known to affect cellular uptake.70,72 Based on literature reports, the optimal size for NP uptake, especially in cancer treatment, is between 70–200 nm and ideally close to 100 nm. However, for specific applications, slightly larger particle sizes may be more advantageous.73–77 Accordingly, 22 nt ssDNA-loaded (HR)3gT MCM-NPs with the mean diameter of 160 ± 18 nm are in range for optimal uptake in contrast to 22 nt ssDNA-loaded H3gT MCM-NPs (241 ± 73 nm). Furthermore, the increased cellular uptake could be facilitated with other internalization mechanisms because of arginine–histidine repeating units that are exposed on the hydrophilic surface of the MCM-NPs. Numerous studies report that arginine residues promote cell penetration and increase internalization efficiency in the delivery of therapeutic macromolecules, in particular nucleic acids.78–86 For example, using a multifunctional envelope-type nanodevice (MEND) that comprises an octa-arginine as a gene delivery system resulted in an improvement in gene expression by more than 300 times compared to an unmodified MEND.87 Besides a larger number of (HR)3gT MCM-NPs taken up per cell, the increased fluorescence intensity is also related to the 5 times higher amount of 22 nt ssDNA loaded per particle compared to H3gT MCM-NPs.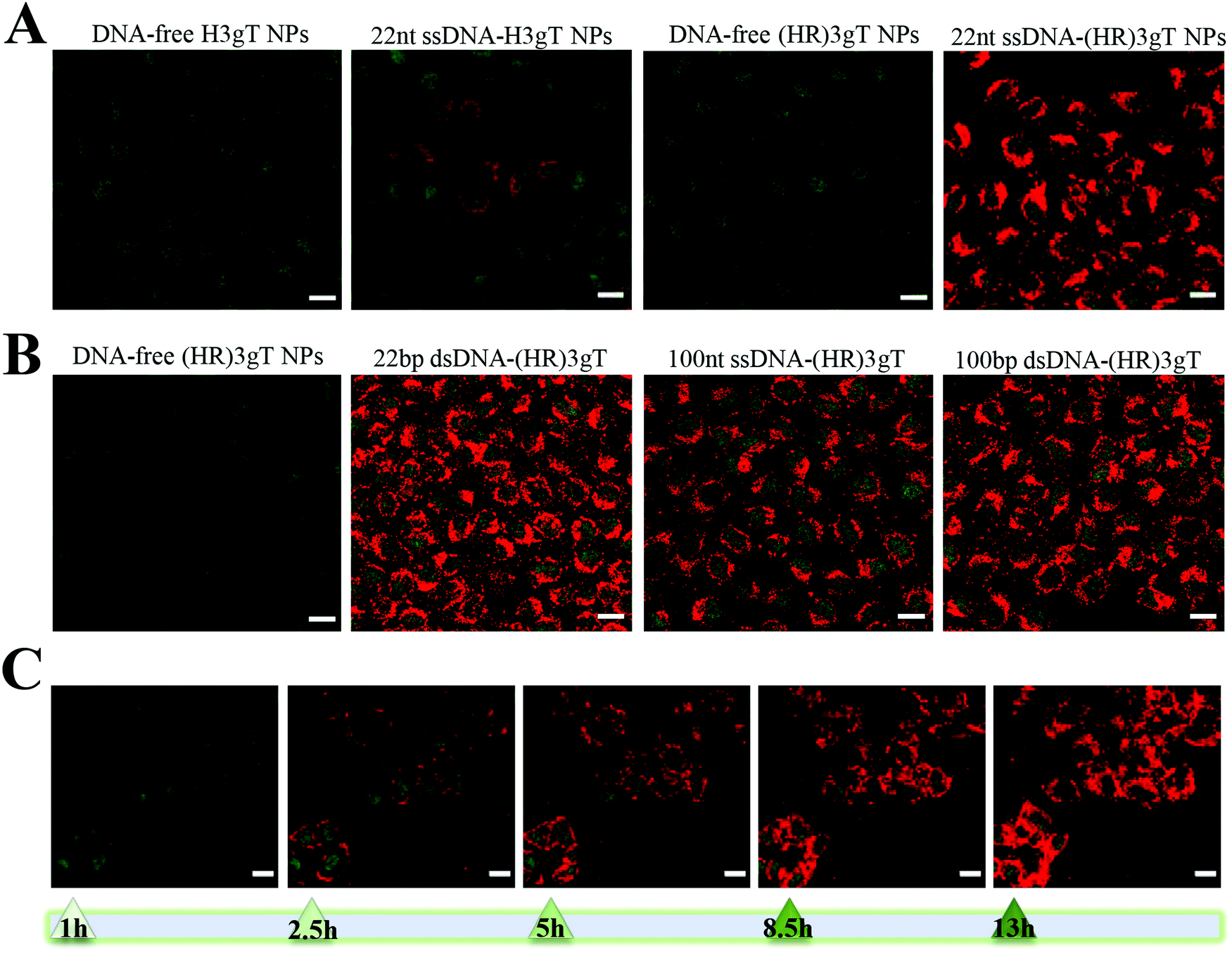 | ||
| Fig. 7 CLSM merged images (GFP and Atto550) of H2B-GFP expressing HeLa cells treated with (A) 22 nt ssDNA-loaded H3gT and (HR)3gT MCM-NPs, and (B) 22 bp and 100 nt/bp ss/dsDNA-loaded (HR)3gT NPs after 24 h. (C) Time course of 100 bp dsDNA-loaded (HR)3gT MCM NP uptake. Scale bars = 20 μm. | ||
Cellular uptake assays with (HR)3gT MCM-NPs loaded with 22 bp dsDNA and longer sequences (100 nt/bp) revealed a similarly high uptake efficiency for all (HR)3gT MCM-NPs (Fig. 7B). As previously mentioned, the number of incorporated dsDNA molecules/particle is approximately half compared to the number of ssDNA molecules in corresponding (HR)3gT MCM-NPs. Thus, under identical imaging conditions, one can expect a lower fluorescence signal for dsDNA-loaded peptide NP uptake. However, CLSM images showed no significant difference between 22 bp dsDNA and 22mer ssDNA-loaded (HR)3gT MCM-NPs (compare Fig. 7A and B), and between 100 bp dsDNA and 100mer ssDNA-loaded MCM-NPs (Fig. 7B). It is possible that small differences in size and surface charge between MCM-NPs loaded with dsDNA versus ssDNA (see Table 1) lead to an increase in cellular uptake and thus, account for the greater fluorescence signal. Furthermore, a time-dependent increase of intracellular fluorescence from 1 to 13h for 100 bp dsDNA-loaded (HR)3gT MCM-NPs is shown in Fig. 7C (the corresponding time-lapse video is available as ESI†). Although in vitro analysis of thermo-responsiveness showed a disassembly of MCM-NPs after 5 h, earlier time points, i.e. 1 and 2 h, revealed that the multicompartment micellar architecture of 100 bp dsDNA-loaded (HR)3gT NPs was still intact (Fig. S10A, ESI†). Moreover, the analysis of cells incubated with dsDNA-loaded (HR)3gT NPs showed that uptake occurred within an hour (Fig. S10B, ESI†), which suggests that at least some of the MCM-NPs are taken up before disassembly. Conceivably, the time-course of MCM-NP thermo-responsiveness adds to the release of DNA inside the cell. Our data suggest that the type of the DNA sequence (i.e. single-stranded or double-stranded) and the length (22 or 100 nucleotides) affect the assembly of MCM-NPs and consequently their cellular uptake properties. Thus, to ultimately achieve a safe and efficient delivery system for protein encoding genes, the size of the DNA will need to be successively increased and tested for its effects on the self-assembly behaviour of the (HR)3gT peptide. It is possible that further modifications of the peptide design might be necessary to accommodate for longer DNA sequences.
