
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Potential and limits of a colloid approach to protein solutions
Anna
Stradner
 *ab and
Peter
Schurtenberger
ab
*ab and
Peter
Schurtenberger
ab
aDivision of Physical Chemistry, Department of Chemistry, Lund University, PO Box 124, SE-221 00 Lund, Sweden. E-mail: anna.stradner@fkem1.lu.se
bLINXS – Lund Institute of advanced Neutron and X-ray Science, Scheelevägen 19, SE-223 70 Lund, Sweden
First published on 5th December 2019
Abstract
Looking at globular proteins with the eyes of a colloid scientist has a long tradition, in fact a significant part of the early colloid literature was focused on protein solutions. However, it has also been recognized that proteins are much more complex than the typical hard sphere-like synthetic model colloids. Proteins are not perfect spheres, their interaction potentials are in general not isotropic, and using theories developed for such particles are thus clearly inadequate in many cases. In this perspective article, we now take a closer look at the field. In particular, we reflect on the fact that modern colloid science has been undergoing a tremendous development, where a multitude of novel systems have been developed in the lab and in silico. During the last decade we have seen a rapidly increasing number of reports on the synthesis of anisotropic, patchy and/or responsive synthetic colloids, that start to resemble their complex biological counterparts. This experimental development is also reflected in a corresponding theoretical and simulation effort. The experimental and theoretical toolbox of colloid science has thus rapidly expanded, and there is obviously an enormous potential for an application of these new concepts to protein solutions, which has already been realized and harvested in recent years. In this perspective article we make an attempt to critically discuss the exploitation of colloid science concepts to better understand protein solutions. We not only consider classical applications such as the attempt to understand and predict solution stability and phase behaviour, but also discuss new challenges related to the dynamics, flow behaviour and liquid–solid transitions found in concentrated or crowded protein solutions. It not only aims to provide an overview on the progress in experimental and theoretical (bio)colloid science, but also discusses current shortcomings in our ability to correctly reproduce and predict the structural and dynamic properties of protein solutions based on such a colloid approach.
 Anna Stradner | Anna is a Professor at the Division of Physical Chemistry at Lund University specializing in biocolloids studied by scattering techniques. After her PhD in Physical Chemistry at the University of Graz/Austria she worked in the Soft Condensed Matter Group in Fribourg/Switzerland on (bio)colloidal suspensions using neutron, X-ray and light scattering. In 2008 she obtained the ‘Venia Legendi’ in Experimental Physics and became head of the Food and Bioscience Group at the Adolphe Merkle Institute/Switzerland until she transferred her activities to Sweden in 2011. Her current research focuses on protein solutions with a particular interest in protein dynamics under crowded conditions. |
 Peter Schurtenberger | Peter Schurtenberger is a Professor in Physical Chemistry at Lund University. He received his PhD from the ETH in Zurich in 1984. After working as a postdoc at Lund University, Sweden, MIT and Harvard University Medical School, he became a senior researcher at the Department of Materials of ETHZ, was appointed as the chair in experimental soft matter physics at the University of Fribourg in 1999, became the founding director of the Adolphe Merkle Institute in 2008, and moved to Lund University in 2010. His research interests focus on soft matter, biophysics, nanotechnology, materials sciences, on the characterization of soft and biological matter with light, X-ray and neutron scattering, and on the development of new instruments for this task. |
1 Introduction
The use of a colloid approach to understand protein solution behavior is not a very recent development. In fact, a number of the early founders of colloid science have treated synthetic and bio-colloids such as proteins alike, adhering thereby to the early definition of colloids solely based on the size of the dispersed or dissolved colloidal matter.1–6 The subsequent enormous advancement of colloid science, or rather soft matter science, was then largely based on fundamental work related to synthetic colloids such as the ubiquitous polystyrene (PS) or poly(methyl methacrylate) (PMMA) spheres.7–10 It is only more recently that colloid science has moved again back into the focus of the protein community. This was primarily driven by the lack of an understanding of the theoretical foundation of protein crystallization and the need to design improved approaches to crystallize proteins and obtain their high resolution structure through X-ray and neutron based crystallography. The pioneering work of George and Wilson11 and Muschol and Rosenberger12 linked the ability of proteins to crystallize with overall measures of their interaction potential such as the second virial coefficient B2, and their findings were then connected to theoretical and simulation work on the relationship between the interaction potential and the resulting phase behavior.13–16 The strong similarities between the available experimental phase diagrams of globular proteins and those obtained for colloids interacting via a combination of a hard sphere repulsion and a short-range attraction were realized,15,17 and a generic phase diagram for globular proteins was proposed that provided general rules for finding optimal crystallization conditions.16,18 Particularly important was also the development of an extended law of corresponding states, which further supported the universality of this generic phase diagram and provided a simple link between the location of the binodal and a reduced second virial coefficient B2* = B2/B2,HS, where B2,HS is the corresponding hard sphere second virial coefficient, as an effective temperature irrespective of the detailed shape of the interaction potential.19Since these pioneering developments in the mid 90's, we have seen a renaissance in the use of the experimental and theoretical toolbox of colloid science in order to understand various solution properties of globular proteins. While protein phase diagrams and crystallization was initially in the focus, this colloid approach to proteins was subsequently extended to a much wider range of topics that included protein self-assembly and aggregation, the stability of crowded protein solutions and mixtures, and the dynamics of strongly interacting protein solutions.20–31 However, this approach has recently been questioned in an article by Sarangapani et al.,32 where the authors concluded that the proteins' capacity to adjust their shape and thus the nature of the interprotein interactions in response to variations in pH and concentration severely limits the usefulness of colloid models to describe the structural and dynamic properties of protein solutions. They also pointed out that proteins are macromolecular entities rather than rigid colloidal particles, and suggested that one should try to consider links to synthetic polymer and polyelectrolyte solutions. The article was enthusiastically received in a “new and notable” article in the same journal by Prausnitz,33 who wrote that “now, after the report of Sarangapani et al., the colloid like theory is dead; the Sarangapani group have delivered a coup de grace. We can take comfort in the remark of Sarangapani et al. that, while scientifically erroneous, the colloid like theory may nevertheless be useful for some purposes in biotechnology. Thank you! That's like saying even a placebo can sometimes cure an illness.”
Does this mean that we no longer should try to use colloid models when attempting to understand and predict various properties of protein solutions? We believe that this is not the case, and that this rather reflects a too narrow and outdated view of what colloids and colloid models are, and in particular neglects the many exciting developments that have happened in the area of synthetic colloids in recent years. While colloidal hard spheres have been very important in creating the foundation of modern colloid science, we have seen a multitude of anisotropic, patchy and/or responsive colloids emerging that we believe are highly relevant as model systems for proteins. It is common knowledge that protein interactions depend on solution parameters such as pH, ionic strength or temperature, but so do colloid interactions. Proteins are indeed biological macromolecules, but one would expect polymer- or polyelectrolyte behavior only for completely denatured proteins, which we do not consider here. While conformational changes in proteins can obviously happen as a function of pH or other external variables, the globular nature of the overall protein (tertiary) structure remains, and will require models that are colloid rather than polymer/polyelectrolyte based. The question of polymer vs. colloid nature will certainly become much more important in the current focus on intrinsically disordered proteins (IDPs).34
However, it is also clear that much more emphasis will have to be given to the anisotropic and patchy nature of protein interactions and its influence on the various structural and dynamic properties of protein solutions. In this article we try to give a brief overview of recent developments in colloid science that may be of importance for protein solutions, and point out a number of topics where such improved colloid models could have a considerable impact in our strive for understanding and predicting protein solutions. We restrict our discussion to a small number of selected topics where we feel a particular need for improvement, and a large potential for profiting from parallel developments in colloid and protein science: (i) improved models for interparticle interactions and the resulting phase behavior; (ii) characterizing and understanding colloid and protein dynamics in dense systems, and the importance of appropriate characterization and simulation tools; (iii) self-association and its consequences on viscosity and dynamical arrest. These topics are chosen for their importance in understanding complex biological systems and processes as well as in view of the needs of pharmaceutical industry for the development of future high concentration drug formulations.
2 Current trends in colloid science – from hard spheres to anisotropic and patchy particles
Much of the progress made in understanding the structural and dynamic properties of colloidal suspensions is a result of their frequent use as ideal model systems to address fundamental issues in condensed matter physics such as liquid ordering, crystallization and glass formation, and the corresponding structural and dynamic properties of the various systems as a function of the interaction potential.8 The initial work had primarily focused on the use of hard sphere-like colloids, which was instrumental in experimentally demonstrating the existence of an entropically driven crystallization in a purely hard sphere-like system,7 and in developing a new research thrust focusing on glass transition, dynamic arrest and jamming.35–38 Subsequently the field has seen a dramatic increase in the complexity of colloids and colloidal interactions, and there were in particular four developments that are especially important for the topic of this article: the importance of short-range attractions on colloid phase behavior;8,13,14,39 equilibrium cluster formation in systems with mixed potentials, combining a long-range soft repulsion and a short-range attraction;25,40–42 the development of anisotropic particles,43–48 and finally the dramatic consequences of patchy interactions on phase behavior, self-assembly and colloid dynamics.31,43,45,49–51 As we will see below, these recent developments are also of prime importance for our understanding of protein solutions, and we therefore briefly summarize the most important features.The investigation of non-equilibrium phenomena in colloidal suspensions has clearly emerged as one of the most important fields of soft matter research. Topics such as dynamical arrest or jamming in suspensions of (weakly) short-range attractive colloids, the interplay between spinodal decomposition and glass formation or the existence of a metastable liquid–liquid binodal have had a considerable attention from the experimental and theoretical soft matter community.39,54,55 A schematic view of our current understanding is given in Fig. 1.
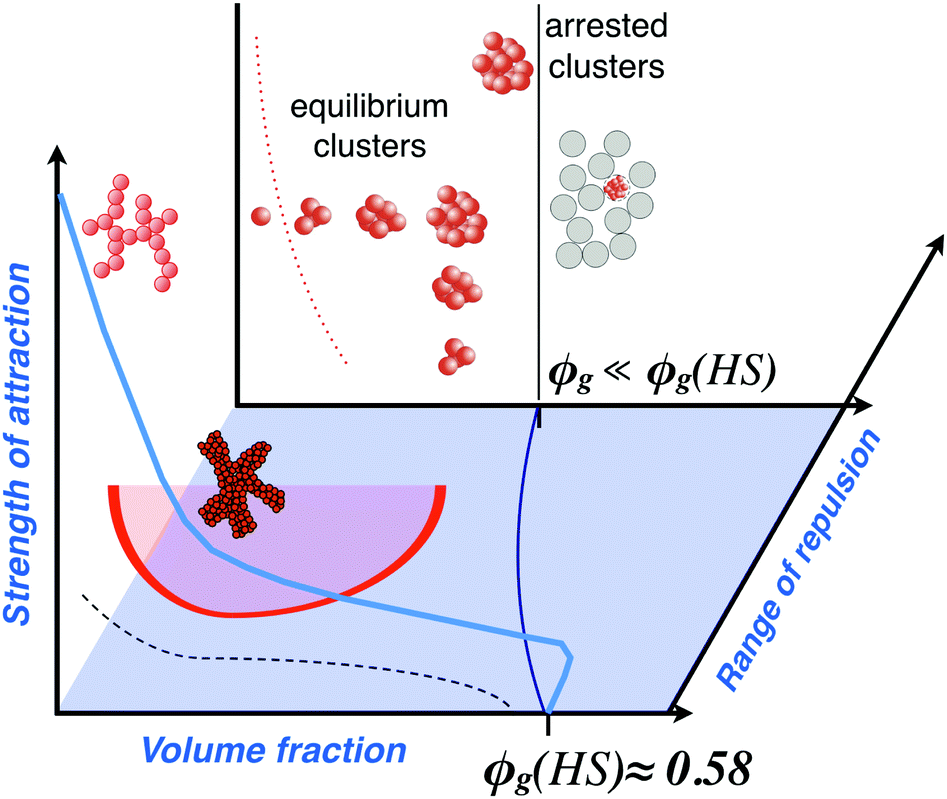 | ||
| Fig. 1 A schematic state diagram for colloids interacting via short-range attractions and long-range (screened Coulomb) repulsions. Shown in the volume fraction/attraction strength plane at zero electrostatic repulsion are the freezing or crystallization line (dashed black line) and the arrest line (solid light blue line), which in the case of no attractions occurs at a volume fraction of about ϕg(HS) ≈ 0.58, where ϕg(HS) corresponds to the glass transition for hard sphere suspensions. Highlighted in red is also the binodal line denoting the metastable liquid–liquid phase separation in short-range attractive systems, which can lead to interesting new arrested states when in competition with the arrest or gel-line. The dark blue line in the volume fraction/range of repulsion plane at zero attractions (blue shaded) denotes the arrest line that moves to lower volume fractions as the electrostatic repulsion is turned on. In the volume fraction/attraction strength plane at long-range electrostatic repulsions, where the range of the repulsion is significantly larger than the attraction range, we find the formation of equilibrium clusters, with a monomer-cluster transition (red dotted line) moving to lower volume fractions as the attraction strength increases, and a cluster glass transition at higher volume fractions (black line). The phase behaviour of systems where the mixed potential consists of attractions and repulsions with comparable range or longer-ranged attractions is even more complex and a recent attempt to describe this in a generic state diagram can be found in ref. 52. Adapted and modified from ref. 53 with permission from The Royal Society of Chemistry. | ||
For ideal hard sphere particles, we first observe a transition from a liquid to a crystal phase, followed by a disordered solid phase, a glass, at volume fractions of approximately ϕ ≈ 0.58.7,35,37 If a weak and short-ranged attraction is now turned on, this leads to the astonishing observation of a melting of the glass, followed by a so-called re-entrant glass or solid formation at even stronger attractions.39,54 For very strong interparticle attractions, we reach the regime of so-called irreversible aggregation, where soft fractal gels form already at very low volume fractions.56–58 At intermediate strength of the attraction, the situation is even more complicated due to the fact that phase separation into a dilute (gas-like) and a concentrated (liquid-like) suspension can occur, and this can subsequently lead to the formation of a long-lived 'interaction network' of particles.59
Colloids frequently carry charges, and thus interact via a screened Coulomb repulsion. When combined with a short-range attraction, new states appear, the metastable liquid–liquid phase separation is suppressed and we observe the formation of (transient) equilibrium clusters.25,40–42 Particularly interesting in this context is the fact that such clusters were found to undergo an arrest transition from a cluster fluid to a cluster glass that occurs at a significantly lower volume fraction when compared to the hard sphere glass at ϕg ≈ 0.58.41,60,61 Phase behavior and the occurrence of dynamical arrest upon an increase in concentration is indeed of prime importance in protein solutions in a wide range of areas, and the contents of Fig. 1 will thus reappear again below.
