
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow polymer microrods of tunable flexibility from dense amphiphilic block copolymer brushes†
Lucille
Chambon
 ab and
Maria
Vamvakaki
*ab
ab and
Maria
Vamvakaki
*ab
aDepartment of Materials Science and Technology, University of Crete, 700 13 Heraklion, Crete, Greece
bInstitute of Electronic Structure and Laser Foundation for Research and Technology – Hellas, 700 13 Heraklion, Crete, Greece. E-mail: vamvakak@iesl.forth.gr
First published on 6th December 2019
Abstract
Polymer microrods of aspect ratio ∼10, and tunable flexibility are attractive model systems to study density and index matched liquid crystalline phases. However, the synthesis of anisotropic polymer particles is arduous, due to the lack of directional polymer growth mechanisms. In this work, non-cross-linked, hollow polymer microrods are developed from a dense block copolymer brush grown from the surface of micron-sized silica rods. The copolymer brush, comprising a hydrophobic inner block and a hydrophilic outer layer, is synthesized by surface-initiated atom transfer radical polymerization, and is exploited in the preparation of robust polymer rod particles in water, following etching of the inorganic core. The solvent-incompatible inner block is crucial for the synthesis of the rod-like polymer particles, in the absence of chemical cross-links, and the block copolymer composition affects the colloidal stability and flexibility of the hollow anisotropic colloids. For shorter hydrophobic block lengths, well-defined, yet flexible, hollow rods are obtained, whereas increasing the hydrophobic content of the copolymer results in rigid, tube-like particles. The approach is generic and could be easily employed to obtain polymer rod particles in any solvent medium, upon the appropriate selection of the solvent-incompatible inner block and the solvent-compatible outer block.
I Introduction
Anisotropic particles have attracted great scientific interest, because of their superior material properties compared to their spherical counterparts. Rod-like particles have been shown to display a strong response to applied fields,1–3 and high biological activity, being internalized more easily by cells,4 and to assemble into percolated networks of improved mechanical properties at low volume fractions.5–7 They also possess a rich phase diagram, being in the isotropic phase at low volume fractions, and forming liquid crystalline phases, with orientational order (nematic phase) and additional positional order (smectic phase), upon increasing the particle concentration.8,9 The liquid crystalline phases can also be accessed or favored by promoting directional interactions.10 The synthesis of inorganic rod-like colloids has dominated the literature, because the crystalline nature of most inorganic precursors favors the one-directional growth.11,12 Iron oxide ellipsoids,13 as well as metallic14 and silica15–17 rods, have been reported, the latter being particularly attractive because of their facile and reproducible synthesis, which yields relatively monodisperse, micrometer-sized particles allowing the direct observation of their liquid crystal phases by confocal microscopy.18 However, inorganic rods generally possess both a high refractive index and high density, and cannot be simultaneously density and index matched in common solvents. Refractive index mismatch increases the scattering of light in confocal microscopy measurements, providing poor optical images, whereas density mismatch leads to sedimentation of the rods with time, preventing their long-term investigation in the dispersed phase.Polymer rod-like particles are highly advantageous to address the above issues, because they possess the low density and refractive index of the polymer, which are easier to match. Furthermore, depending on the chosen polymer, these rod-like colloids can attain additional features, such as softness, stimuli-response and encapsulation and release of active molecules. However, the synthesis of rod-like polymer particles is rather challenging, because of the lack of directional growth mechanisms. The most extensively reported rod polymer particles in the literature are the worm-like micelles or vesicles, obtained by the self-assembly of block copolymers of appropriate composition and size.19,20 These rods possess nanometer-size dimensions, and generally cannot be observed by optical microscopy, while they are inherently flexible. In another approach, anisotropic polymer particles have been obtained by the mechanical elongation of spherical poly(methyl methacrylate) particles,21 or SU-8 droplets,22,23 however rigid rods have only been obtained and functional polymers are difficult to access. An attractive strategy has employed an inorganic template and seeded dispersion/emulsion polymerization or a layer-by-layer assembly process, followed by the etching of the inorganic core, to obtain hollow, rod-like polymer particles.24–26 All reported examples to date, required the dense cross-linking of the polymer layer to retain the rod-like shape of the colloids, after the removal of the core, whereas the length and aspect ratio of the particles were limited to maximum values of 2 μm and 6, respectively, to accommodate the high surface energy, non-monotonous surface curvature and low symmetry of the anisotropic particles.
In the present study, we report for the first time the synthesis of water dispersible, non-cross-linked, hollow polymer rod-like colloids of length between 2 and 10 μm, with tunable flexibility. The aspect ratio of the hollow rods synthesised herein is ∼10, which is relatively high for micron-sized particles, and has not been reported for hollow particles before. Hard silica rod-like particles of similar dimensions were used as the template in a surface-initiated atom transfer radical polymerization (SI-ATRP) to prepare a dense amphiphilic block copolymer brush onto the surface of the inorganic colloids. Next, the silica cores were removed with a concentrated hydrofluoric acid solution in water, and the morphology and flexibility of the remaining polymer were assessed using scanning and transmission electron microscopies. The presence of a hydrophobic inner block of a minimum length was found to be crucial for the polymer particles to retain the rod-like shape, whereas the hydrophilic outer block stabilized the particles against aggregation. Next, varying the copolymer composition allowed the flexibility of the rod particles to be tuned. Fluorescently labelled hollow polymer rods were obtained by utilizing a functional comonomer that enabled the binding of fluorescent dye molecules onto the particles. The significantly lower density of the hollow polymer rods prepared herein, compared to their silica precursor particles, and their tunable flexibility, are highly advantageous for liquid crystalline phase behavior studies. Moreover, the ability of the rods to disintegrate in selective solvents for the two blocks was shown, rendering them promising candidates in phase transformation and delivery applications.