Conclusion
In this study, we addressed the challenge of developing an efficient and safe non-viral delivery system for DNA sequences larger than the average antisense oligonucleotide. Specifically, we established for the first time a purely peptidic self-assembling MCM-NP that is able to entrap single- and double-stranded DNA of 100 nucleotides/base pairs. We rationally designed and synthesized the amphiphilic (HR)3gT peptide where we significantly modified a short H3gT peptide by (i) increasing the charge of the hydrophilic domain and (ii) extending the hydrophobic domain to achieve a ratio that typically leads to the self-assembly of micellar nanostructures. The comprehensive analysis of the physicochemical characteristics of (HR)3gT MCM-NPs in the presence of different DNA cargoes indicates that the entrapment is primarily governed by electrostatic interactions between DNA and peptide, and the solvent conditions used for self-assembly. Based on the amino acid modifications, (HR)3gT peptide was able to entrap single- and double-stranded DNA sequences of 100 nucleotides whereas the short H3gT MCM-NPs could only entrap 22 nt ssDNA. A thermodynamically stable system was obtained, in which the sum of different factors including chain stretching, interfacial tension, and repulsive interactions between hydrophilic domains, especially in the presence of negatively charged DNA are minimized. Moreover, the addition of arginine residues facilitated the crossing of biological barriers, which points to a great potential of (HR)3gT MCM-NPs for non-viral gene-delivery applications. (HR)3gT MCM-NPs with different DNA loads maintained a stable size and multi-compartment micellar structure over five months at 4 °C, but disintegrated into smaller MCMs and/or individual micelles at 37 °C. The thermo-responsiveness, together with the low cell toxicity and rapid cellular uptake, constitute essential features in the efficient delivery of the DNA segments. In addition, the possibility of chemical modification by variation of their amino acid sequence and composition renders peptide nanoparticles easily tunable compared to lipid nanoparticles. Taking into account these advantages, our (HR)3gT non-viral DNA delivery system provides the first stepping stone towards developing a purely peptide-based self-assembling nanostructure for the delivery of entire genes. Future designs will need to address several parameters, e.g. nuclear targeting, to optimize the delivery efficiency of peptidic MCM-NPs for medical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of this project provided by the Swiss Nanoscience Institute (SNI), the University of Basel and by ETH Zürich is highly appreciated. The authors thank G. Persy (University of Basel) for TEM measurements.References
- S. L. Ginn, I. E. Alexander, M. L. Edelstein, M. R. Abedi and J. Wixon, J. Gene Med., 2013, 15, 65–77 CrossRef CAS PubMed.