Another very important aspect in this context is shape anisotropy, as proteins in general are far from being perfect spheres. In recent years we have seen a wealth of colloid synthesis strategies, leading to a spectacular array of colloids of different shapes, compositions and functionalities (see Fig. 2 for some key examples).43,46,62,65–69 Together with similar advances in computer simulations of anisotropic particles, they have improved our understanding of the influence of anisotropy in both shape and interactions on aggregation, self-assembly and phase behaviour in colloidal suspensions.43,65,69–72 However, most of the work has been devoted to phase behavior and self assembly, and there is still a lack of knowledge on the dynamics of anisotropic particles, and the influence of shape anisotropy on dynamical arrest. Particles with moderate shape anisotropy such as ellipsoids with axial ratios of order 1–4 are particularly relevant for comparisons with globular proteins. While there are some simulation as well as experimental studies focusing on various aspects of the dynamics of ellipsoids at high concentrations, most of the experimental work is on (quasi-) 2D systems, with only a few notable exceptions on 3D bulk behavior.73–79 Progress on the experimental side is primarily hindered by the fact that most existing model systems for such ellipsoidal particles are highly turbid at high concentrations, thus making an investigation of dynamic properties such as the collective or self diffusion coefficient or the particle mean square displacement very difficult or impossible with standard optical tools. It is only through the use of recently introduced techniques such as differential dynamic microscopy or X-ray photon correlation spectroscopy that these properties have become accessible at high concentrations close to the glass transition for these model ellipsoids also.63,79
 | ||
| Fig. 2 Examples for the recent developments in the synthesis of anisotropic particles. Top: Examples of anisotropic colloids. Ellipsoidal core shell and cubic hematite particles (reprinted and adapted with permission from ref. 62; Copyright (2008) Schweizerische Chemische Gesellschaft); peanut-shaped core–shell particles (reprinted and adapted with permission from ref. 63. Copyright (2018) WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim); faceted and bowl-shaped core–shell microgel particles (reprinted and adapted from ref. 46 with permission from The Royal Society of Chemistry). Bottom: Patchy colloids and colloidal molecules. Bottom left: Colloidal clusters made of different thermoresponsive particles that form structures similar to multi-patch particles, where the different patches can change from overall repulsive to overall attractive as a function of temperature (reprinted with permission from Peng64). Bottom right: Examples of so-called reconfigurable colloidal molecules formed by thermoresponsive particles with complementary shapes that can reversibly assemble into molecule-like clusters, and change configuration as a function of temperature. Shown are confocal laser scanning micrographs (CLSM) and schematic representations illustrating the specific self-assembly in colloidal molecules with a valency of 4 at 20 °C (methane, CH4; top). Increasing the temperature to 40 °C, the valency of the assembly decreases to 2 (carbon dioxide, CO2; middle). At 48 °C, the assembly resembles a dihydrogen, H2, configuration (bottom) (reprinted from ref. 48). | ||
During the last 2 decades, enormous experimental and theoretical effort has also been devoted to the design and synthesis and to the understanding of the phase behaviour of so-called patchy colloidal particles, i.e. particles with site-specifically engineered surfaces (see Fig. 2 for some key examples).43,51,82–85 As a consequence of their surface patchiness these particles can interact via directional and specific interactions. Both experiments and simulations demonstrate that this can result in a considerably more complex phase and self-organization behaviour than in colloids exhibiting purely isotropic interactions. This has recently been discussed in a comprehensive review article on the programmed self-assembly of patchy particles by Duguet et al.,84 where the enormous variety of available particles and the resulting phase behavior is illustrated.
However, while there is a large and still rapidly increasing number of publications focusing on patchy particles, the vast majority are either simulation studies of their self-assembly behavior, descriptions of new synthesis routes or of the self-assembly patterns found with these particles. There is a clear lack of studies providing an experimental verification of the many predictions on the phase behavior of patchy colloids, and virtually no work at all is devoted to the consequences of patchy interactions on the dynamics of these suspensions. This is particularly important for an extension of the colloid approach to protein solutions beyond classical spherical particles with centrosymmetric potentials, as this currently limits our ability to use less coarse-grained models that are based on the molecular structure of proteins for a calculation of key structural and dynamic properties and their dependence on solution parameters such as the ionic strength, temperature, pH or protein concentration.
With these recent developments in colloid science in mind, we will now survey the current trends in the application of concepts and models from colloid science to proteins, and point out the current limits and future prospects of such an approach. This discussion basically centers on the question of the optimal coarse-graining strategy necessarily required to allow for a quantitative and predictive understanding of the various aspects of protein solutions. As schematically described in Fig. 3, this requires a strategy where sufficient molecular details are retained in order to allow for a quantitative reproduction of the particular quantity aimed for, while limiting the computational costs to make the necessary calculations or simulations feasible.
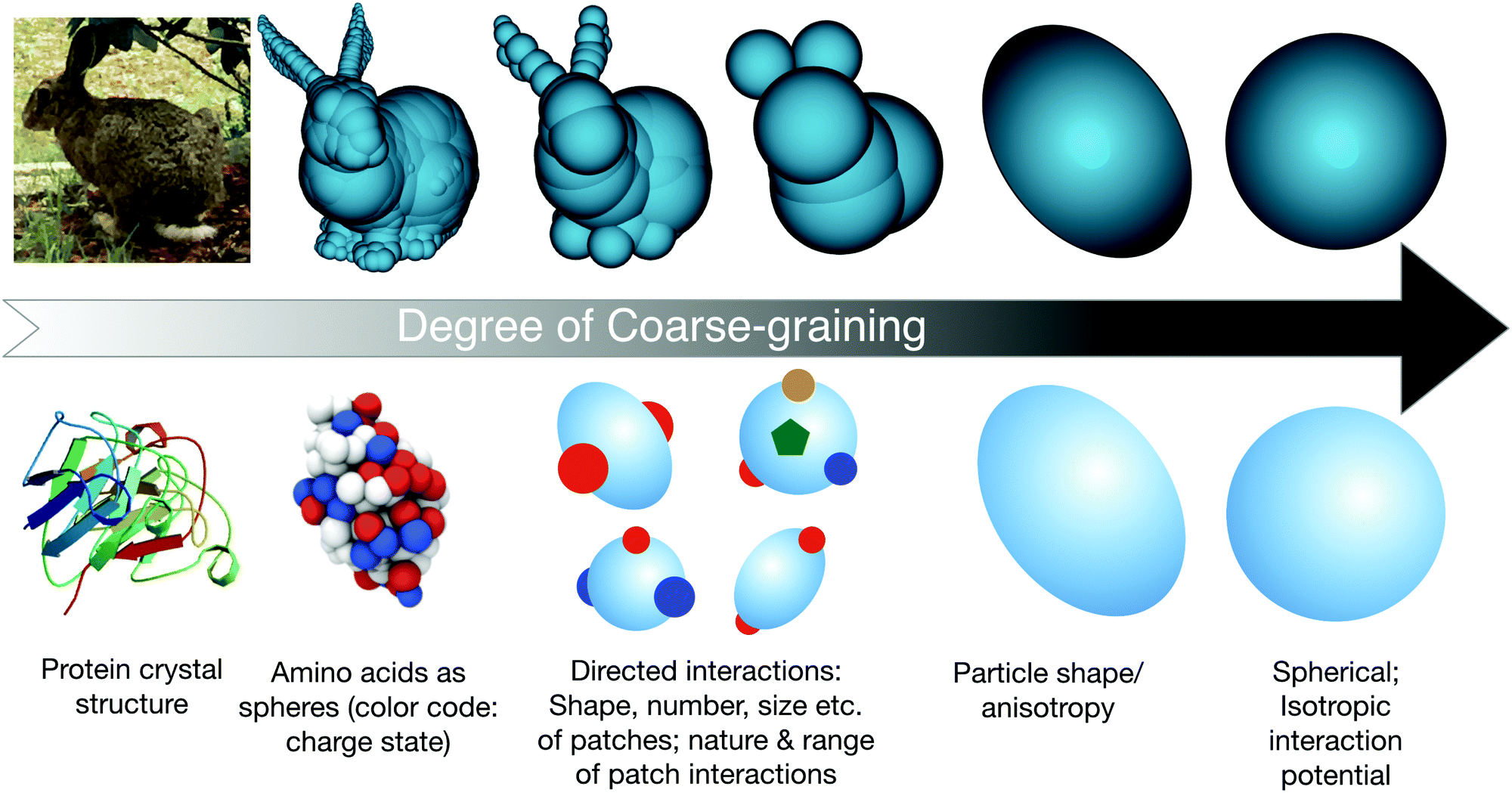 | ||
| Fig. 3 Schematic view of potential steps in the coarse-graining procedure for modeling globular proteins. The bunny figures (top row, sketch 2, 3 and 4 from left to right) are taken from the website: Gareth Bradshaw, February 2003, “Sphere-Tree Construction Toolkit”, http://isg.cs.tcd.ie/spheretree/. Bottom row, first sketch from left: image of the bovine eye lens protein γB-crystallin from the RCSB PDB (rcsb.org) of PDB ID 1AMM from ref. 80. Bottom row, second sketch from left: reprinted with permission from ref. 81. Copyright (2012) American Chemical Society. | ||
3 Protein phase behavior
The stability of protein solutions is vital for biological functionality or drug formulation using so-called biologics (or biopharmaceuticals), and globular proteins are generally well optimized against non-functional interactions.86 Although there is recent and exciting evidence that phase transitions in living cells may in fact represent an important mechanism in cellular organization,87 protein phase separation has often been related to the occurrence of so-called protein condensation diseases.88,89 The connection between liquid–liquid phase separation in solutions of eye lens proteins and the formation of cataract, still one of the leading causes of blindness worldwide, has initially spurred a systematic study of the phase diagrams of various representatives of the γ-crystallin family by Benedek and collaborators.88,90–96 At the same time, this group also extended the investigation of protein liquid–liquid phase separation to another key model protein, lysozyme, for which early work had already indicated the existence of critical phenomena,97 and found very similar behavior.98 In addition to providing a first systematic study of liquid–liquid phase separation in globular proteins, these studies also provided evidence that the resulting binodal is actually metastable with respect to the liquid–crystal transition.91,99In parallel, work on the phase behavior of colloid–polymer mixtures indicated that the gas–liquid transition could become metastable with respect to the liquid–solid transition, provided that the size ratio polymer/colloid was sufficiently small (≲0.25) to render the depletion interaction between the colloids short-range.13,14,100 The analogy between the phase behavior of colloids with short-range attractions and globular proteins exhibiting metastable liquid–liquid phase separation was quickly realized, and immediately explored to advance the difficult and tedious search for solution conditions favorable for growing protein crystals.15,16,20,21,29
This development was motivated by the pressing need for high quality protein crystals for determining protein structures through X-ray crystallography, and the notorious difficulties in finding appropriate solution conditions for the growth of such crystals. The striking similarity of the known phase diagrams of different globular proteins, and the discovery of George and Wilson11 that the second virial coefficient B2 of proteins that could successfully be crystallized were all found in a narrow range of slightly negative values, provided the initial scientific basis.18,101 Another key ingredient was the “extended law of corresponding states” proposed by Noro and Frenkel,19 which indicated that the reduced second virial coefficient B2* could be used as an effective parameter to quantify the strength of the short-range attraction and locate the gas–liquid and liquid–solid phase boundaries.102 All these individual puzzle stones were then put together to a picture, in which globular proteins were believed to interact in a way well described by a (centrosymmetric) short-range attractive potential. For such potentials, B2* then largely determines the location of the binodal and the liquid–solid phase boundary for a given protein system, and crystallization is favoured in a crystallization window between the liquid–solid boundary and the binodal, and at concentrations below an arrest or gel line expected above the critical concentration (Fig. 4).16,20,29 This scenario provides a rational approach to protein crystallization, where a measurement of B2* and its dependence upon various solution conditions would allow for a relatively rapid screening to find optimal nucleation and growth conditions. However, has this advancement in understanding of the physics of protein phase behavior and crystal formation indeed changed the picture? Unfortunately, growing high quality protein crystals still largely happens via a trial-and-error approach, and we lack predictive understanding of the link between the protein composition and the choice of the optimal solution composition in order to control the kinetics of nucleation and growth. Reality is obviously much more complex, and a lower degree of coarse-graining in the models used to describe protein solutions is clearly required. While fully atomistic simulations of a few hundred proteins have been performed,103,104 determining phase diagrams requires hundreds of simulations, which is not yet feasible. We therefore need to find an appropriate coarse-graining strategy that is optimized for the problem at hand.