II Materials and methods
A Materials
Chloroform (99.99%, Fischer Scientific), tetrahydrofuran (THF, 99.5%, AppliChem), acetone (99.99%, LachNer), acetonitrile (99.99%, Scharlab), 1-pentanol (98%, Alfa Aesar), absolute ethanol (EtOH, 98%, Aldrich), hydrogen peroxide (H2O2, 30 wt%, ChemLab), sulfuric acid (H2SO4, 96%, PENTA), polyvinylpirrolidone (PVP 40 kDa, Aldrich), ammonium hydroxide solution (25 wt%, Aldrich), sodium citrate dihydrate (>99.0%, Aldrich), tetraethylorthosilicate (TEOS, 98%, Aldrich), hydrofluoric acid solution (HF, 40 wt%, Aldrich), 4.4′-dinonyl-2,2′-bipyridine (dNbpy, 97%, Aldrich), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%, Aldrich), copper(II) bromide (CuBr2, 99%, Aldrich), triethoxysilane (95%, Aldrich), 1,2-bis(2-iodoethoxy)ethane (BIEE, 96%, Aldrich), rhodamine B (95%, Alfa Aesar), and ethyl 2-bromoisobutyrate (EBiB, 98%, Alfa Aesar) were used as received. The monomers, methyl methacrylate (MMA, 99%, Alfa Aesar) and glycidyl methacrylate (GlyMA, 97%, Aldrich) were passed through a neutral aluminium oxide column to remove the inhibitor, and were left to stir overnight over CaH2, to dry, before use. The monomer, 2-(dimethylamino)ethyl methacrylate (DMA, 98%, Aldrich) was passed through a basic aluminium oxide column to remove the inhibitor and was stirred for 15 min over CaH2 before being filtered into the reaction. The catalysts, copper(I) bromide (CuBr, 99.999%) and coper(I) chloride (CuCl, 99.99%), were supplied by Sigma-Aldrich and were purified by stirring overnight in acetic acid, followed by washing with EtOH and diethyl ether and drying under vacuum, before use. Milli-Q water, obtained from Millipore apparatus with a resistivity of 18.2 MΩ cm at 298 K, was used for all experiments. Dialysis membranes of molecular weight cut-off 12–14 Da were supplied by Medicell membranes Ltd and were washed in water at 40 °C before use to remove the glycerol from the pores.B Synthesis of silica rods
The protocol used for the synthesis of the silica rods was adapted from a previously published work.16,18 In a typical synthesis, 120 g PVP was dissolved in 1.2 L pentanol by sonication for 4 h. Then, 32.4 mL Milli-Q water, 7.2 mL of a 0.18 M sodium citrate solution and 128 mL EtOH were added to the polymer solution. The mixture was stirred with an overhead stirrer for 5 min, before the addition of 24.3 mL ammonium hydroxide solution (25 wt%) and 10.8 mL TEOS. After 30 s, the stirring was stopped, and 4 hours later, 5.4 mL TEOS was added and the flask was gently shaken by hand. The reaction was stopped after 15 h and the reaction medium was centrifuged at 4400g for 90 min. The recovered rods were washed, and size separated by successive centrifugation and redispersion cycles (4 times in EtOH at 736g for 12 min, 4 times in water at 736g for 12 min and 8 times in water at 304g for 12 min). The precipitate was kept and the smaller rods in the supernatant were discarded. Finally, very large rods were removed after two centrifugation cycles in water at 60g, the supernatant being kept, and the precipitates discarded.C Initiator immobilization
The silica rods were calcined at 800 °C for 60 min, using a temperature ramp of 5 °C min−1, under air, to remove the PVP polymer left in their structure from the synthesis. Then they were treated with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v H2SO4/H2O2 piranha solution (Caution! Piranha solution reacts violently with organic matter). Typically, 2 g of rods was dispersed in 30 mL H2SO4 using sonication, followed by the dropwise addition of 10 mL H2O2, and the solution was left stirring overnight before being diluted with water. The rods were recovered by centrifugation at 11000g for 12 min, were washed with water until the pH of the supernatant was around 6, and were dried under vacuum at 60 °C overnight.
:1 v/v H2SO4/H2O2 piranha solution (Caution! Piranha solution reacts violently with organic matter). Typically, 2 g of rods was dispersed in 30 mL H2SO4 using sonication, followed by the dropwise addition of 10 mL H2O2, and the solution was left stirring overnight before being diluted with water. The rods were recovered by centrifugation at 11000g for 12 min, were washed with water until the pH of the supernatant was around 6, and were dried under vacuum at 60 °C overnight.