- R. M. Levine, C. M. Scott and E. Kokkoli, Soft Matter, 2013, 9, 985–1004 RSC.
- L. Naldini, Nature, 2015, 526, 351 CrossRef CAS PubMed.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346 CrossRef CAS PubMed.
- N. Nayerossadat, T. Maedeh and P. A. Ali, Adv. Biomed. Res., 2012, 1, 27 CrossRef PubMed.
- M. Foldvari, D. W. Chen, N. Nafissi, D. Calderon, L. Narsineni and A. Rafiee, J. Controlled Release, 2016, 240, 165–190 CrossRef CAS PubMed.
- I. Slivac, D. Guay, M. Mangion, J. Champeil and B. Gaillet, Expert Opin. Biol. Ther., 2017, 17, 105–118 CrossRef CAS PubMed.
- X. Liang, L. Liu, Y.-Q. Wei, G.-P. Gao and X.-W. Wei, Hum. Gene Ther., 2018, 29, 1227–1234 CrossRef CAS PubMed.
- L. W. Seymour and A. J. Thrasher, Nat. Biotechnol., 2012, 30, 588 CrossRef CAS PubMed.
- J. M. Kaminski, M. R. Huber, J. B. Summers and M. B. Ward, FASEB J., 2002, 16, 1242–1247 CrossRef CAS PubMed.
- G. Lin, L. Li, N. Panwar, J. Wang, S. C. Tjin, X. Wang and K.-T. Yong, Coord. Chem. Rev., 2018, 374, 133–152 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541 CrossRef CAS PubMed.
- M. Nakanishi, A. Eguchi, T. Akuta, E. Nagoshi, S. Fujita, J. Okabe, T. Senda and M. Hasegawa, Curr. Protein Pept. Sci., 2003, 4, 141–150 CrossRef CAS PubMed.
- M. E. Martin and K. G. Rice, AAPS J., 2007, 9, E18–E29 CrossRef CAS PubMed.
- S. d. M. Barros, L. A. Avila, S. K. Whitaker, K. E. Wilkinson, P. Sukthankar, E. I. C. Beltrão and J. M. Tomich, Langmuir, 2017, 33, 7096–7104 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- L. A. Avila, L. R. M. M. Aps, N. Ploscariu, P. Sukthankar, R. Guo, K. E. Wilkinson, P. Games, R. Szoszkiewicz, R. P. S. Alves, M. O. Diniz, Y. Fang, L. C. S. Ferreira and J. M. Tomich, J. Controlled Release, 2016, 241, 15–24 CrossRef CAS PubMed.
- S. D. Patil, D. G. Rhodes and D. J. Burgess, AAPS J., 2005, 7, E61–E77 CrossRef CAS PubMed.
- G. Lambert, E. Fattal and P. Couvreur, Adv. Drug Delivery Rev., 2001, 47, 99–112 CrossRef CAS PubMed.
- D. R. Scoles and S. M. Pulst, RNA Biol., 2018, 15, 707–714 Search PubMed.
- D. Di Fusco, V. Dinallo, I. Marafini, M. M. Figliuzzi, B. Romano and G. Monteleone, Front. Pharmacol., 2019, 10, 305–312 CrossRef CAS.
- A. M. Rossor, M. M. Reilly and J. N. Sleigh, Pract. Neurol., 2018, 18, 126–131 CrossRef PubMed.