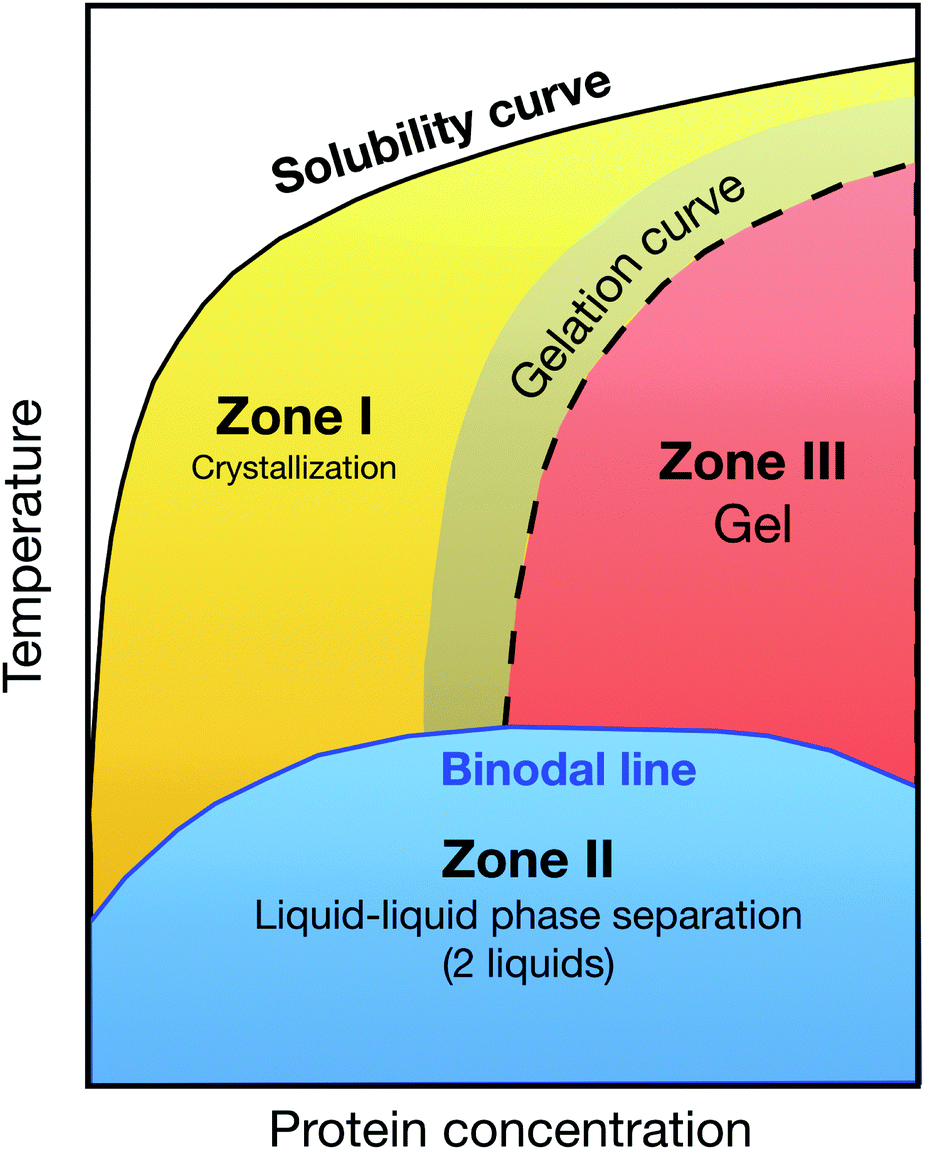 | ||
| Fig. 4 Generic state diagram for globular proteins postulated by Muschol and Rosenberger in their pioneering work from 1997:16 the coloured zones below the solubility curve are metastable with respect to crystallization. Below the (metastable) binodal line, protein solutions phase separate into a concentrated and dilute solution (zone II, blue shaded area). The optimal region for protein crystallization is the yellow shaded area in zone I, while zone III is prone to gelation. Redrawn after ref. 16. | ||
At this point it is thus helpful to step back and look again more carefully at the existing data. While the available data on experimental phase diagrams is limited to few proteins such as lysozyme and different variants of γ-crystallin only, it covers a variety of different solution conditions. Overall, the similarities between binodals of short-range attractive colloid models such as a square-well (SW) fluid are striking.106 However, a closer look has revealed distinct differences between the measured and calculated binodals for a SW model, and required the use of patchy colloid models with directional interactions to achieve much more quantitative agreement with experimental data.105,107,108 This is illustrated with data for lysozyme shown in Fig. 5 (top), which clearly shows that the isotropic SW potential cannot reproduce the relatively wide binodal, which is much better approximated by the patchy colloid model.105 Given the complex surface of globular proteins, this is not really surprising. Similarly, while colloids with isotropic potential form dense crystal structures such as face-centered cubic (fcc), body-centered cubic (bcc) or hexagonal close-packed (hcp), protein crystal structures are generally much more open.109 Again, these type of structures can indeed be reproduced using patchy models.110 On the other hand, almost quantitative agreement with the ELCS was found when using an effective protein size σeff that includes contributions from repulsive interactions such as screened charges in the normalization of the measured B2 values, as shown in Fig. 5 (bottom).106 Where does that leave us in our attempt to design a coarse-graining strategy?
 | ||
| Fig. 5 Phase behavior of globular proteins. Top: Experimental phase diagram of lysozyme at high ionic strength (pH = 7.8; 500 mM NaCl) compared to the predictions from a centrosymmetric and patchy colloid model, respectively. The symbols indicate the experimental data for the fluid-crystal coexistence (or solubility) curve (filled triangles), the metastable binodal (open circles) and the metastable spinodal (open squares). The two dashed curves show the calculated binodal and spinodal from a centrosymmetric colloid model, while the two solid curves describe the calculated binodal and spinodal applying a patchy colloid model (for details of the model see ref. 105). Region I corresponds to a stable fluid phase, region II to a fluid-crystal coexistence, region III a metastable gas–liquid coexistence and region IV a crystalline phase. Reprinted and adapted from ref. 105, with the permission of AIP Publishing. Bottom: A comparison of the experimentally determined phase behavior of lysozyme for a broad set of experimental conditions with predictions for an adhesive hard sphere (AHS) and a short-range attractive square well (SW) potential, and a test of the applicability of the extended law of corresponding states, ELCS, as proposed by Noro and Frenkel19 (NF). Note that here a reduced second virial coefficient b2* instead of the normalized second virial coefficient B2* is used for the y-axis, where the protein size is replaced by an effective diameter σeff in the calculation of the hard sphere value B2,HS in order to also include other repulsive interactions caused by screened charges. Reprinted and adapted from ref. 106, with the permission of AIP Publishing. | ||
Given the large effects that patch number, size and strength can have on the phase diagram of patchy particles,83 one would not expect that the corresponding binodals for different proteins and solution conditions would follow an ELCS even remotely. However, dramatic changes occur for low numbers of strongly attractive and small patches.83,112 In proteins, this primarily corresponds to conditions where strong specific interactions lead to the formation of dimers, larger oligomers or other molecular complexes, which may in fact suppress phase separation that would otherwise proceed via weaker non-specific attractions.113,114 On the other hand, simulations for patchy particles with different patch interactions show that the binodals of particles with the same patch distribution, but different types of patches, indeed collapse when plotted as a function of the normalized second virial coefficient B2*.110,115 This indicates that we may indeed be able to use a simple measurement of B2* as a quite robust parameter to estimate the location of a critical point and binodal for globular proteins, and their dependence upon variations in solution conditions. This is of course good news for example for formulation science. On the other hand, however, when looking at the behavior of patchy particles, the situation of crystallographers is much less fortunate.29,116 We obviously need much more detail in the anisotropic interaction potential and the underlying coarse-grained structural model of the protein to successfully reproduce or predict protein crystal formation. There were a number of attempts to develop a detailed angularly dependent interaction potential based on known crystal structures in order to calculate B2*, investigate protein assembly, or gain insight into protein crystallization.81,117–121 However, this requires already that the protein structure is known. Moreover, using fully detailed all-atomistic simulations is still out of question for studies that require a larger number of proteins such as in attempts to calculate phase diagrams, or study structural and dynamic properties of concentrated solutions and mixtures. Here we need improved approaches to define the salient features of interactions between given proteins and translate them into an appropriately coarse-grained patchy model. Moreover, we also need much more work on various processes such as transient complex formation, nucleation and growth in patchy systems, and the influence of internal flexibility in protein crystal formation.
So far we have concentrated on equilibrium phase diagrams for colloids and proteins. As discussed previously, non-equilibrium phenomena such as glass and gel formation are of key importance in these systems, and have attracted considerable attention from the community (Fig. 1). In the context of model colloid studies, it has also been suggested that the arrest line for short-range attractive colloids should also scale with B2*, and a measurement of B2* should thus allow for an estimate of the volume fraction at which arrest should occur for a given temperature or strength of attraction.122 Dynamical arrest as a result of an arrested spinodal decomposition in globular proteins has been well studied for lysozyme.53,59,111 However, while the binodal lines for lysozyme at different ionic strength can be rescaled according to the Noro–Frenkel ELCS, the arrest lines that characterize the location of the attractive glass or gel line below the binodal do not follow the ELCS, but seem to depend on temperature only (Fig. 6).111 This is in contrast to both the experimental work in ref. 122 as well as Brownian and Newtonian computer simulations in ref. 123. In both cases, the particles chosen were purely short-range attractive, whereas lysozyme under the conditions chosen to look at an arrested spinodal decomposition has a mixed potential, with a short-range attraction as well as a screened Coulomb repulsion (Fig. 6). In fact, using idealized mode coupling theory (MCT) with such a mixed potential indeed reveals a behavior where the attractive glass transition is temperature-dependent only, and thus does not exhibit a B2* scaling.111 This indicates that for interactions typical for proteins in aqueous solutions we may not be able to construct and use a generic state diagram that captures both equilibrium phase behavior as well as non-equilibrium arrest transitions.
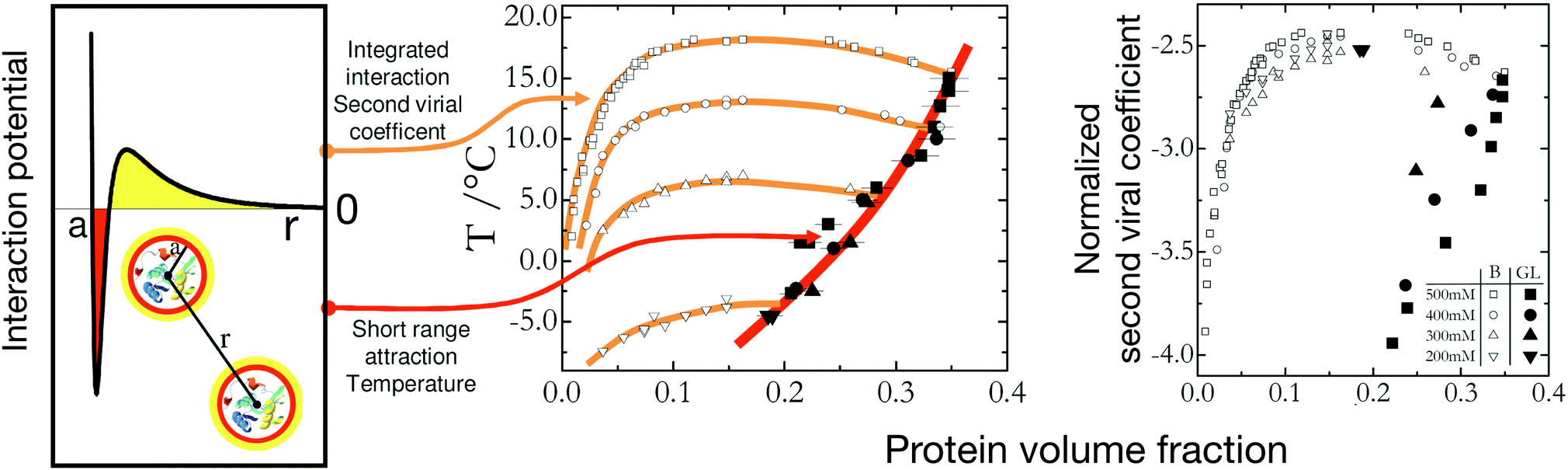 | ||
| Fig. 6 Effect of interaction potential details on the liquid–liquid phase separation and dynamical arrest in lysozyme solutions. The lysozyme state diagram with the metastable binodal (open symbols) and the dynamical arrest lines (full symbols) for different ionic strengths (from 200 mM (inverse triangles) to 500 mM (squares)) is represented in two different ways. Middle: Plotted as temperature versus protein volume fraction the dynamical arrest lines for different ionic strengths all overlap, indicating that they are primarily determined by the contact potential (indicated in red in the potential on the left) and rather insensitive to the longer-ranged repulsive part of the potential. Right: In agreement with the ELCS, the binodal lines all overlap when plotted using the normalized second virial coefficient as an effective temperature and measure for the integral features of the interaction potential. Lines are guides to the eye. Reprinted and adapted from ref. 111 with permission from The Royal Society of Chemistry. | ||
Until now we have concentrated on globular proteins only. However, other classes of proteins have gained enormous attention from the community, primarily for reasons connected to the formulation of novel biopharmaceuticals. Prime examples are monoclonal antibodies (mAbs) such as immunoglobulin gamma (IgG), which are considered as a major platform for potential drug candidates.124,125 However, successful mAb applications require stable and low viscosity high concentration formulations, which are often difficult to achieve as mAbs are prone to exhibit reversible self-association at high concentrations that result in enhanced viscosity, and are also known to exhibit high turbidity due to the presence of liquid–liquid phase separation.45,126–130 A number of studies have made attempts to characterize cluster formation in mAb solutions, and to interpret antibody solution properties through analogies with colloids.129–137 This is by no means straightforward due to the non-spherical shape and internal flexibility of mAbs, since interactions between proteins are frequently treated based on spherical approximations, and in particular the enormous effect that specific, directional interactions can have are often not considered.