(3-(2-Bromoisobutyryl)propyl)triethoxysilane (BIBPTES) was synthesized following the protocol of Von Werne et al.27 Next, 2 g of the piranha-treated rods was dispersed in 160 mL dry chloroform using sonication for 3 h, before the addition of 0.7 mL BIBPTES, and the reaction was allowed to proceed under nitrogen for 48 h. The rods were recovered by centrifugation at 11000g for 12 min, and were washed by successive centrifugation/redispersion cycles, twice with chloroform and three times with THF. Finally, they were dried overnight under reduced pressure.
D Surface-initiated polymerizations from the initiator-functionalized rods
200 mg of initiator-functionalized rods was dispersed in 42 mL (396.4 mmol) MMA and 13 mL (99.1 mmol) GlyMA using sonication for 5 min at 5 °C. Then, 194.2 mg (0.48 mmol) of the ligand, dNbpy, and 29 μL (0.2 mmol) of “free” initiator, EBiB, were added under nitrogen. The reaction medium was de-oxygenized by three freeze–pump–thaw cycles, before the addition of 28.4 mg (0.2 mmol) CuBr and 8.8 mg (0.04 mmol) CuBr2. The reaction mixture was placed in an oil bath at 60 °C for 2 to 6 h. The polymerization was stopped by opening the reaction flask to air and diluting the reaction medium with THF. The polymer grafted rods were recovered by centrifugation at 11000g for 12 min and were washed by three successive centrifugation/redispersion cycles in THF and one cycle in acetone. To effectively remove all the non-grafted polymer from the hybrid rods, the latter were left to stir in acetone at 50 °C for 48 h. The grafted rods were then recovered by centrifugation at 11000g for 12 min and washed by three centrifugation/redispersion cycles in acetone. Once the rods were isolated, the reaction medium was passed through a neutral aluminum oxide column to remove the copper catalyst and was precipitated in hexane, to recover the “free” polymer grown in solution, which was kept for GPC analysis. Next, the PMMA grafted rods were dispersed in acetone and were solvent exchanged to DMA monomer using four centrifugation/redispersion cycles. They were then placed in a reaction flask and 56 mL (326.4 mmol) of DMA monomer, 108.5 μL (0.4 mmol) of the ligand, HMTETA, and 9.8 μL (0.07 mmol) of the “free” initiator, EBiB, were added under nitrogen. The reaction medium was de-oxygenized using three freeze–pump–thaw cycles, followed by the addition of 19.8 mg CuCl (0.2 mmol). The reaction was allowed to proceed at 26 °C for 4 to 19 h. The polymerization was stopped by opening the reaction flask to air and diluting with THF. The copolymer-grafted rods were recovered by centrifugation at 11000g for 12 min and were washed by centrifugation/redispersion cycles, three times with THF and once with acetonitrile. To clean the polymer-grafted rods from the “free” polymer, the particles were left to stir in acetonitrile at 50 °C for 48 h. They were recovered by centrifugation and were washed by three centrifugation/redispersion cycles in acetonitrile. The reaction medium, separated from the rods, was passed through a neutral aluminum oxide column and was precipitated in hexane, to recover the non-grafted polymer, which was kept for GPC analysis.
For comparison, one sample grafted with a PDMA homopolymer brush was synthesized, following the same protocol as the PDMA chain extension reaction described above, using initiator-functionalized rods. The PDMA brush was cross-linked by the quaternization of the tertiary amine groups, using a bifunctional quaternization agent, BIEE, and targeting to cross-link 50% of the DMA units in the brush. In detail, 50 mg of PDMA-grafted rods, bearing 0.16 mmol DMA units, were dispersed in 250 mL acetonitrile, before the addition of 7.3 μL (0.04 mmol) cross-linker, BIEE. The reaction was allowed to proceed for 48 h before the purification of the rods by three consecutive centrifugation/redispersion cycles in acetonitrile.
E Dye binding on the rods
Rhodamine molecules were chemically bound onto the polymer functionalized rods by reacting them with the epoxy groups of the GlyMA comonomer. 30 mg of polymer-grafted rods, bearing 0.006 mmol GlyMA units, were dispersed in acetonitrile and were solvent-exchanged to THF by centrifugation/redispersion cycles. The rods were dispersed in 50 mL dry THF, 22 mg (0.05 mmol) rhodamine B was added and the reaction was allowed to proceed overnight, at 60 °C, in the dark. The rods were then recovered by centrifugation and were washed from any unreacted rhodamine using eight centrifugation/redispersion cycles in THF.F Core etching
30 mg of polymer grafted rods, dispersed in acetonitrile, were solvent-exchanged to water, and were finally dispersed in 2 mL of water, before the addition of 1 mL of a 30% HF solution (Caution! HF solution is highly corrosive and pungent fumes are produced). The rods were left to stir for 2 h, before the addition of a 2.5 M NaOH solution to increase the pH of the suspension to 7. The solution was then transferred to a dialysis membrane and was dialyzed against nanopure water for at least one week.G Characterization techniques
The morphology of the samples was determined using a field emission scanning electron microscope (FE-SEM) JEOL JSM-7000F at 15 kV and elemental analysis was performed by EDS, on the same instrument. The samples for imaging were prepared by drop-casting a particle dispersion in water onto glass substrates followed by drying overnight in air. All samples were coated with a 10 nm Au/Pd layer before analysis. The samples for EDS were drop-cast onto GaAs/Au substrates. TEM images were recorded using a JEOL JEM-2100 microscope at 80 kV. The samples were prepared by allowing a drop of the suspension of the rods in water to evaporate on a carbon coated copper grid, before analysis.TGA was performed using a PerkinElmer Pyris Diamond TG/DTA instrument. A temperature ramp from 30 °C to 600 °C, at 10 °C min−1, was applied under nitrogen.