- K. S. Frazier, Toxicol. Pathol., 2015, 43, 78–89 CrossRef.
- C. Hardee, L. Arévalo-Soliz, B. Hornstein and L. Zechiedrich, Genes, 2017, 8, 65 CrossRef PubMed.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671 CrossRef CAS PubMed.
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Adv. Drug Delivery Rev., 2017, 115, 115–154 CrossRef CAS PubMed.
- Z. Yang, Z. Jiang, Z. Cao, C. Zhang, D. Gao, X. Luo, X. Zhang, H. Luo, Q. Jiang and J. Liu, Nanoscale, 2014, 6, 10193–10206 RSC.
- Z.-Y. He, F. Deng, X.-W. Wei, C.-C. Ma, M. Luo, P. Zhang, Y.-X. Sang, X. Liang, L. Liu, H.-X. Qin, Y.-L. Shen, T. Liu, Y.-T. Liu, W. Wang, Y.-J. Wen, X. Zhao, X.-N. Zhang, Z.-Y. Qian and Y.-Q. Wei, Sci. Rep., 2016, 6, 23764 CrossRef CAS.
- T. Boulikas, Encapsulation of plasmid DNA (lipogenes™) and therapeutic agents with nuclear localization signal/fusogenic peptide conjugates into targeted liposome complexes, Google Pat., 2016 Search PubMed.
- A. Dinari, T. T. Moghadam, M. Abdollahi and M. Sadeghizadeh, Sci. Rep., 2018, 8, 8112 CrossRef.
- H. A. Kim, K. Nam and S. W. Kim, Biomaterials, 2014, 35, 7543–7552 CrossRef CAS PubMed.
- H. O. McCarthy, J. McCaffrey, C. M. McCrudden, A. Zholobenko, A. A. Ali, J. W. McBride, A. S. Massey, S. Pentlavalli, K.-H. Chen and G. Cole, J. Controlled Release, 2014, 189, 141–149 CrossRef CAS PubMed.
- A. S. Massey, S. Pentlavalli, R. Cunningham, C. M. McCrudden, E. M. McErlean, P. Redpath, A. A. Ali, S. Annett, J. W. McBride, J. McCaffrey, T. Robson, M. E. Migaud and H. O. McCarthy, Mol. Pharmaceutics, 2016, 13, 1217–1228 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- J. Cheng, N. Gabrielson and Y. Lichen, Biomaterials, 2010, 31(34), 9117–9127 CrossRef.
- L.-Y. Wong, B. Xia, E. Wolvetang and J. Cooper-White, Biomacromolecules, 2018, 19, 353–363 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef CAS PubMed.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244 CrossRef CAS PubMed.
- V. Mikhalevich, I. Craciun, M. Kyropoulou, C. G. Palivan and W. Meier, Biomacromolecules, 2017, 18, 3471–3480 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- Y. Zhao, W. Yang, C. Chen, J. Wang, L. Zhang and H. Xu, Curr. Opin. Colloid Interface Sci., 2018, 35, 112–123 CrossRef CAS.
- D. Mandal, A. N. Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC.
- M. Rad-Malekshahi, L. Lempsink, M. Amidi, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2016, 27, 3–18 CrossRef CAS PubMed.
- S. M. Barros, S. K. Whitaker, P. Sukthankar, L. A. Avila, S. Gudlur, M. Warner, E. I. C. Beltrão and J. M. Tomich, Arch. Biochem. Biophys., 2016, 596, 22–42 CrossRef CAS PubMed.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Nano Lett., 2018, 18, 6832–6841 CrossRef CAS PubMed.
- D. de Bruyn Ouboter, T. B. Schuster, A. Mantion and W. Meier, J. Phys. Chem. C, 2011, 115, 14583–14590 CrossRef CAS.
- Q. Meng, Y. Kou, X. Ma, Y. Liang, L. Guo, C. Ni and K. Liu, Langmuir, 2012, 28, 5017–5022 CrossRef CAS PubMed.
- S. J. Sigg, V. Postupalenko, J. T. Duskey, C. G. Palivan and W. Meier, Biomacromolecules, 2016, 17, 935–945 CrossRef CAS PubMed.
- E. Haustein and P. Schwille, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 151–169 CrossRef CAS PubMed.
- N. Pal, S. D. Verma, M. K. Singh and S. Sen, Anal. Chem., 2011, 83, 7736–7744 CrossRef CAS PubMed.
- K. Koynov and H.-J. Butt, Curr. Opin. Colloid Interface Sci., 2012, 17, 377–387 CrossRef CAS.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- G. Gunkel-Grabole, S. Sigg, M. Lomora, S. Lörcher, C. Palivan and W. Meier, Biomater. Sci., 2015, 3, 25–40 RSC.