The binodals observed for mAbs differ substantially from those observed from globular proteins. Their critical concentrations (Cc) and temperatures (Tc) typically are lower than those of globular proteins, and the binodal and spinodal is wider, resembling those of low valency patchy particles. This is illustrated in Fig. 7 (top), which shows a comparison between the measured binodals for a mAb, two different γ-crystallins and lysozyme.138 All binodals are rescaled with their values of Tc and Cc, respectively. There are indeed a number of quite successful attempts to use patchy particle theory to model protein interactions and the resulting self-assembly and phase behavior.129,130,135 This is demonstrated in Fig. 7 (bottom), which shows a comparison between calculated and measured binodals for two different mAbs.129 However, there is still need for significant improvements in relating model parameters to the actual composition and structure of the mAb and the solution, and we also need more work on establishing the impact of the high flexibility of the mAb structure.
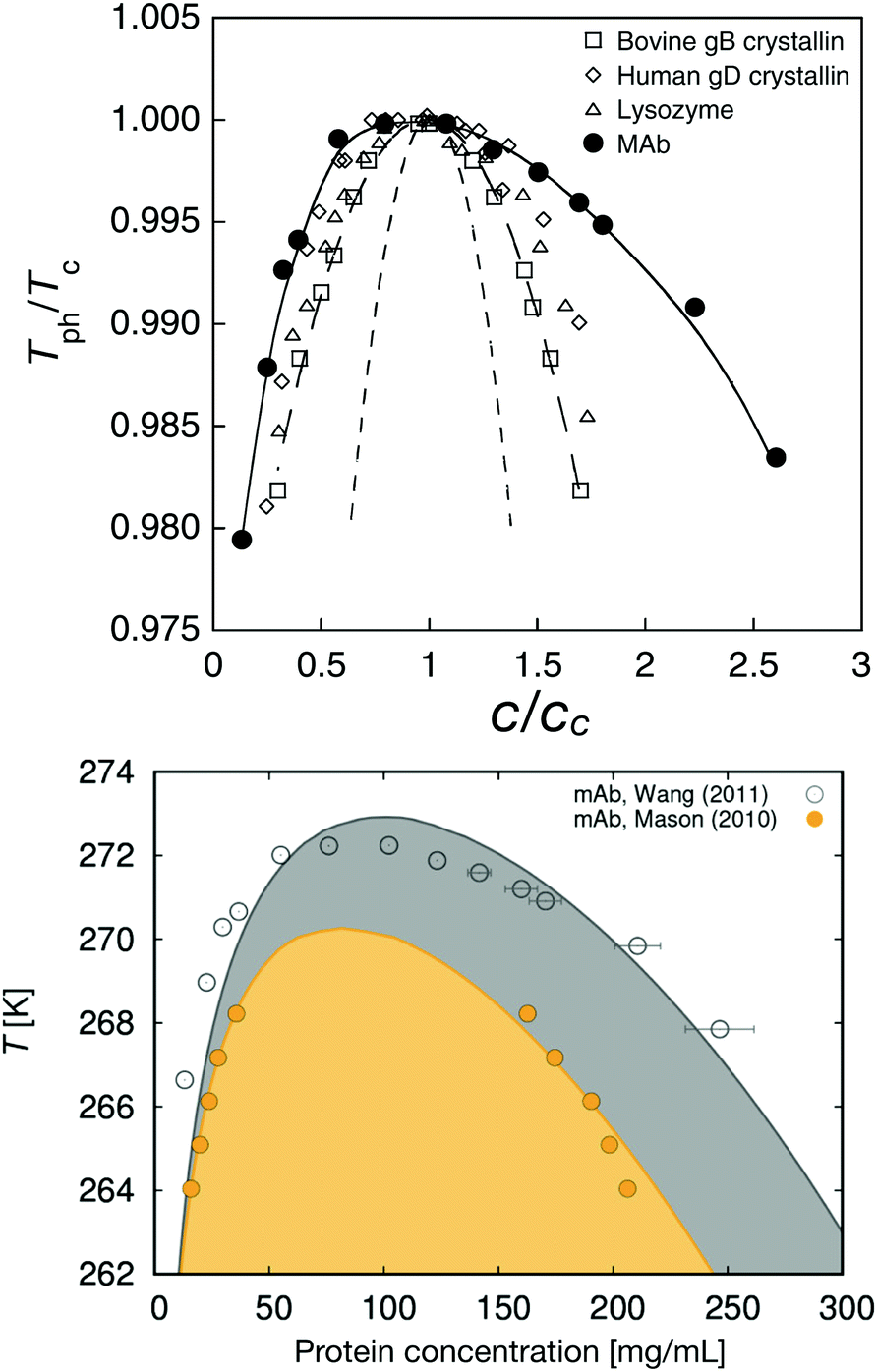 | ||
| Fig. 7 Phase behavior of monoclonal antibodies. Top: Rescaled coexistence curves for four different proteins where the phase separation temperature Tph is scaled with the critical temperature Tc, and the protein concentration c is scaled with the critical concentration cc. The short-dashed line shows the theoretical coexistence curve of spherical particles using a mean-field approximation for the attraction and the Carnahan-Starling expression for the hard sphere entropy contribution. The long-dashed line shows the theoretical fit for the data of bovine γB-crystallin (open squares) while the solid line is a guide to the eye for the coexistence curve of a monoclonal antibody (mAb; solid circles). Data points on the coexistence curves of human γD-crystallin (open diamonds) and chicken egg white lysozyme (open triangles) are also shown. Reprinted with permission from ref. 138. Bottom: Coexistence curves as a function of temperature (in K) versus protein concentration for two monoclonal antibodies, the one shown in the top figure138 and another monoclonal antibody.128 The symbols denote experimental data, while the full lines are calculations. The two-phase region is indicated by the shaded area. Reprinted with permission from ref. 129. Copyright (2018) American Chemical Society. | ||
4 Protein dynamics and crowding effects in cells and pharmaceutical formulations
While analogies to colloids have primarily been used in order to understand protein interactions and characterize and predict structural properties and phase behavior of protein solutions, we have recently seen an increasing amount of work also devoted to the dynamics of protein solutions. The internal motion, the diffusion of proteins in crowded solutions and their macroscopic flow properties are key issues related to drug design, drug delivery formulations and our understanding of the cellular machinery, yet we lack both adequate theoretical models with predictive power as well as the necessary experimental data needed to validate these models on the required broad range of length and time scales. We therefore also briefly discuss the current status of investigation of protein dynamics in highly concentrated or crowded environments, and how analogies to colloids can help in this field.Life sciences today are at a crossroads. Current approaches towards understanding the cellular machinery predominantly focus on the molecular level.139 However, creating an inventory of detailed molecular structures of and interactions between biological macromolecules is simply not enough to fully understand complex systems such as living cells. While the molecules of biology do remarkable and surprising things, they still obey the laws of physics. It is important to realize that collective properties such as the entropy and free energy represent driving forces for chemical reactions, self-assembly, and phase transitions within cells. A quantitative understanding of a cellular system using an integrative systems biology approach will thus have to start from collecting information about molecular interactions, and then devise a statistical physics-based model to arrive at a mathematical/numerical modelling with predictive power. This also requires a sound understanding of dynamic processes such as internal motion and diffusion of proteins, and their dependence on the various interactions present in a crowded environment. This not only poses enormous theoretical challenges, but their experimental characterization on the relevant time and length scales requires novel approaches. It is clear that significant progress in this area can only be achieved on the basis of a concerted long-term effort that combines traditionally disparate intellectual disciplines. It is here where analogies to colloids and the use of the experimental and theoretical toolbox developed for colloids can help.
The interior of living cells is a highly concentrated or crowded medium that can contain thousands of proteins up to very high volume fractions of 40% or more. Diffusion of proteins in cells is essential, as it strongly influences the cellular machinery through numerous processes such as signal transmission or reactions between proteins. In a dense and crowded environment such as the cell, an individual protein will feel the presence and interaction potential of all the surrounding proteins. Direct and hydrodynamic interactions strongly alter diffusion already on length scales comparable to the protein size, and we thus expect that they significantly influence reactions between different proteins compared to the situation encountered in typical in vitro model experiments. It is thus vital to measure, understand and predict the diffusion of proteins in crowded media.23,140–143
There is an increasing number of studies devoted to this topic, but experimentally these studies primarily rely on the use of potentially disturbing labelling combined with techniques such as fluorescence recovery after photobleaching (FRAP) or fluorescence correlation spectroscopy (FCS). These techniques measure long time tracer diffusion over distances much larger than the protein size.142,143 Other popular techniques to characterize the self and collective diffusion of proteins at high concentration are NMR diffusion measurements145 and dynamic light scattering (DLS).28,144,146–148 Once again, these techniques characterize self and collective diffusion over distances much larger than the protein size. Short and long time diffusion over distances of order the protein size that are highly important for cellular processes, however, are very difficult to investigate. Here neutron and X-ray based methods such as neutron spin echo (NSE) measurements or X-ray photon correlation spectroscopy (XPCS) in principle are ideally suited as they are capable of characterizing diffusive processes on these short characteristic length scales.
The importance of the appropriate choice of length scales is demonstrated in Fig. 8, which shows a comparison of DLS and NSE results for the concentration and temperature dependence of the short time collective diffusion coefficient of γB-crystallin. This globular protein is known to exhibit liquid–liquid phase separation, with a critical concentration around cc ≈ 220–240 mg ml−1, or a critical volume fraction of ϕc ≈ 0.15–0.17.91,92,144 The DLS data shows a dramatic effect of both temperature and concentration on the collective short time diffusion coefficient Dsc(q) (Fig. 8 top). DLS measures the decay of large length scale density or concentration fluctuations, and for globular proteins not too far away from a critical point these are entirely dominated by critical fluctuations. What we thus observe in DLS is critical slowing down when approaching the critical point or a spinodal.146,147 Due to the universal nature of critical phenomena, the only thing that matters here is the reduced temperature or distance away from the critical point or the spinodal, and simple coarse grained colloid models of particles with short-range attractive potentials that can reproduce the binodal and spinodal will also yield the concentration and temperature dependence of Dsc. This changes only when approaching the arrest line at very high concentrations close to the arrest line, where the measured correlation functions show an unusual almost logarithmic decay due to the competition between critical slowing down and dynamical arrest, and where more work that also takes into account the effects of patchy interactions on diffusion will be needed.144
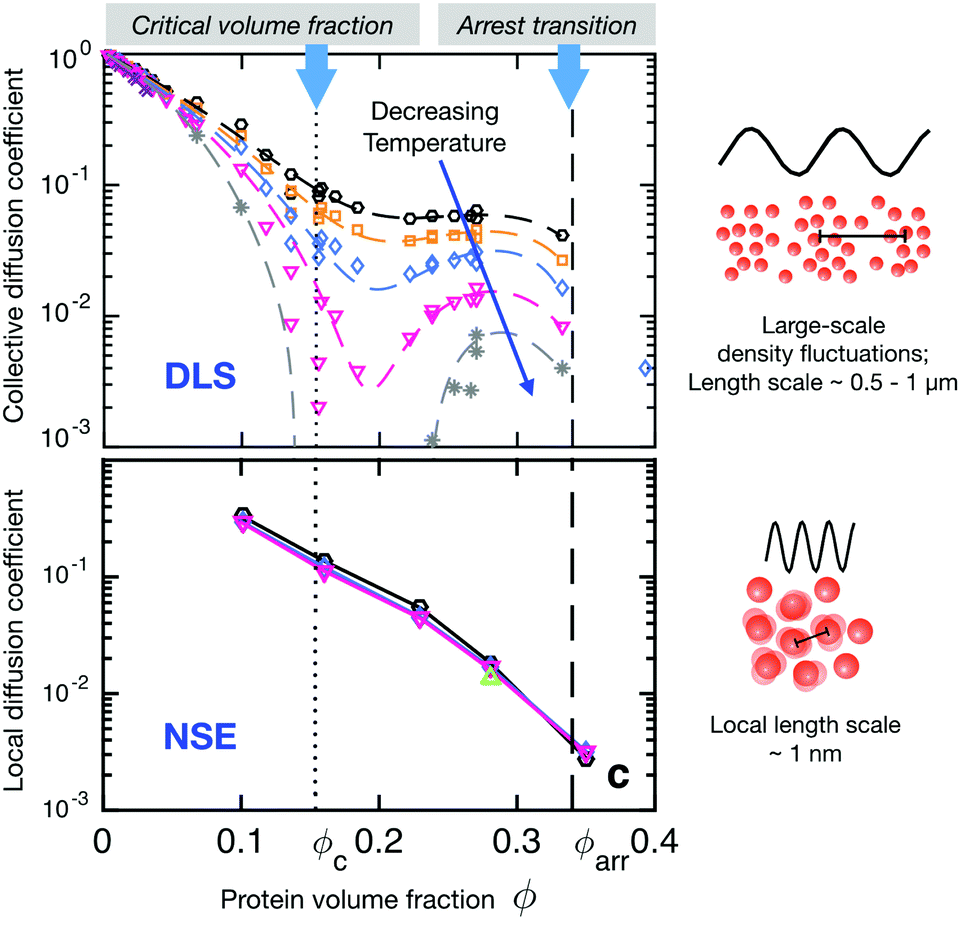 | ||
| Fig. 8 Short-time diffusion coefficients (rescaled by the respective free diffusion coefficient D0) of γB-crystallin probed at different length scales as a function of volume fraction and temperature. Colour code of the symbols denotes different temperatures in the range between 10 and 35 °C. Coloured dashed and full lines are guides to the eye. The vertical black dotted and dashed lines mark the critical volume fraction and the macroscopic arrest line, respectively. Also shown are schematic descriptions of the length-scale dependent processes seen by DLS and NSE. Top: Rescaled short-time collective diffusion coefficient in the long wavelength limit as obtained from DLS. The diffusion coefficient shows a strong temperature dependence particularly close to the critical volume fraction resulting from so-called critical slowing down that becomes more pronounced when approaching the critical point or spinodal line by decreasing the temperature. Bottom: Rescaled short-time local (or cage) diffusion coefficient as obtained from NSE experiments at the nearest neighbour peak q* in the respective static structure factors, corresponding to the nearest neighbour distance, and thus probing local dynamics on length scales of the protein size. The local dynamics is not affected by the vicinity to the critical concentration nor does it show any significant temperature dependence. Reprinted and adapted with permission from ref. 144, https://pubs.acs.org/doi/10.1021/acs.jpclett.5b02092. Further permissions related to the material excerpted should be directed to the ACS. | ||
The situation is very different for NSE, where collective diffusion is characterized at much larger q-values or smaller characteristic length scales. The NSE data shown in Fig. 8 has been obtained at a q-value corresponding to q*, the nearest neighbor peak of the structure factor, or a length scale of the nearest neighbor distance or protein diameter.144 At this large q-value, critical contributions to the structure factor have decayed completely, and S(q) is now dominated by the local structure, i.e., by the nearest neighbor cage formed by the surrounding proteins. On these length scales, collective dynamics characterized by Dsc(q*) corresponds to the relaxation of structural correlations between neighboring proteins or cage diffusion.31,149 As the interaction potential is only weakly dependent or independent on temperature, the resulting rescaled cage diffusion coefficient Dsc(q*)/D0, where D0 is the free diffusion coefficent of γB-crystallin in the absence of interaction effects, shows no detectable temperature dependence (Fig. 8 bottom). However, the NSE data also shows a dramatic decrease of cage diffusion with increasing concentration, with Dsc(q*)/D0 decreasing by one order of magnitude already for a volume fraction of around 15%. This slowing down of collective short time diffusion is much more pronounced than what one would expect for example for excluded volume effects, where Dsc(q*)/D0 should decrease by only about 30%.31
There are indeed a number of attempts in the literature to use colloid theory in order to interpret protein diffusion in crowded solutions measured by neutron-based quasi-elastic scattering methods such as NSE or neutron backscattering.27,31,60,150,151 While for proteins with dominating repulsive interactions such as described by screened Coulomb and effective hard sphere-like potentials agreement is very good,27,31 this is clearly not the case for proteins with weak short-range attractive potentials such as γB-crystallin. Attempts to use a coarse-grained colloid model with an isotropic short-ranged attractive potential completely failed in particular at low volume fractions, dramatically underestimating the slowing down of local collective short-time diffusion due to crowding effects. Using a patchy model where the overall strength of the effective pair potential as characterized by B2* is comparable, demonstrated that weak attractive patches can indeed strongly influence Dsc(q*)/D0 due to the formation of transient clusters and networks (Fig. 9).31 The importance of transient cluster formation on short time dynamics for short-range attractive particles has also been pointed out in other simulation studies.152,153 With the extreme sensitivity of the local collective short-time diffusion to the exact interaction potential in patchy systems, it is clear that understanding and predicting protein dynamics in crowded environments on these short length scales requires a considerable future effort for conditions where the interactions are not primarily repulsive, but influenced by directional attractive contributions.