Porosimetry measurements were conducted by nitrogen adsorption and desorption isotherms at 77 K using a NOVA 3200e gas sorption analyzer (Quantachrome, USA). Before the measurements, the samples were degassed at 80 °C under vacuum (<10−5 Torr) for 12 h. The specific surface area values of the samples were calculated by applying the Brunauer–Emmett–Teller (BET) method to the adsorption branch of the N2 isotherms in the 0.05–0.25 relative pressure (P/P0) range.
Molecular weights and molecular weight distributions of the polymers synthesized in solution were determined using a GPC set-up comprising a Thermo Finnigan TSP P1000 pump, two Mixed-D and Mixed-E (Polymer Labs) columns at 40 °C and a refractive index detector (model ERC-RI 101). The eluent was THF with 2 vol% triethylamine at a 1 mL min−1 flow rate. Eight narrow molecular weight linear PMMA standards ranging from 850 to 342900 g mol−1 were used for calibration. In a typical measurement, a 2 wt% solution of the polymer in THF was injected into the system.
1H NMR spectra were recorded on a 500 MHz Avance Bruker NMR spectrometer. The samples were prepared by dissolving the polymers at 20 mg mL−1 in CDCl3, and tetramethylsilane was used as the internal reference.
III Results and discussion
Hard silica rods of average length and diameter of 4.5 ± 2 μm and 330 ± 40 nm, respectively were synthesized by modifying the protocol of Kuijk et al. (Fig. S1, ESI†).16 After calcination and immobilization of initiator molecules on the surface of the rods, the particles were used in the surface-initiated atom transfer radical copolymerization (SI-ATRP) of methyl methacrylate (MMA) with glycidyl methacrylate (GlyMA), using ethyl-2-bromoisobutyrate (EBiB) as a free initiator to control the polymerization. In the second polymerization step, the poly(MMA-co-GlyMA) block was chain extended, due to the living character of ATRP, to grow a second hydrophilic poly(2-(dimethylamino)ethyl methacrylate) (PDMA) block on the outer surface of the colloids (Fig. 1).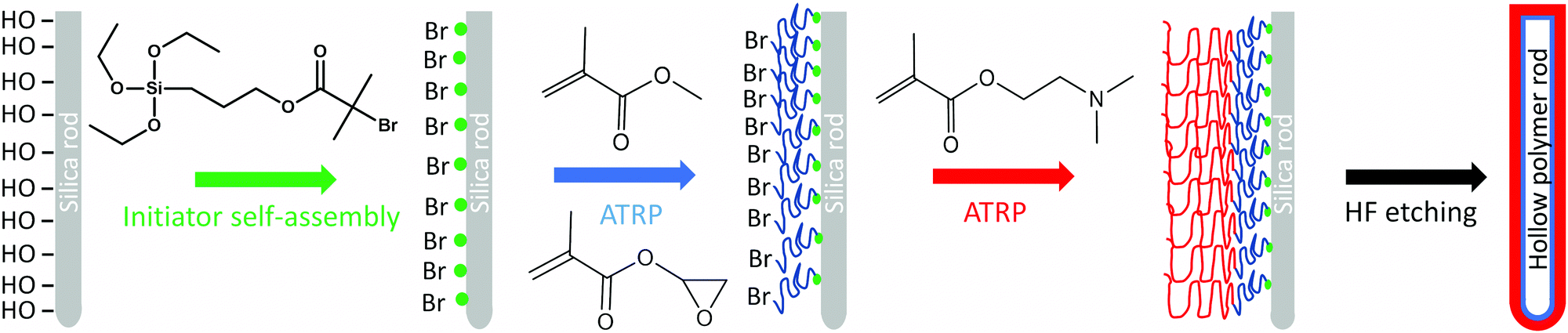 | ||
| Fig. 1 Schematic representation of the synthetic procedure followed for the preparation of the hollow polymer rod colloids. | ||
Silica rods grafted with block copolymer chains of different copolymer compositions were obtained by varying the polymerization time of the two blocks (samples SiP-1 to SiP-3, Table 1). GPC was used to determine the macromolecular characteristics (molecular weights and molecular weight distributions) of the “free” polymer chains, grown in solution from the EBiB initiator, assumed to be similar to those of the grafted polymer chains.28 The molecular weight of the hydrophobic block increased from 35000 g mol−1 to 65000 g mol−1 to 130000 g mol−1, whereas the molecular weight of the PDMA block decreased from 160000 g mol−1 to 90000 g mol−1 to 70000 g mol−1 for SiP-1, SiP-2 and SiP-3, respectively (Table 1).