- L. E. Euliss, J. A. DuPont, S. Gratton and J. DeSimone, Chem. Soc. Rev., 2006, 35, 1095–1104 RSC.
- K. Y. Win and S.-S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS PubMed.
- K. McAllister, P. Sazani, M. Adam, M. J. Cho, M. Rubinstein, R. J. Samulski and J. M. DeSimone, J. Am. Chem. Soc., 2002, 124, 15198–15207 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- P. Prabu, A. A. Chaudhari, N. Dharmaraj, M. Khil, S. Park and H. Kim, J. Biomed. Mater. Res., Part A, 2009, 90, 1128–1136 CrossRef CAS PubMed.
- A. E. Nkodo, J. M. Garnier, B. Tinland, H. Ren, C. Desruisseaux, L. C. McCormick, G. Drouin and G. W. Slater, Electrophoresis, 2001, 22, 2424–2432 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- A. J. Convertine, D. S. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- A. M. Mohsen, M. H. Asfour and A. A. Salama, Drug Dev. Ind. Pharm., 2017, 43, 2043–2054 CrossRef CAS PubMed.
- N. Bishop, Rev. Med. Virol., 1997, 7, 199–209 CrossRef PubMed.
- D. Oupický, R. Carlisle and L. Seymour, Gene Ther., 2001, 8, 713 CrossRef PubMed.
- M. Thomas and A. Klibanov, Appl. Microbiol. Biotechnol., 2003, 62, 27–34 CrossRef CAS PubMed.
- M. Ogris, S. Brunner, S. Schüller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595 CrossRef CAS PubMed.
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316–5323 CrossRef CAS PubMed.
- T. B. Schuster, D. de Bruyn Ouboter, E. Bordignon, G. Jeschke and W. Meier, Soft Matter, 2010, 6, 5596–5604 RSC.
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko and M. Jayaraman, Nat. Nanotechnol., 2012, 7, 389 CrossRef CAS PubMed.
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Eur. J. Pharm. Biopharm., 2008, 69, 1–9 CrossRef CAS PubMed.
- L. Huang and E. Wagner, Non-viral Vectors for Gene Therapy, A Division of Harcourt, Academic Press, San Diego, 1999, p. 3 Search PubMed.
- S. Yamano, J. Dai, S. Hanatani, K. Haku, T. Yamanaka, M. Ishioka, T. Takayama, C. Yuvienco, S. Khapli and A. M. Moursi, Biomaterials, 2014, 35, 1705–1715 CrossRef CAS PubMed.
- R. Kircheis, L. Wightman and E. Wagner, Adv. Drug Delivery Rev., 2001, 53, 341–358 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- H. L. Åmand, K. Fant, B. Nordén and E. K. Esbjörner, Biochem. Biophys. Res. Commun., 2008, 371, 621–625 CrossRef PubMed.
- J. B. Rothbard, E. Kreider, C. L. VanDeusen, L. Wright, B. L. Wylie and P. A. Wender, J. Med. Chem., 2002, 45, 3612–3618 CrossRef CAS PubMed.
- C.-H. Tung and R. Weissleder, Adv. Drug Delivery Rev., 2003, 55, 281–294 CrossRef CAS PubMed.
- A. El-Sayed, S. Futaki and H. Harashima, AAPS J., 2009, 11, 13–22 CrossRef CAS PubMed.
- Y. Gao, Z. Xu, S. Chen, W. Gu, L. Chen and Y. Li, Int. J. Pharm., 2008, 359, 241–246 CrossRef CAS PubMed.
- X. Liu, C. Liu, J. Zhou, C. Chen, F. Qu, J. J. Rossi, P. Rocchi and L. Peng, Nanoscale, 2015, 7, 3867–3875 RSC.
- V. Bagnacani, V. Franceschi, M. Bassi, M. Lomazzi, G. Donofrio, F. Sansone, A. Casnati and R. Ungaro, Nat. Commun., 2013, 4, 1721 CrossRef PubMed.
- H. Qin, Y. Jiang, J. Zhang, C. Deng and Z. Zhong, Mol. Pharmaceutics, 2019, 16(8), 3711–3719 CrossRef CAS PubMed.
- E. R. Silva, G. Cooney, I. W. Hamley, W. A. Alves, S. Lee, B. F. O'Connor, M. Reza, J. Ruokolainen and D. Walls, Soft Matter, 2016, 12, 9158–9169 RSC.
- A. El-Sayed, I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2008, 283, 23450–23461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01990a |
| This journal is © The Royal Society of Chemistry 2020 |