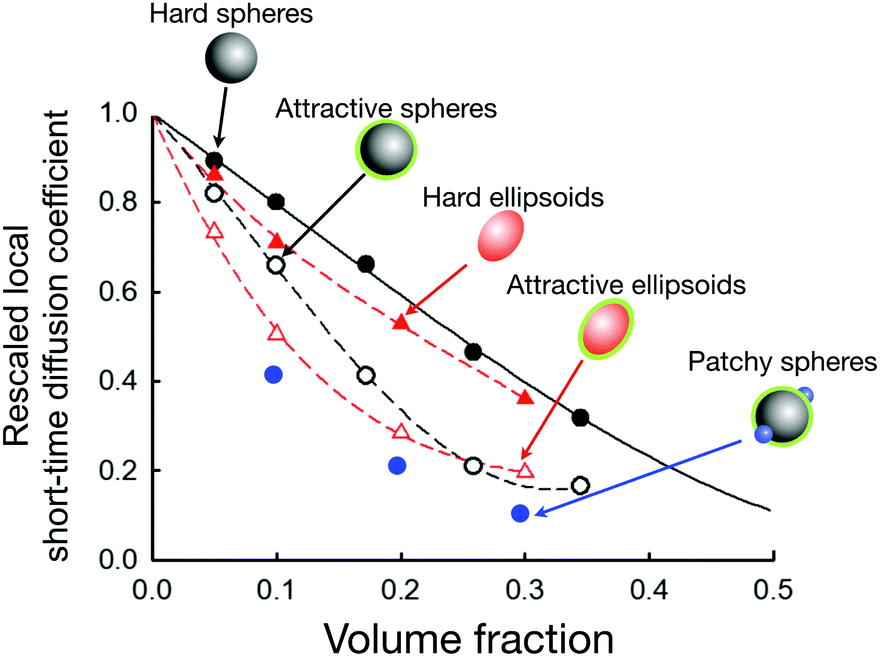 | ||
Fig. 9 Simulations of the rescaled local short-time diffusion coefficient (or short-time cage diffusion coefficient) of particles with slightly different shapes and interaction potentials (data taken from ref. 31 and 154) as a function of the volume fraction. Symbols denote the simulation results for hard spheres (filled black circles), short-range weakly attractive spheres (open black circles), hard ellipsoids (filled red triangles), short-range weakly attractive ellipsoids (open red triangles), and short-range weakly attractive spheres with two additional attractive patches (filled blue circles). The dashed lines are guides to the eye. The black line is the theoretical prediction for hard spheres.149 The axial ratio of the ellipsoids is roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The weakly attractive potential (for the attractive spheres, attractive ellipsoids and patchy spheres) is chosen such to maintain approximately the same position in the phase diagram (i.e. a comparable distance to the critical temperature) for all three systems. This figure demonstrates the enormous effect of a short-range weak attraction on the local short-time diffusion when compared to the purely hard sphere behaviour. It also reveals the tremendous effect of attractive patches (at constant overall attraction strength), and demonstrates the considerable influence of a weak shape anisotropy on the local short-time diffusion. :2. The weakly attractive potential (for the attractive spheres, attractive ellipsoids and patchy spheres) is chosen such to maintain approximately the same position in the phase diagram (i.e. a comparable distance to the critical temperature) for all three systems. This figure demonstrates the enormous effect of a short-range weak attraction on the local short-time diffusion when compared to the purely hard sphere behaviour. It also reveals the tremendous effect of attractive patches (at constant overall attraction strength), and demonstrates the considerable influence of a weak shape anisotropy on the local short-time diffusion. | ||
There is however a word of caution needed. The characteristic length scales over which motion is measured in quasi-elastic neutron scattering (QENS) experiments such as NSE or backscattering is not only relevant because of the underlying mechanisms that determine collective diffusion in strongly interacting particle systems. For proteins, this also has other consequences, related to their internal structure and flexibility. At short length scales, smaller than the protein size, QENS also becomes sensitive to rotational diffusion and internal motion.27,155–159 Analysing protein diffusion at q-values beyond the nearest neighbor peak is thus often more complex than for synthetic hard colloids due to these additional contributions that need to be taken into account. However, protein conformational dynamics as measured by QENS at larger q-values is interesting in its own right, as it is essential for their biological function: a protein must adopt specific active conformations in order to properly function. In particular large-scale domain motions, which can be induced by ligand binding or are intrinsically related to the protein domain structure, can for example modulate the entry of substrates into active sites and control enzymatic reactions. Neutron spin echo (NSE) spectroscopy is ideally suited for probing domain motions on the required length (nm) and time (ns–μs) scales without the need of any perturbing labelling, and has indeed been used successfully in a few cases already.155–159 It is however also important to point out that this requires an analysis where contributions from translational and rotational diffusion and local internal domain motions can be decoupled. This can be achieved in principle using molecular dynamics simulations (MD) based on the known protein structure.158 However, this becomes much more difficult at higher concentrations, where direct and hydrodynamic interactions between proteins complicate the resulting dynamics.27
There is another aspect of proteins that has frequently been neglected when using a colloid analogy to understand and predict diffusion of proteins in crowded environments. Globular proteins are rarely perfect spheres, but rather posses an anisotropic shape. γB-crystallin for example has a shape that resembles a triaxial ellipsoid, with axial ratios of about 1.7 and 2.144 The importance of shape anisotropy in the interpretation and simulation of short time diffusion in crowded solutions has been discussed before, but the prevailing assumption has been that for small axial ratios such as those in γB-crystallin an effective sphere approximation160 should be adequate to treat interaction effects for these systems.26,27 However, a recent simulation study using molecular dynamics simulations that includes hydrodynamic interactions based on a mesoscale hybrid approach has clearly shown a strong effect from shape anisotropy for shapes such as those resembling γB-crystallin, which is particularly pronounced for weakly attractive interactions.154 Moreover, not only are these effects quite strong, but they also contribute in a non-monotonic way as a function of concentration, as shown in Fig. 9. In order to understand and predict the dynamics of crowded protein solutions, it is thus not sufficient to perform in vitro experiments under dilute conditions and use estimates of the overall strength of interparticle interactions together with standard effective sphere colloid models. There is a clear need for an extension of the often used simple colloid models and to incorporate more molecular features into such coarse-grained models when attempting to describe local short-time diffusion in crowded solutions.
The ability to understand and predict dynamic properties of concentrated protein solutions and mixtures is not only vital for an understanding of cellular mechanisms, but also important for the formulation of future protein drugs. The development and use of mAb-based human therapeutics has for example rapidly evolved during the last 20 years and led to an almost exponential growth in market value.161 Together with the need for high concentration formulations that allow for facile home administration, this creates the need for an advanced predictive understanding of the stability and viscosity of concentrated protein solutions.127,162 In particular the frequently observed propensity of mAbs to reversibly aggregate, and the consequences of this concentration dependent cluster formation on the resulting sample viscosity significantly adds to the effects of particle interactions on the flow properties of concentrated formulations.127,132,163 There is thus a need to improve our analytical capability to investigate concentrated formulations, and to disentangle the effects of interprotein interactions, concentration dependent reversible aggregation, and degradation on the long-term stability and viscosity. The question is whether we can use the different routes towards dynamical arrest discussed in connection with short-range attractive colloids and summarized in Fig. 1 for this purpose?
We can look at this by comparing the available information for three proteins that have been used as well-defined model proteins: lysozyme, γB-crystallin and α-crystallin. Given the detrimental nature of an increased viscosity in high concentration biologics formulations due to the existence of a dynamical arrest transition, this is a question that is not only of interest for basic research on protein solutions, but of enormous practical importance for pharmaceutical industry.127,162 Similar to attempts made to use the ELCS in order to link easily measurable experimental parameters such as B2* with the thermodynamic stability with respect to a possible liquid–liquid phase separation, B2* or its dynamic analogue kD, obtained from DLS measurements, could in principle allow for an early prediction of the location of such an arrest line. This would be particularly attractive as such measurements require small amounts of sample only, which is another key concern in the early stages of drug development. However, when looking at the B2* values for these three proteins, the picture looks as follows: lysozyme is strongly repulsive (B2* ≫ 1) at these low ionic strengths;25,60 γB-crystallin is overall weakly attractive (B2* ≈ −1);165 and α-crystallin shows hard sphere behavior, i.e. B2* = 1.28 On the other hand, as shown in Fig. 10, the arrest transition occurs at ϕg(lysozyme) ≈ 0.28 ≤ ϕg(γB-crystallin) ≈ 0.35 ≪ ϕg(α-crystallin ≈ 0.58. As discussed previously, there is good evidence that for these more complex mixed and/or patchy potentials found for proteins, the ELCS does not hold for the location of the arrest line, and we need to consider the actual arrest mechanisms that act for these three proteins.