| Sample | 1st block: P(MMA-co-GlyMA) | 2nd block: PDMA | Hybrid particles | PDMA content by GPCd (wt%) | PDMA content by TGAe (wt%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| GlyMA contenta (mol%) | M n (g mol−1) | M w/Mnb | M n (g mol−1) | M w/Mnb | 1st block contentc (wt%) | Copolymer contentc (wt%) | |||
| a Determined by 1H NMR spectroscopy. b Determined by GPC using PMMA standards. c Determined with TGA. d Calculated using eqn (S1) (ESI). e Calculated using eqn (S2) (ESI). | |||||||||
| SiP-0 | — | — | — | 160000 |
1.1 | — | 42 | 100 | 100 |
| SiP-1 | 0 | 35000 |
1.2 | 160000 |
1.2 | 25 | 57 | 82 | 75 |
| SiP-2 | 20 | 65000 |
1.1 | 90000 |
1.3 | 14 | 31 | 58 | 64 |
| SiP-3 | 20 | 130000 |
1.1 | 70000 |
1.2 | 36 | 38 | 34 | 8 |
Selected polymer-grafted rod samples were dyed with rhodamine B, by the reaction of the glycidyl group of GlyMA with the carboxylic moiety of rhodamine B. A reference sample was also prepared by growing a PDMA homopolymer brush from the surface of the silica colloids (sample SiP-0, Table 1). The hydrophilic PDMA chains in the polymer shell were cross-linked, using 1,2-bis(2-iodoethoxy)ethane as a bifunctional quaternization agent.29
The polymer content of the hybrid rods, following the growth of each block, was assessed by TGA. Characteristic TGA curves are shown in Fig. S3 (ESI†), and the corresponding weight losses are listed in Table 1. The PDMA content of the copolymers was calculated from the molecular weights measured by GPC (eqn (S1), ESI†) and was compared to that obtained by TGA (eqn (S2), ESI†). For SiP-1 and SiP-2, the PDMA content by GPC was found to be 82 and 58 wt%, respectively, in good agreement with the values determined by TGA (75 and 64 wt%). However, for SiP-3, the PDMA content calculated by GPC was 34 wt%, almost 5 times higher than that found by TGA (8 wt%). A plausible explanation for this, is that for the longer PMMA block, some chains became terminated and were therefore not extended with the PDMA block, leading to a lower PDMA content of the hybrid particles, by TGA. Despite their lower PDMA content, the SiP-3 rods could be readily dispersed in water, allowing their further characterization by electron microscopy. The effective grafting density of the polymer chains on the surface of the silica rods was calculated using eqn (S3) and (S4) (ESI†). Given the polydispersity of the rods, an exact value of their specific surface could not be calculated from geometric terms. Instead, the specific surface of the particles was determined by BET nitrogen adsorption experiments and was found to be 11 m2 g−1, which gave a calculated polymer grafting density of ∼0.3 chains per nm2, suggesting the synthesis of dense grafted polymer brushes on the surface of the rods.30
Next, the silica cores were removed with a concentrated hydrofluoric acid solution in water, to yield hollow polymer particles. Following the removal of the silica core, the samples were obtained in water and were labelled HR-0 to HR-3, corresponding to the polymer-grafted rods SiP-0 to SiP-3, respectively. Both the polymer-grafted silica rods and the samples after core etching were observed by SEM to assess their morphology and size. Typical SEM images of the polymer-grafted rods are presented in Fig. S4 (ESI†), verifying that the silica particles were completely and homogeneously coated with the polymer layer.
Fig. 2a shows the SEM image of the hollow polymer particles (HR-0) obtained from the silica rods grafted with the 50% cross-linked PDMA layer. Spherical, donut-like structures with size 450 ± 100 nm were obtained, verifying the hollow nature of the particles. It is worth noting that despite the high degree of cross-linking of the hydrophilic polymer layer, the hollow particles could not retain the highly anisotropic shape and rearranged into the energetically favorable spherical structures. Next, the sample comprising a short hydrophobic PMMA block of Mn = 35000 g mol−1, and a long PDMA block of Mn = 160000 g mol−1 (HR-1, PDMA content 75 wt% by TGA), showed shapeless aggregates (Fig. 2b) and only a few, completely flat, rod-like particles, with size much smaller than that of the precursor silica rods (Fig. 2b inset), suggesting the disruption of the hollow polymer colloids in the aqueous medium.
 | ||
| Fig. 2 SEM images of the hollow polymer samples (a) HR-0 and (b) HR-1. The inset shows the particles at a higher magnification. | ||
However, upon increasing the molecular weight of the hydrophobic block to Mn = 65000 g mol−1 and simultaneously decreasing the molecular weight of the PDMA block to Mn = 90000 g mol−1 (HR-2, PDMA content 64 wt% by TGA), well-defined rod-like particles were observed in SEM (Fig. 3a), which retained the size and shape of the precursor silica particles. The complete removal of the silica core was confirmed by the disappearance of the Si peak in the EDS analysis of the samples deposited on a GaAs/Au substrate (Fig. S5, ESI†). This result is remarkable and reveals that a dense, solvent incompatible polymer layer can prevent the disruption of the large size and aspect ratio hollow polymer rods in the solvent medium, even in the absence of chemical cross-links. The HR-2 rods appeared bent and flat on the substrate, indicating the formation of quite flexible polymer rod particles (Fig. 3a arrows and inset). The dimensions of the HR-2 particles were compared to those of the precursor SiP-2 rods (Fig. 3b and Table S1, ESI†). The particle length and diameter increased from 4.5 ± 2 μm and 390 ± 40 nm for the SiP-2 rods to 5 ± 2 μm and 550 ± 80 nm for the HR-2 rods, which was attributed to the removal of the hard core, and the flattening of the hollow polymer particles on the substrate. Finally, upon further increasing the molecular weight of the hydrophobic block to Mn = 130000 g mol−1 and decreasing the molecular weight of the PDMA block to Mn = 70000 g mol−1 (HR-3, PDMA content 8 wt% by TGA), rigid, tube-like, hollow rod colloids were obtained, which did not deform and flatten upon drying (Fig. 3c). The hollow nature of the colloids was confirmed by SEM (Fig. 3c, inset) and the diameter and length of the HR-3 rods were found to be 420 ± 110 nm and 4.5 ± 2 μm, respectively (Fig. 3d and Table S1, ESI†), in good agreement with the size of the SiP-3 particles (410 ± 40 nm and 4.5 ± 2 μm), suggesting that the overall dimensions of the hollow polymer shell were not affected by the removal of the silica core.
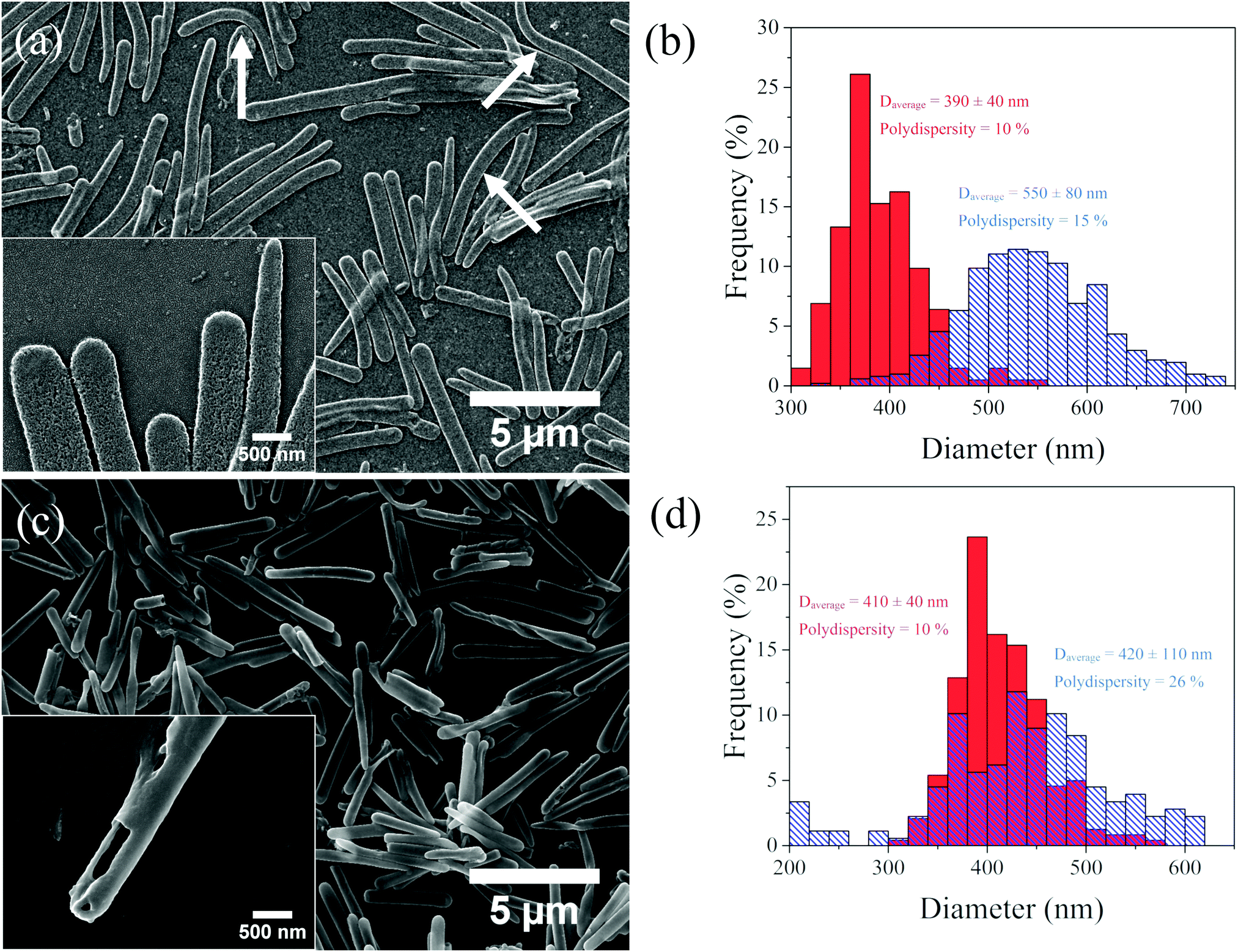 | ||
| Fig. 3 SEM images of the (a) HR-2 and (c) HR-3 hollow polymer microrods. Insets present higher magnification images of the rods. Diameter distributions of (b) core–shell rods SiP-2 (red, filled) and hollow rods HR-2 (blue, line pattern) and (d) core–shell rods. | ||
The hollow polymer rods, HR-2 and HR-3, were also observed by TEM, and were compared to the precursor SiP-2 and SiP-3 particles to assess the thickness of the polymer layer and possible changes in its size following the elimination of the silica core. Before the removal of the core (Fig. 4a and b), the dry thickness of the polymer layer for SiP-2 and SiP-3 was found to be 23 ± 4 nm and 33 ± 5 nm, respectively, in good agreement with the values estimated by SEM (30 nm for SiP-2 and 40 nm for SiP-3) as the difference of the diameter of the bare rods and the core–shell rods (Table S1, ESI†). The complete removal of the silica in the hollow particles was confirmed by the disappearance of the dark colored core in TEM (Fig. 4c and d). For HR-2, light grey and very flexible rod particles were observed, which often folded and twisted, forming multiple kinks along the rod length (Fig. 4c), in good agreement with the SEM results discussed above. The shell of the rods was very light in color and prohibited the determination of its thickness (Fig. 4c, inset). For HR-3, TEM verified the rigid structure of the rods (Fig. 4d), whereas the polymer shell was clearly visible, allowing the determination of the wall thickness and the diameter of the central lumen, found to be 80 ± 10 nm and 260 nm ± 100 nm, respectively (Fig. 4d, inset). When comparing the thickness of the polymer layer for the SiP-3 and HR-3 particles, an increase from 33 nm to 80 nm was found, following the removal of the core, whereas simultaneously the diameter of the central lumen decreased from 330 nm to 260 nm. This increase of the polymer shell thickness and the shrinkage of the central lumen, at a constant overall particle diameter, suggests that the polymer shell undergoes a relaxation process towards the interior of the rod upon removal of the core.