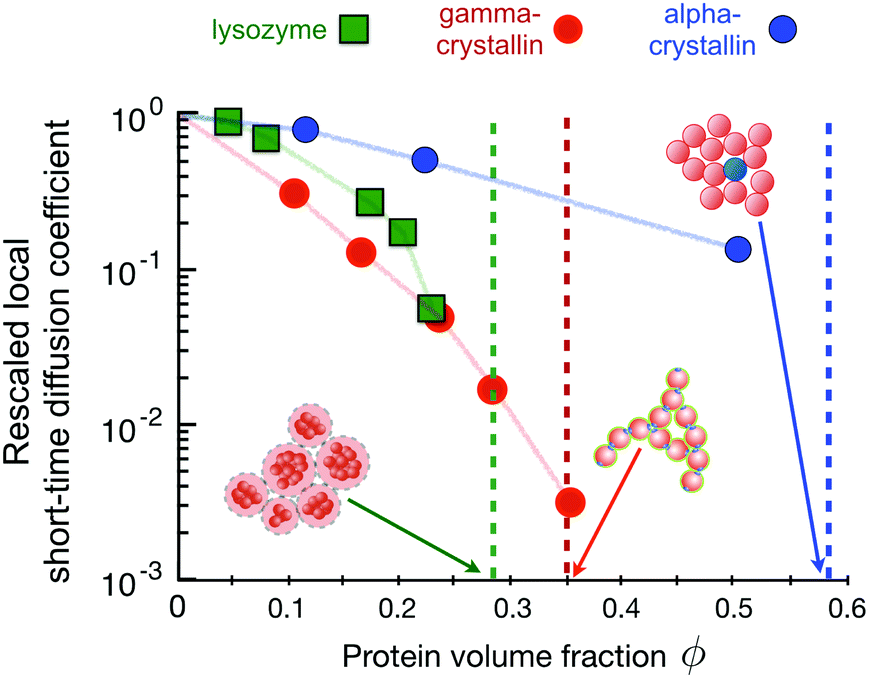 | ||
| Fig. 10 The connection between the concentration dependence of the rescaled local short-time diffusion (or short-time cage diffusion149) as obtained from Neutron Spin Echo (NSE) measurements and dynamical arrest for three different proteins (data taken from ref. 31 and 60). The dashed vertical lines (same colour code as for the symbols) indicate the macroscopic arrest transitions for the proteins as determined by macro- and microrheology28,60,164 and DLS experiments.144 The three proteins show very different arrest scenarios, as showcased in the cartoons: lysozyme, which is investigated here under conditions where the combination of a short-range attraction and a long-range repulsion (low ionic strength, positive net charge (pH ≈ 7.8), T = 5 °C) leads to the formation of equilibrium clusters.25 These charged clusters form a cluster glass, with a liquid–solid transition at volume fractions as low as ϕ ≈ 0.28.60 Solutions of the bovine eye lens protein γB-crystallin (at T = 25 °C) macroscopically arrest at a volume fraction of ϕ ≈ 0.35. Computer simulations together with experimental static and dynamic light and neutron scattering data indicate that this is due to a combination of weak short-range attractions and additional patch-interactions, facilitating the formation of large, open clusters and space-spanning networks already at relatively low volume fractions.144 Finally, the bovine eye lens protein α-crystallin shows an arrest transition at ϕ ≈ 0.58, reminiscent of colloidal hard sphere systems where at this high volume fraction the particles get trapped in their nearest neighbour cages.28 | ||
Lysozyme at low ionic strength at a pH value where it carries a net positive charge interacts via a combination of a short-range attraction and a long-range screened Coulomb repulsion, which leads to the formation of equilibrium clusters.25 These charged clusters increase in size with increasing protein concentration and form a cluster glass.60,61 Due to the relatively open structure and the long-range repulsion between the clusters, the corresponding arrest transition occurs already at volume fractions as low as ϕg ≈ 0.28.60 Increasing the temperature reduces the cluster growth slightly, and thus also shifts the arrest transition to somewhat larger volume fractions.61 It is interesting to note that the concentration dependence of the relative (zero-shear) viscosity ηr = ηs/η0 where ηs is the zero shear viscosity of the sample and η0 that of the solvent, is well described by the Quemada relation ηr = (1 − ϕ/ϕg) also found for hard sphere systems, indicating that the clusters act as effective hard spheres, with a sphere size given by the combination of the cluster radius and the Debye length. The onset of dynamical arrest is also reflected in the rescaled local short-time collective diffusion coefficient measured at the cluster–cluster peak in the structure factor, and we see that the cluster growth at higher concentrations results in values of Dsc(q*)/D0 that strongly decrease at higher ϕ-values, where cluster growth becomes important (Fig. 10).60
At the solution conditions discussed here, the bovine eye lens protein γB-crystallin exhibits short time collective diffusion indicative of weak patchy attractions,31,144 where Dsc(q*)/D0 decreases dramatically with increasing concentration (Fig. 10). Computer simulations of patchy colloids have indicated that this unusually strong decrease is linked to the formation of transient clusters, which become much more pronounced for patchy than for isotropic potentials.31 These open transient clusters are also responsible for the macroscopic arrest that occurs already at low volume fractions ϕg ≈ 0.35, where these networks become space spanning, and where the network then is load-bearing despite the transient nature of the individual bond.144
Finally, the bovine eye lens protein α-crystallin has been shown to interact via a hard-sphere-like potential, resulting in structural and dynamic properties that are quantitatively described by a colloid model of polydisperse hard spheres (Fig. 10).28 This hard sphere nature of α-crystallin is also reflected in an arrest transition that occurs at the expected value for hard sphere colloids, ϕg ≈ 0.58. Here the mechanism is due to the formation of cages by the nearest neighbor particles, resulting in a two step decay of the intermediate scattering function.28,36 While the long-time diffusion coefficient, which is related to the escape of particles from the transient cages is not visible with NSE, the short-time Dsc(q*)/D0 is well characterized by the weaker concentration dependence of pure hard spheres.31,149
The three examples shown in Fig. 10 not only document the dramatically different arrest lines and arrest scenarios for these globular proteins that differ in their effective interaction potential. The figure moreover shows that the measured collective short-time diffusion on length scales of the nearest neighbor distance is also strongly influenced by the potential, and that Dsc(q*)/D0 is extremely sensitive to the actual potential details. While the structural features of these protein solutions can be well reproduced by a strongly coarse-grained model with an isotropic potential (mixed potential for lysozyme, short-range attractive for γB-crystallin, hard sphere for α-crystallin), this does completely fail for the calculation of short time diffusion in the case of the patchy potential of γB-crystallin.31 There are two lessons to be learned, one utterly inconvenient and one rather convenient from the viewpoint of those who are interested in describing and predicting properties of protein solutions. On the one hand, there is unfortunately no easy way of predicting the existence and location of an arrest transition for concentrated solutions of globular proteins from simple low-concentration measurements such as a measurement of a second virial coefficient. On the other hand, however, measuring short-time collective diffusion on length scales of the nearest neighbor distance (or the protein diameter) offers an exquisitely sensitive test for specific interaction potentials through a comparison of the concentration dependence of the measured and simulated Dsc(q*)/D0. At the same time, Dsc(q*)/D0 is providing unique insight into the underlying mechanisms of a given arrest transition. We believe that in combination with appropriate simulation schemes, the method has enormous potential for evaluating interactions between proteins and their dependence on small variations in composition such as for example induced by point mutations. The drawback is, however, that the field currently suffers from the very limited availability of the technique, with only a few neutron spin echo instruments existing worldwide.
At first sight, these findings also seem quite discouraging when considering the current challenge to develop stable high concentration and low viscosity biologics formulations. As described before, a key problem in antibody formulation is that mAbs can exhibit enhanced self-association, causing too high viscosities for administering the drug. There have again been numerous studies where researchers tried to determine the molecular origins of self-association, and to predict enhanced self-association and viscosity based on simple low concentration parameters such as B2* and kD, albeit mostly with limited success.130,134,166–172 Given the difficulties in trying to reproduce Dsc(q*)/D0 or the location of the arrest line for globular proteins, it seems out of question to arrive at some prediction of the location of an arrest line or the concentration dependence of the relative viscosity ηr for mAbs that exhibit a concentration-dependent self-association and a corresponding strong increase of ηr at higher concentrations. However, the observation previously made for lysozyme cluster fluids, where ηr was found to follow the Quemada relation,61 clearly indicates that there is hope! If we can use an effective hard sphere model to describe the concentration dependence of the zero shear viscosity for protein cluster fluids, the same may be possible also for self-associating mAbs.
This has indeed been demonstrated for an IgG system, where the problem of enhanced self-association in high concentration antibody solutions and the concomitant high viscosities was tackled using a succession of coarse graining steps.130 In this study, mAb self-association and viscosity as a function of concentration was characterized using a combination of experiments (static and dynamic light scattering and microrheology), theory and simulations using analogies to patchy colloids. In a first step, self-association and cluster formation due to attractive electrostatic interactions between oppositely charged points on the arms (antigen binding or FAB domains) and on the tail (constant or FC domain) was identified as the driving force for the ionic strength dependence of the concentration-induced increase of the relative viscosity. A model that takes into account the anisotropic character of both the mAb shape (Y-shaped particle) and of the mAb–mAb interactions (patchy attraction) was then constructed, based on electrostatic calculations of a single antibody molecule in buffer, and MC simulations and analytical calculations were performed, the latter using an analytic solution based on Wertheim173 theory. A comparison between calculated and measured values of the concentration dependence of the isothermal compressibility or apparent molecular weight provided quantitative values for the strength of the attractive patches. In combination with so-called hyperbranched polymer theory174 this directly provided the cluster size distribution as a function of concentration.
To further predict the dynamic properties, an additional coarse-graining step was made, by considering mAb clusters rather than single mAb molecules as effective hard or sticky hard spheres. Here the cluster size distribution from the initial fit to the static light scattering data was used together with the corresponding theories for the concentration dependence of the apparent collective diffusion coefficient or hydrodynamic radius for these two models. The only additional free parameters in this step were a constant required for the rescaling of the volume fraction from the one based on individual mAbs to the one describing the cluster fluid, and the stickiness parameter for the sticky hard sphere model. In a final step, the relative viscosity ηr was calculated for each concentration without any additional free parameter using the Mooney relation ηr = exp[2.5ϕHS/(1 − ϕHS/ϕg)] often applied for polydisperse hard spheres,175 and assuming ϕg = 0.71 based on literature values for hard spheres with comparable polydispersity. Remarkably, the measured data – the apparent molecular weight as obtained by static light scattering, the apparent hydrodynamic radius from dynamic light scattering and the zero-shear viscosity from microrheology measurements – were in good agreement with the model predictions, showing that excluded volume interactions between the assembled clusters are indeed at the origin of the strong increase of the viscosity at high mAb concentrations. Moreover, the attraction strength of the patch–patch interactions calculated from the Wertheim fit to the static light scattering data agreed remarkably well with an estimate based on screened Coulomb interactions and a charge distribution on the IgG based on electrostatic calculations for the specific type of immunoglobulin.
This is indeed quite encouraging, and indicates that this could be a suitable approach to investigate and quantitatively assess the effects of additional excipients or chemical modifications on the antibody interaction. Such a model could then be used to estimate their effect on antibody stability and the resulting viscosity from molecular information, which would be vital for an advanced formulation strategy where unpromising candidates could be discarded at an early stage. Another interesting feature of such a successive coarse-graining procedure with appropriate patchy models is that it allows to determine the interaction strength and the cluster size distribution as a function of concentration for a given mAb system, which can subsequently be tested with DLS and (micro)rheology measurements without additional free parameters other than a rescaling of the volume fraction. The development of theoretical models to describe mAb self-association into clusters, and its consequences on other easily measurable structural and dynamic properties, has been in the focus of the community. With this approach, such models can be submitted to a critical assessment. Obviously this type of approach could easily be extended to other proteins that also display patchy interactions, and would allow for example to investigate various arrest scenarios for such systems as a function of key solution parameters.
5 Conclusions
This perspective article was initially largely motivated by the notion described in ref. 33, questioning altogether the usefulness of applying colloid-like theory to protein solutions. Having spent many years in trying to gain understanding of the structural and dynamic properties of protein solutions with such an approach, this asked for a re-evaluation of the situation. With the discussion presented above, we hope that it becomes clear that the statements made in ref. 33 rather reflect a too narrow view of what colloids are than a serious game stopper. Colloids are more than simple hard spheres, and in particular the recent trends in synthesizing, characterizing and simulating patchy, responsive and anisotropic particles have provided a wealth of information and new models that can be harnessed to learn much more about protein solutions. However, we have also seen that while the currently existing models indeed allow us to make fairly precise predictions about the phase behavior and the static properties of proteins under well-defined solution compositions (keeping in mind that quantities such as the interaction potential, the size and the shape of synthetic colloids also do not necessarily remain invariant as a function of pH, temperature or concentration), the situation already changes unfavourably when aiming at processes such as crystallization where angularly resolved interaction potentials and local dynamics matter. We clearly lack well-defined procedures to extract the relevant details from a 3D structure of a particular protein and then create a maximally coarse-grained patchy model that retains just the necessary anisotropic interaction features for understanding or predicting a specific property.This becomes even worse when interested in dynamic properties such as length-scale dependent collective and self diffusion and viscosity. Here we suffer primarily from two deficiencies: first of all, the existing work on patchy colloids has primarily focused on self-assembly and phase behavior. Very little attention has however been given to dynamic properties, and we thus find a lack of suitable theoretical models that could help us to understand the dynamics of proteins at higher concentrations. While there have been isolated attempts to harvest the significant progress made in the understanding of diffusion in concentrated colloidal suspensions and apply the same concepts and theoretical models to protein solutions, we are still unable to predict local and global dynamic properties of dense protein solutions based on the known molecular structure of the individual components, or to assess the effect of single point mutations on them. The existing theoretical models for (spherical and isotropically interacting) colloids cannot be directly transferred to protein solutions due to their complex structure and internal flexibility, and there is correspondingly a considerable lack of quantitative characterization and understanding of protein diffusion in crowded environments on the required molecular level. While for example our understanding of polymer dynamics has advanced tremendously since the early 1980's by the availability of neutron spin echo and other quasielastic neutron techniques and the simultaneous progress in polymer theory and multi-scale modelling techniques,174,176 the corresponding field of protein dynamics has clearly lacked similar progress. There is thus an urgent need for improving our ability to characterize, interpret and predict the dynamic properties of individual proteins as well as of their concentrated solutions and mixtures.
Moreover, the experimental toolbox of colloid physics has primarily been developed for investigating larger colloids with sizes of several hundred nanometers, and the wealth of existing data on colloid dynamics comes mainly from techniques such as DLS or confocal laser scanning microscopy (CLSM). Proteins are much smaller, and there are only a few techniques that can for example deliver measurements of short and long time collective and self diffusion on length scales of the protein size. Neutron and X-ray based scattering methods have the potential to deliver the required experimental data; however, we lack a theoretical and simulation toolbox similar to what already exists for structural properties and the interpretation of SAXS and SANS data. While detailed near-atomistic simulations and a comparison with neutron and X-ray based spectroscopic data is feasible for reproducing the internal dynamics of individual proteins, a similar approach is not viable for crowded solutions and mixtures. We need significant methodological development, including developing both advanced coarse-graining approaches in simulation and a combination of several experimental methods such as NSE and X-ray photon correlation spectroscopy (XPCS).177 This needs to go hand in hand with the development of new sample environments to account for the very limited quantities of proteins typically available for such investigations, and the design of new measurement and data analysis procedures to overcome the notorious beam damage problem when working with intense synchrotron radiation.