 | ||
| Fig. 4 TEM images of the (a) SiP-2 and (b) SiP-3 core–shell rods and the (c) HR-2 and (d) HR-3 hollow polymer microrods. Insets show higher magnification images of the hollow rods. | ||
Owing to their large size, the hollow polymer rods were visualized by optical microscopy (Fig. S6a, ESI†). Optical microscopy images and a video (Video S1, ESI†) of a dispersion of the HR-2 rods showed several bent rods, attesting to their flexibility in solution. Moreover, the binding of Rhodamine B to the GlyMA units of the block copolymer facilitated the observation of the hollow rods by fluorescence microscopy (Fig. S6b and Video S2, ESI†) and confirmed their potential for colloidal assembly studies.
Overall, we have demonstrated that by grafting a dense amphiphilic block copolymer brush onto inorganic rod particles, and varying the length of the two blocks and the block copolymer composition, well-defined hollow polymer rods of high aspect ratio and very different characteristics can be obtained following core etching. The hollow rods with a lower PMMA content were flat on the solid substrate and highly flexible, whereas the hollow particles with longer PMMA chains were stiff, and their dimensions were similar to those of the precursor core–shell particles. This is explained by considering the increase in the hydrophilicity of the polymer layer when increasing the PDMA content,31 which renders the rods more flexible and deformable in water, whereas for very high PDMA contents (sample HR-1) the block copolymer becomes soluble in water and the rod-like hollow particles are disrupted by the aqueous medium.
Previous reports on the synthesis of hollow polymer rods using the template-assisted method employed hydrophilic or hydrophobic highly cross-linked polymer layers.24–26 In these studies, chemical cross-links were required to retain the rod-like shape of the shell after core etching. In the present work, we demonstrate for the first time, that a non-cross-linked, densely grafted polymer brush, bearing an insoluble inner block, is sufficient to maintain the rod-like shape, after core etching. The colloidal stability is conferred by the hydrophobic PMMA block, which forms a dense, water-incompatible layer in which the chains cannot rearrange and disrupt the rod-like shape, even for a rod length as large as 10 μm.
Finally, the absence of cross-links within the polymer layer was exploited in the disruption of the hollow polymer rods when varying the solvent quality for the block copolymer. For this, an equal volume of THF, which is a good solvent for both the PMMA and the PDMA blocks, was added to an aqueous dispersion of the HR-2 rods, and the colloidal stability of the particles was followed as a function of time, by SEM. Immediately after solvent mixing, the rod-like polymer particles were clearly observed (Fig. 5a), while after 24 h in the solvent mixture, fewer rods, embedded in a polymer film, were found, suggesting the gradual disruption of the anisotropic colloids to the constituent polymer chains (Fig. 5b). After 7 days in the water/THF medium, the rods completely disappeared, and a polymer film was observed on the substrate (Fig. 5c). On the other hand, the hollow polymer rods dispersed in water were found stable for up to 7 months (Fig. S7, ESI†).
 | ||
| Fig. 5 SEM images of the HR-2 hollow polymer rods in a 50/50 v/v water/THF mixture, (a) immediately after solvent mixing, and after (b) one day and (c) seven days in the solvent mixture. | ||
The tunable disruption of the polymer rods in selective solvents could be utilized in switchable anisotropic colloidal phases or in the controlled release of encapsulated active compounds from the lumen of the hollow rods.
IV Conclusions
In conclusion, a novel approach for the synthesis of non-cross-linked, hollow polymer rod-like colloids of large size and aspect ratio, with tunable shell thickness and flexibility has been proposed. The hollow polymer microrods were synthesized using a template-assisted method, by grafting dense P(MMA-co-GlyMA)-b-PDMA block copolymer brushes from the surface of silica rods, followed by etching of the inorganic core. The presence of an inner dense hydrophobic block, of sufficient thickness, allowed the polymer brush to retain the rod-like shape, even in the absence of chemical cross-links, while the second hydrophilic block enabled the facile dispersion of the particles in water. The colloidal stability and flexibility of the hollow polymer rods were successfully tuned by varying the block copolymer composition and the sizes of the two blocks. Moreover, due to the large size and aspect ratio of the colloids and the ability to bind a fluorescent dye onto the polymer layer, the particles could be observed by optical and fluorescence microscopy. Finally, the disruption of the hollow polymer rods in a good solvent for both blocks was demonstrated, which could be utilized in delivery systems or phase transformation applications. A systematic study to precisely define the influence of the block sizes and/or copolymer composition on the stability and flexibility of the hollow polymer rods is underway. The proposed approach is generic and could be easily extended to any solvent medium by the appropriate selection of the solvent incompatible inner block, whereas the second block could be exploited to diversify the particles’ properties, inducing for example stimuli-response to access novel classes of colloidal microstructures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no. 641839. The authors thank Aleka Manousaki, Dimitrios Theodoridis and Ioannis Vamvasakis for assistance with the SEM, TEM and nitrogen absorption measurements, respectively.Notes and references
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- A. Satoh and Y. Sakuda, Mol. Phys., 2007, 105, 3145–3153 CrossRef CAS.
- J. Noh, C.-M. Yoon and J. Jang, J. Colloid Interface Sci., 2016, 470, 237–244 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- S. T. Knauert, J. F. Douglas and F. W. Starr, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1882–1897 CrossRef CAS.
- A. S. Lubansky, D. V. Boger and J. J. Cooper-White, J. Non-Newtonian Fluid Mech., 2005, 130, 57–61 CrossRef CAS.
- C. Zhou, Q. Wu, Y. Yue and Q. Zhang, J. Colloid Interface Sci., 2011, 353, 116–123 CrossRef CAS PubMed.
- M. J. Stephen and J. P. Straley, Rev. Mod. Phys., 1974, 46, 617–704 CrossRef CAS.
- E. Grelet, Phys. Rev. X, 2014, 4, 021053 Search PubMed.
- A. Repula, M. Oshima Menegon, C. Wu, P. van der Schoot and E. Grelet, Phys. Rev. Lett., 2019, 122, 128008 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- G. Ambrožič, S. D. Škapin, M. Žigon and Z. C. Orel, J. Colloid Interface Sci., 2010, 346, 317–323 CrossRef PubMed.
- M. Ocaña, M. P. Morales and C. J. Serna, J. Colloid Interface Sci., 1999, 212, 317–323 CrossRef PubMed.
- V. Sharma, K. Park and M. Srinivasarao, Mater. Sci. Eng., R, 2009, 65, 1–38 CrossRef.
- N. Hijnen and P. S. Clegg, Chem. Mater., 2012, 24, 3449–3457 CrossRef CAS.
- A. Kuijk, A. van Blaaderen and A. Imhof, J. Am. Chem. Soc., 2011, 133, 2346–2349 CrossRef CAS.
- M. P. B. V. Bruggen, Langmuir, 1998, 14, 2245–2255 CrossRef.
- A. Kuijk, A. Imhof, M. H. W. Verkuijlen, T. H. Besseling, E. R. H. van Eck and A. van Blaaderen, Part. Part. Syst. Charact., 2014, 31, 706–713 CrossRef CAS.
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS PubMed.
- M.-H. Li and P. Keller, Soft Matter, 2009, 5, 927 RSC.
- Z. Zhang, P. Pfleiderer, A. B. Schofield, C. Clasen and J. Vermant, J. Am. Chem. Soc., 2011, 133, 392–395 CrossRef CAS PubMed.
- R. G. Alargova, K. H. Bhatt, V. N. Paunov and O. D. Velev, Adv. Mater., 2004, 16, 1653–1657 CrossRef CAS.
- C. Fernández-Rico, T. Yanagishima, A. Curran, D. G. A. L. Aarts and R. P. A. Dullens, Adv. Mater., 2019, 31, 1807514 CrossRef PubMed.
- Q. Li, S. Jin and B. Tan, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- G. Liu, X. Yang and Y. Wang, Langmuir, 2008, 24, 5485–5491 CrossRef CAS PubMed.
- O. Shimoni, Y. Yan, Y. Wang and F. Caruso, ACS Nano, 2013, 7, 522–530 CrossRef CAS PubMed.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS PubMed.
- C. Kang, R. Crockett and N. D. Spencer, Polym. Chem., 2016, 7, 302–309 RSC.
- V. Bütün, A. B. Lowe, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1999, 121, 4288–4289 CrossRef.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143–9150 CrossRef CAS.
- F. L. Baines, S. P. Armes, N. C. Billingham and Z. Tuzar, Macromolecules, 1996, 29, 8151–8159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01868a |
| This journal is © The Royal Society of Chemistry 2020 |