However, having discussed all what is still missing, we should nevertheless not end without pointing out the enormous progress already made. We believe that a colloid approach to protein solutions has indeed an enormous potential in many areas as diverse as cell and systems biology, pharmaceutical formulation and materials sciences. The essential point in applications of concepts from colloid physics to proteins, however, has to be a case by case reflection on the required level of coarse graining needed for a given problem, and a critical choice of the experimental techniques and data chosen for a meaningful test of model predictions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Knut and Alice Wallenberg Foundation (Project Grant KAW 2014.0052), the Swedish Research Council (VR Grants 2016-03301 and 2018-04627), the European Research Council (ERC-339678-COMPASS), and the Faculty of Science at Lund University.References
- T. Graham, Philos. Trans. R. Soc. London, 1861, 151, 183–224 CrossRef.
- W. Pauli, Pflüger's Archiv für die Gesammte Physiologie des Menschen und der Tiere, 1910, 136, 483–501 CrossRef.
- W. Pauli, Kolloid-Z., 1922, 31, 252–256 CrossRef CAS.
- J. Loeb, Science, 1920, 52, 449–456 CrossRef CAS PubMed.
- J. Loeb, J. Gen. Physiol., 1918, 1, 237–254 CrossRef CAS PubMed.
- H. S. Wells, J. B. Youmans and D. G. Miller, J. Clin. Invest., 1933, 12, 1103–1117 CrossRef CAS PubMed.
- P. N. Pusey and W. Van Megen, Nature, 1986, 320, 340–342 CrossRef CAS.
- V. J. Anderson and H. N. Lekkerkerker, Nature, 2002, 416, 811–815 CrossRef CAS PubMed.
- D. Frenkel, Science, 2006, 314, 768–769 CrossRef CAS PubMed.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- A. George and W. W. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 361–365 CrossRef CAS PubMed.
- M. Muschol and F. Rosenberger, J. Chem. Phys., 1995, 103, 10424–10432 CrossRef CAS.
- M. H. Hagen and D. Frenkel, J. Chem. Phys., 1994, 101, 4093–4097 CrossRef CAS.
- H. N. W. Lekkerkerker, W. C.-K. Poon, P. N. Pusey, A. Stroobants and P. B. Warren, Europhys. Lett., 1992, 20, 559–564 CrossRef CAS.
- N. Asherie, A. Lomakin and G. B. Benedek, Phys. Rev. Lett., 1996, 77, 4832–4835 CrossRef CAS.
- M. Muschol and F. Rosenberger, J. Chem. Phys., 1997, 107, 1953–1962 CrossRef CAS.
- D. Rosenbaum, P. C. Zamora and C. F. Zukoski, Phys. Rev. Lett., 1996, 76, 150–153 CrossRef CAS PubMed.
- P. R. Ten Wolde and D. Frenkel, Science, 1997, 277, 1975–1978 CrossRef CAS PubMed.
- M. G. Noro and D. Frenkel, J. Chem. Phys., 2000, 113, 2941–2944 CrossRef CAS.
- N. Asherie, Methods, 2004, 34, 266–272 CrossRef CAS PubMed.
- R. Piazza, Curr. Opin. Colloid Interface Sci., 2000, 5, 38–43 CrossRef CAS.
- R. Piazza, Curr. Opin. Colloid Interface Sci., 2004, 8, 515–522 CrossRef CAS.
- R. P. Sear, J. Phys.: Condens. Matter, 2005, 17, S3587–S3595 CrossRef CAS.
- R. Sear, Curr. Opin. Colloid Interface Sci., 2006, 11, 35–39 CrossRef CAS.
- A. Stradner, H. Sedgwick, F. Cardinaux, W. C. K. Poon, S. U. Egelhaaf and P. Schurtenberger, Nature, 2004, 432, 492–495 CrossRef CAS PubMed.
- T. Ando and J. Skolnick, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18457–18462 CrossRef CAS PubMed.
- F. Roosen-Runge, M. Hennig, F. Zhang, R. M. J. Jacobs, M. Sztucki, H. Schober, T. Seydel and F. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11815–11820 CrossRef CAS PubMed.
- G. Foffi, G. Savin, S. Bucciarelli, N. Dorsaz, G. M. Thurston, A. Stradner and P. Schurtenberger, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16748–16753 CrossRef CAS PubMed.
- D. Fusco and P. Charbonneau, Colloids Surf., B, 2016, 137, 22–31 CrossRef CAS PubMed.
- J. J. McManus, P. Charbonneau, E. Zaccarelli and N. Asherie, Curr. Opin. Colloid Interface Sci., 2016, 22, 73–79 CrossRef CAS.
- S. Bucciarelli, J. S. Myung, B. Farago, S. Das, G. A. Vliegenthart, O. Holderer, R. G. Winkler, P. Schurtenberger, G. Gompper and A. Stradner, Sci. Adv., 2016, 2, e1601432 CrossRef PubMed.
- P. S. Sarangapani, S. D. Hudson, R. L. Jones, J. F. Douglas and J. A. Pathak, Biophys. J., 2015, 108, 724–737 CrossRef CAS PubMed.
- J. Prausnitz, Biophys. J., 2015, 108, 453–454 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS PubMed.
- P. N. Pusey and W. van Megen, Phys. Rev. Lett., 1987, 59, 2083–2086 CrossRef CAS PubMed.
- W. van Megen, J. Phys.: Condens. Matter, 2002, 14, 310 Search PubMed.
- G. L. Hunter and E. R. Weeks, Rep. Prog. Phys., 2012, 75, 066501 CrossRef PubMed.
- A. J. Liu and S. R. Nagel, Annu. Rev. Condens. Matter Phys., 2010, 1, 347–369 CrossRef.
- K. A. Dawson, Curr. Opin. Colloid Interface Sci., 2002, 7, 218–227 CrossRef CAS.
- A. I. Campbell, V. J. Anderson, J. S. Van Duijneveldt and P. Bartlett, Phys. Rev. Lett., 2005, 94, 1–4 Search PubMed.
- J. C. F. Toledano, F. Sciortino and E. Zaccarelli, Soft Matter, 2009, 5, 2390 RSC.
- B. Ruzicka and E. Zaccarelli, Soft Matter, 2011, 7, 1268 RSC.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- J. J. Crassous, H. Dietsch, P. Pfleiderer, V. Malik, A. Diaz, L. A. Hirshi, M. Drechsler and P. Schurtenberger, Soft Matter, 2012, 8, 3538 RSC.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–55 CrossRef CAS.
- J. J. Crassous, A. M. Mihut, L. K. Månsson and P. Schurtenberger, Nanoscale, 2015, 7, 15971–15982 RSC.
- P. Schurtenberger, in Soft Matter Self-Assembly, ed. C. Likos, F. Sciortino, P. Ziherl and E. Zaccarelli, Ios Press, Nieuwe Hemweg 6B, 1013 BG Amsterdam, The Netherlands, 2016, pp. 81–136 Search PubMed.
- A. M. Mihut, B. Stenqvist, M. Lund, P. Schurtenberger and J. J. Crassous, Sci. Adv., 2017, 3, e1700321 CrossRef PubMed.
- E. Bianchi, J. Largo, P. Tartaglia, E. Zaccarelli and F. Sciortino, Phys. Rev. Lett., 2006, 97, 168301 CrossRef.
- E. Bianchi, P. Tartaglia, E. Zaccarelli and F. Sciortino, J. Chem. Phys., 2008, 128, 144504 CrossRef.
- F. Romano and F. Sciortino, Nat. Commun., 2012, 3, 975 CrossRef PubMed.
- K. van Gruijthuijsen, M. Obiols-Rabasa, P. Schurtenberger, W. G. Bouwman and A. Stradner, Soft Matter, 2018, 14, 3704–3715 RSC.
- T. Gibaud, N. Mahmoudi, J. Oberdisse, P. Lindner, J. S. Pedersen, C. L. P. Oliveira, A. Stradner and P. Schurtenberger, Faraday Discuss., 2012, 158, 267 RSC.
- K. N. Pham, A. Puertas, J. Bergenholtz, S. Egelhaaf, A. Moussaid, P. Pusey, A. Schofield, M. Cates, M. Fuchs and W. Poon, Science, 2002, 296, 104–106 CrossRef CAS PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- V. Trappe and P. Sandkühler, Curr. Opin. Colloid Interface Sci., 2004, 8, 494–500 CrossRef CAS.
- S. Romer, F. Scheffold and P. Schurtenberger, Phys. Rev. Lett., 2000, 85, 4980–4983 CrossRef CAS PubMed.
- S. Romer, H. Bissig, P. Schurtenberger and F. Scheffold, EPL, 2014, 108, 48006 CrossRef.
- F. Cardinaux, T. Gibaud, A. Stradner and P. Schurtenberger, Phys. Rev. Lett., 2007, 99, 1–4 CrossRef PubMed.
- F. Cardinaux, E. Zaccarelli, A. Stradner, S. Bucciarelli, B. Farago, S. U. Egelhaaf, F. Sciortino and P. Schurtenberger, J. Phys. Chem. B, 2011, 115, 7227–7237 CrossRef CAS PubMed.
- M. J. Bergman, T. Garting, P. Schurtenberger and A. Stradner, J. Phys. Chem. B, 2019, 123, 2432–2438 CrossRef CAS.
- H. Dietsch, V. Malik, M. Reufer, C. Dagallier, A. Shalkevich, M. Saric, T. Gibaud, F. Cardinaux, F. Scheffold, A. Stradner and P. Schurtenberger, CHIMIA Int. J. Chem., 2008, 62, 805–814 CrossRef CAS.
- A. Pal, T. Zinn, M. A. Kamal, T. Narayanan and P. Schurtenberger, Small, 2018, 14, 1802233 CrossRef PubMed.
- F. Peng, PhD thesis, Lund University, 2019.
- M. J. Solomon and P. T. Spicer, Soft Matter, 2010, 6, 1391 RSC.
- S. Sacanna, W. T. M. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575–578 CrossRef CAS PubMed.
- S. Sacanna and D. J. Pine, Curr. Opin. Colloid Interface Sci., 2011, 16, 96–105 CrossRef CAS.
- L. Rossi, S. Sacanna, W. T. M. Irvine, P. M. Chaikin, D. J. Pine and A. P. Philipse, Soft Matter, 2011, 7, 4139–4142 RSC.
- K. Zhao and T. G. Mason, Rep. Prog. Phys., 2018, 81, 126601 CrossRef CAS PubMed.
- P. Bolhuis and D. Frenkel, J. Chem. Phys., 1997, 106, 666–687 CrossRef CAS.
- A. Haji-Akbari, M. Engel, A. S. Keys, X. Zheng, R. G. Petschek, P. Palffy-Muhoray and S. C. Glotzer, Nature, 2009, 462, 773–777 CrossRef CAS PubMed.
- G. Bautista-Carbajal, A. Moncho-Jordá and G. Odriozola, J. Chem. Phys., 2013, 138, 064501 CrossRef PubMed.
- R. Schilling and T. Scheidsteger, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 2932–2949 CrossRef CAS.
- M. J. Solomon and D. V. Boger, J. Rheol., 1998, 42, 929–949 CrossRef CAS.
- M. Letz, R. Schilling and A. Latz, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 5173–5178 CrossRef CAS PubMed.
- C. De Michele, R. Schilling and F. Sciortino, Phys. Rev. Lett., 2007, 98, 265702 CrossRef PubMed.
- Z. Zheng, F. Wang and Y. Han, Phys. Rev. Lett., 2011, 107, 065702 CrossRef PubMed.
- Z. Zheng, R. Ni, F. Wang, M. Dijkstra, Y. Wang and Y. Han, Nat. Commun., 2014, 5, 3829 CrossRef CAS PubMed.
- A. Pal, V. Martinez, T. Ito, J. Arlt, J. Crassous, W. C. Poon and P. Schurtenberger, Sci. Adv., 2020 Search PubMed , in press.
- V. S. Kumaraswamy, P. F. Lindley, C. Slingsby and I. D. Glover, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 611–622 CrossRef CAS PubMed.
- A. Kurut, B. A. Persson, T. Åkesson, J. Forsman and M. Lund, J. Phys. Chem. Lett., 2012, 3, 731–734 CrossRef CAS PubMed.
- N. Kern and D. Frenkel, J. Chem. Phys., 2003, 118, 9882–9889 CrossRef CAS.
- E. Bianchi, R. Blaak and C. N. Likos, Phys. Chem. Chem. Phys., 2011, 13, 6397–6410 RSC.
- É. Duguet, C. Hubert, C. Chomette, A. Perro and S. Ravaine, C. R. Chim., 2016, 19, 173–182 CrossRef.
- E. Bianchi, B. Capone, I. Coluzza, L. Rovigatti and P. D. Van Oostrum, Limiting the valence: advancements and new perspectives on patchy colloids, soft functionalized nanoparticles and biomolecules, 2017 Search PubMed.
- J. Gsponer and M. M. Babu, Cell Rep., 2012, 2, 1425–1437 CrossRef CAS PubMed.
- J. Berry, C. P. Brangwynne and M. Haataja, Rep. Prog. Phys., 2018, 81, 046601 CrossRef PubMed.
- G. B. Benedek, Invest. Ophthalmol. Visual Sci., 1997, 38, 1911–1921 CAS.
- J. D. Gunton, A. Shiryayev and D. L. Pagan, Protein condensation: kinetic pathways to crystallization and disease, 2007 Search PubMed.
- R. J. Siezen, M. R. Fisch, C. Slingsby and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1701–1705 CrossRef CAS PubMed.
- J. A. Thomson, P. Schurtenberger, G. M. Thurston and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7079–7083 CrossRef CAS PubMed.
- P. Schurtenberger, R. A. Chamberlin, G. M. Thurston, J. A. Thomson and G. B. Benedek, Phys. Rev. Lett., 1989, 63, 2064–2067 CrossRef CAS PubMed.
- M. L. Broide, C. R. Berland, J. Pande, O. O. Ogun and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 5660–5664 CrossRef CAS.
- J. Pande, C. Berland, M. Broide, O. Ogun, J. Melhuish and G. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4916–4920 CrossRef CAS PubMed.
- C. Liu, A. Lomakin, G. M. Thurston, D. Hayden, A. Pande, J. Pande, O. Ogun, N. Asherie and G. B. Benedek, J. Phys. Chem., 1995, 99, 454–461 CrossRef CAS.
- G. B. Benedek, J. Pande, G. M. Thurston and J. I. Clark, Prog. Retinal Eye Res., 1999, 18, 391–402 CrossRef CAS.
- C. Ishimoto and T. Tanaka, Phys. Rev. Lett., 1977, 39, 474–477 CrossRef CAS.
- V. G. Taratuta, A. Holschbach, G. M. Thurston, D. Blankschtein and G. B. Benedek, J. Phys. Chem., 1990, 94, 2140–2144 CrossRef CAS.
- C. R. Berland, G. M. Thurston, M. Kondo, M. L. Broide, J. Pande, O. Ogun and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1214–1218 CrossRef CAS.
- A. Gast, C. Hall and W. Russel, J. Colloid Interface Sci., 1983, 96, 251–267 CrossRef CAS.
- P. R. ten Wolde and D. Frenkel, Theor. Chem. Acc., 1999, 101, 205–208 Search PubMed.
- G. A. Vliegenthart and H. N. W. Lekkerkerker, J. Chem. Phys., 2000, 112, 5364–5369 CrossRef CAS.
- S. R. McGuffee and A. H. Elcock, J. Am. Chem. Soc., 2006, 128, 12098–12110 CrossRef CAS.
- P. Mereghetti, R. R. Gabdoulline and R. C. Wade, Biophys. J., 2010, 99, 3782–3791 CrossRef CAS PubMed.
- C. Gögelein, G. Nägele, R. Tuinier, T. Gibaud, A. Stradner and P. Schurtenberger, J. Chem. Phys., 2008, 129, 085102 CrossRef PubMed.
- F. Platten, N. E. Valadez-Pérez, R. Castañeda-Priego and S. U. Egelhaaf, J. Chem. Phys., 2015, 142, 174905 CrossRef PubMed.
- A. Lomakin, N. Asherie and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9465–9468 CrossRef CAS PubMed.
- M. Kastelic, Y. V. Kalyuzhnyi, B. Hribar-Lee, K. A. Dil and V. Vlachy, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6766–6770 CrossRef CAS.
- S. W. Wukovitz and T. O. Yeates, Nat. Struct. Biol., 1995, 2, 1062–1067 CrossRef CAS PubMed.
- N. Dorsaz, L. Filion, F. Smallenburg and D. Frenkel, Faraday Discuss., 2012, 159, 9 RSC.
- T. Gibaud, F. Cardinaux, J. Bergenholtz, A. Stradner and P. Schurtenberger, Soft Matter, 2011, 7, 857–860 RSC.
- A. Giacometti, F. Lado, J. Largo, G. Pastore and F. Sciortino, J. Chem. Phys., 2010, 132, 1–15 CrossRef PubMed.
- C. J. Roberts and M. A. Blanco, J. Phys. Chem. B, 2014, 118, 12599–12611 CrossRef CAS PubMed.
- W. M. Jacobs, D. W. Oxtoby and D. Frenkel, J. Chem. Phys., 2014, 140, 204109 CrossRef PubMed.
- G. Foffi and F. Sciortino, J. Phys. Chem. B, 2007, 111, 9702–9705 CrossRef CAS PubMed.
- I. Staneva and D. Frenkel, J. Chem. Phys., 2015, 143, 194511 CrossRef PubMed.
- B. Neal, D. Asthagiri, O. Velev, A. Lenhoff and E. Kaler, J. Cryst. Growth, 1999, 196, 377–387 CrossRef CAS.
- M. Hloucha, J. Lodge, A. Lenhoff and S. Sandler, J. Cryst. Growth, 2001, 232, 195–203 CrossRef CAS.
- A. Kurut and M. Lund, Faraday Discuss., 2013, 160, 271–278 RSC.
- G. Pellicane, G. Smith and L. Sarkisov, Phys. Rev. Lett., 2008, 101, 248102 CrossRef PubMed.
- A. Taudt, A. Arnold and J. Pleiss, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2015, 91, 033311 CrossRef PubMed.
- A. P. R. Eberle, R. Castañeda-Priego, J. M. Kim and N. J. Wagner, Langmuir, 2012, 28, 1866–1878 CrossRef CAS PubMed.
- G. Foffi, C. De Michele, F. Sciortino and P. Tartaglia, Phys. Rev. Lett., 2005, 94, 1–4 CrossRef PubMed.
- A. L. Nelson, E. Dhimolea and J. M. Reichert, Nat. Rev. Drug Discovery, 2010, 9, 767–774 CrossRef CAS PubMed.
- J. M. Reichert, mAbs, 2012, 4, 413–415 CrossRef PubMed.
- C. Narasimhan, H. Mach and M. Shameem, Ther. Delivery, 2012, 3, 889–900 CrossRef CAS PubMed.
- S. J. Shire, Curr. Opin. Biotechnol., 2009, 20, 708–714 CrossRef CAS PubMed.
- B. D. Mason, J. Zhang-vanEnk, L. Zhang, R. L. Remmele and J. Zhang, Biophys. J., 2010, 99, 3792–3800 CrossRef CAS PubMed.
- M. Kastelic and V. Vlachy, J. Phys. Chem. B, 2018, 122, 5400–5408 CrossRef CAS PubMed.
- N. Skar-Gislinge, M. Ronti, T. Garting, C. Rischel, P. Schurtenberger, E. Zaccarelli and A. Stradner, Mol. Pharmaceutics, 2019, 16, 2394–2404 CrossRef CAS PubMed.
- E. J. Yearley, I. E. Zarraga, S. J. Shire, T. M. Scherer, Y. Gokarn, N. J. Wagner and Y. Liu, Biophys. J., 2013, 105, 720–731 CrossRef CAS PubMed.
- E. J. Yearley, P. D. Godfrin, T. Perevozchikova, H. Zhang, P. Falus, L. Porcar, M. Nagao, J. E. Curtis, P. Gawande, R. Taing, I. E. Zarraga, N. J. Wagner and Y. Liu, Biophys. J., 2014, 106, 1763–1770 CrossRef CAS PubMed.
- M. M. Castellanos, J. A. Pathak, W. Leach, S. M. Bishop and R. H. Colby, Biophys. J., 2014, 107, 469–476 CrossRef CAS PubMed.
- P. D. Godfrin, I. E. Zarraga, J. Zarzar, L. Porcar, P. Falus, N. J. Wagner and Y. Liu, J. Phys. Chem. B, 2016, 120, 278–291 CrossRef CAS PubMed.
- C. Calero-Rubio, A. Saluja and C. J. Roberts, J. Phys. Chem. B, 2016, 120, 6592–6605 CrossRef CAS PubMed.
- D. Corbett, M. Hebditch, R. Keeling, P. Ke, S. Ekizoglou, P. Sarangapani, J. Pathak, C. F. V. D. Walle, S. Uddin, C. Baldock, C. Avendaño and R. A. Curtis, J. Phys. Chem. B, 2017, 121, 8276–8290 CrossRef CAS PubMed.
- C. Calero-Rubio, A. Saluja, E. Sahin and C. J. Roberts, J. Phys. Chem. B, 2019, 123, 5709–5720 CrossRef CAS PubMed.
- Y. Wang, A. Lomakin, R. F. Latypov and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16606–16611 CrossRef CAS PubMed.
- D. Plewczyński and K. Ginalski, The interactome: predicting the protein-protein interactions in cells, 2009 Search PubMed.
- R. J. Ellis, Curr. Opin. Struct. Biol., 2001, 11, 114–119 CrossRef CAS PubMed.
- R. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- J. A. Dix and A. Verkman, Annu. Rev. Biophys., 2008, 37, 247–263 CrossRef CAS PubMed.
- M. Weiss, in International Review of Cell and Molecular Biology, Elsevier Inc., 1st edn, 2014, vol. 307, pp. 383–417 Search PubMed.
- S. Bucciarelli, L. Casal-Dujat, C. De Michele, F. Sciortino, J. Dhont, J. Bergenholtz, B. Farago, P. Schurtenberger and A. Stradner, J. Phys. Chem. Lett., 2015, 6, 4470–4474 CrossRef CAS PubMed.
- W. S. Price, F. Tsuchiya and Y. Arata, J. Am. Chem. Soc., 1999, 121, 11503–11512 CrossRef CAS.
- B. M. Fine, J. Pande, A. Lomakin, O. O. Ogun and G. B. Benedek, Phys. Rev. Lett., 1995, 74, 198–201 CrossRef CAS PubMed.
- B. M. Fine, A. Lomakin, O. O. Ogun and G. B. Benedek, J. Chem. Phys., 1996, 104, 326–335 CrossRef CAS.
- M. Heinen, F. Zanini, F. Roosen-Runge, D. Fedunová, F. Zhang, M. Hennig, T. Seydel, R. Schweins, M. Sztucki, M. Antalík, F. Schreiber and G. Nägele, Soft Matter, 2012, 8, 1404–1419 RSC.
- A. J. Banchio and G. Nägele, J. Chem. Phys., 2008, 128, 104903 CrossRef PubMed.
- F. Roosen-Runge, M. Hennig, T. Seydel, F. Zhang, M. W. Skoda, S. Zorn, R. M. Jacobs, M. Maccarini, P. Fouquet and F. Schreiber, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 68–75 CrossRef CAS PubMed.
- M. Grimaldo, F. Roosen-Runge, M. Hennig, F. Zanini, F. Zhang, M. Zamponi, N. Jalarvo, F. Schreiber and T. Seydel, J. Phys. Chem. Lett., 2015, 6, 2577–2582 CrossRef CAS PubMed.
- S. von Bülow, M. Siggel, M. Linke and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 201817564 CrossRef PubMed.
- S. Das, J. Riest, R. G. Winkler, G. Gompper, J. K. G. Dhont and G. Nägele, Soft Matter, 2018, 14, 92–103 RSC.
- J. S. Myung, F. Roosen-Runge, R. G. Winkler, G. Gompper, P. Schurtenberger and A. Stradner, J. Phys. Chem. B, 2018, 122, 12396–12402 CrossRef CAS PubMed.
- M. Monkenbusch, D. Richter and R. Biehl, ChemPhysChem, 2010, 11, 1188–1194 CrossRef CAS PubMed.
- R. Biehl, M. Monkenbusch and D. Richter, Soft Matter, 2011, 7, 1299–1307 RSC.
- R. Biehl and D. Richter, J. Phys.: Condens. Matter, 2014, 26, 503103 CrossRef CAS PubMed.
- N. Smolin, R. Biehl, G. Kneller, D. Richter and J. Smith, Biophys. J., 2012, 102, 1108–1117 CrossRef CAS PubMed.
- D. J. E. Callaway, B. Farago and Z. Bu, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 76 CrossRef PubMed.
- B. R. Jennings and K. Parslow, Proc. R. Soc. A, 1988, 419, 137–149 CrossRef CAS.
- A. Beck, T. Wurch, C. Bailly and N. Corvaia, Nat. Rev. Immunol., 2010, 10, 345–352 CrossRef CAS PubMed.
- S. J. Shire, Z. Shahrokh and J. Liu, J. Pharm. Sci., 2004, 93, 1390–1402 CrossRef CAS PubMed.
- C. J. Roberts, Curr. Opin. Biotechnol., 2014, 30, 211–217 CrossRef CAS PubMed.
- T. Garting and A. Stradner, Small, 2018, 14, 1801548 CrossRef PubMed.
- S. Bucciarelli, N. Mahmoudi, L. Casal-Dujat, M. Jéhannin, C. Jud and A. Stradner, J. Phys. Chem. Lett., 2016, 7, 1610–1615 CrossRef CAS PubMed.
- M. Kastelic, K. A. Dill, Y. V. Kalyuzhnyi and V. Vlachy, J. Mol. Liq., 2018, 270, 234–242 CrossRef CAS PubMed.
- M. S. Neergaard, D. S. Kalonia, H. Parshad, A. D. Nielsen, E. H. Møller and M. Van De Weert, Eur. J. Pharm. Sci., 2013, 49, 400–410 CrossRef CAS PubMed.
- B. D. Connolly, C. Petry, S. Yadav, B. Demeule, N. Ciaccio, J. M. R. Moore, S. J. Shire and Y. R. Gokarn, Biophys. J., 2012, 103, 69–78 CrossRef CAS PubMed.
- S. Saito, J. Hasegawa, N. Kobayashi, N. Kishi, S. Uchiyama and K. Fukui, Pharm. Res., 2012, 29, 397–410 CrossRef CAS PubMed.
- P. M. Buck, A. Chaudhri, S. Kumar and S. K. Singh, Mol. Pharmaceutics, 2014, 12, 127–139 CrossRef PubMed.
- D. S. Tomar, S. Kumar, S. K. Singh, S. Goswami and L. Li, mAbs, 2016, 8, 216–228 CrossRef CAS PubMed.
- M. A. Woldeyes, W. Qi, V. I. Razinkov, E. M. Furst and C. J. Roberts, J. Pharm. Sci., 2019, 108, 142–154 CrossRef CAS PubMed.
- M. S. Wertheim, J. Stat. Phys., 1984, 35, 19 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- M. Mooney, J. Colloid Interface Sci., 1951, 6, 162–170 CrossRef CAS.
- D. Richter, M. Monkenbusch, A. Arbe and J. Colmenero, Neutron Spin Echo in Polymer Systems, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005 Search PubMed.
- J. Möller, M. Sprung, A. Madsen and C. Gutt, IUCrJ, 2019, 6, 794–803 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |