DOI:
10.1039/C9SM01847F
(Review Article)
Soft Matter, 2020,
16, 54-63
Dynamic surface-assisted assembly behaviours mediated by external stimuli
Received
12th September 2019
, Accepted 21st October 2019
First published on 21st October 2019
Abstract
Supramolecular self-assembly behaviors on solid substrates have been widely investigated in the last few decades. Owing to the complexity of interfacial assembly systems, the precise regulation of supramolecular nanostructures is still challenging and waits to be solved. The supramolecular nanostructures are governed by non-covalent bonds, so they can be disrupted and influenced by an external environment. In this review, the dynamic supramolecular nanostructures that are mediated by external stimuli containing guest species, light irradiation, temperature and electric field are discussed in detail. The research studies mentioned in this article are all accomplished by STM, and the effects of these external stimuli on the assembled nanostructures have been elucidated exhaustively here.
1. Introduction
Supramolecular self-assembly is widely regarded as a promising nanotechnology due to its potential advantages in creating desirable functional nanomaterials/surfaces through precisely regulating nanostructures.1–4 Directed by non-covalent bonds comprising hydrogen bonds, halogen bonds and metal–ligand interactions, building units spontaneously aggregate into well-organized architectures during the self-assembly period.5–12 Currently, 2D surface-assisted self-assembly has attracted much attention and series of research achievements have been made. However, complete control over self-assembled systems remains to be resolved. The major forces involved in 2D self-assembly mainly consist of intermolecular and interfacial interactions. In general, self-assembled nanostructures are the balanced outcome of the molecular adsorption–desorption process arising from their physisorption on substrates and intermolecular non-covalent driven forces. Therefore, slight changes in the adsorbates,13,14 substrates15,16 or solvents17,18 can even exert a remarkable influence on the interactions among them, finally accounting for the rearrangement of adsorbates on the substrate. It is noteworthy that the nanostructures of absorbates are susceptible to external stimuli such as guest molecules and light irradiation.19–22 For instance, guest molecules can co-locate with adsorbates on substrates by interacting with adsorbates or substrates, and light stimuli are capable of triggering the conversion of adsorbate conformation, which is helpful in interfacial structural transitions. Hence, the proper control of an external environment is expected to manipulate the interfacial nanostructures, and should be highly-focused in the self-assembly process.
Since its invention in the 1980s, scanning tunnelling microscopy (STM) has extensively been applied for surface analysis by virtue of its sub-nanometre resolution, non-destructive test over the sample and various operating environments. In particular, it has been demonstrated that STM under ambient conditions is a powerful technique for investigating the dynamic assembly behaviours at the solid/solution interface caused by external stimuli. Assisted by STM, information of the real-space, real-time distribution of molecules and atoms on a surface can be provided. The local structure of the surface monolayer instead of the average nature of the bulk or the entire surface can be obtained via STM, resulting in the direct observation of surface defects. Besides, various environmental conditions including ultra-high vacuum, ambient conditions and a solution environment are suitable for STM, and special methods of preparing samples are not required. However, it is difficult using STM under ambient conditions to characterize molecules physisorbed on poorly conductive substrates such as insulators. Simultaneously, molecules that possess poor planarity and poor ability to self-assemble into stable nanostructures are not favourable for their STM investigations. In addition, regarding the self-assembly systems at the liquid/solid interface, solutions with good conductivity can impede STM imaging since they can seriously affect the true resolution of molecules adsorbed on a surface. Remarkably, STM can also act as a suitable external stimulant to the interfacial self-assembly systems as well as a molecular imaging tool.23,24 A localized control of reversible electric field can be provided by STM, which has an important effect on the molecular orientation and packing nanostructures. In the review, the responsiveness of the surface-assisted assembly systems to four external stimuli, guest species, light irradiation, temperature and electric field investigated by STM under ambient conditions, will be introduced in detail separately. Guest species can form co-assembly systems with host molecules stabilized by the immobilization of cavities or host–guest non-covalent interactions, thereby manipulating the host architectures. With regard to light irradiation, the isomerization and chemical reactions including [2+2] photodimerization reactions and polymerization of photo-sensitive molecules can be induced. Furthermore, the elevated temperature is helpful for fabricating a thermodynamically stable architecture through overcoming the kinetic barrier. Finally, the electric field can directly control the orientation of polar/charged molecules on substrates affecting the final nanoarchitectures, induce the construction of a bilayer architecture, and even trigger the transformation from supramolecular networks to covalent organic networks.
2. Regulatory effect of guest species
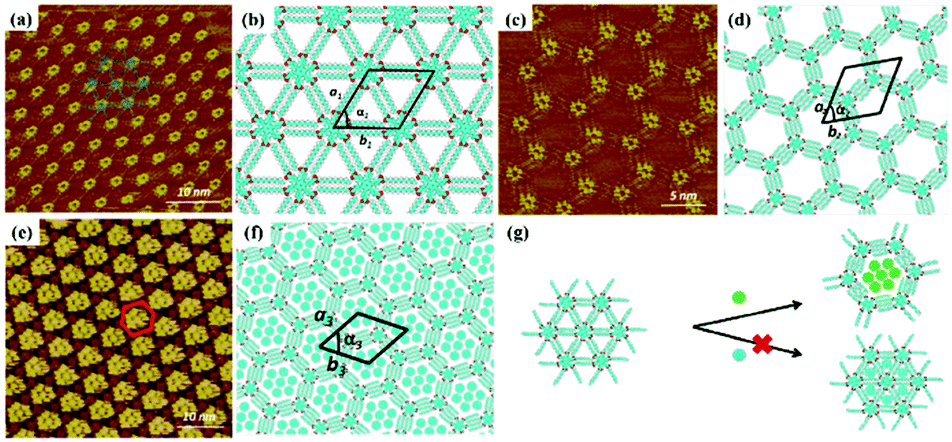
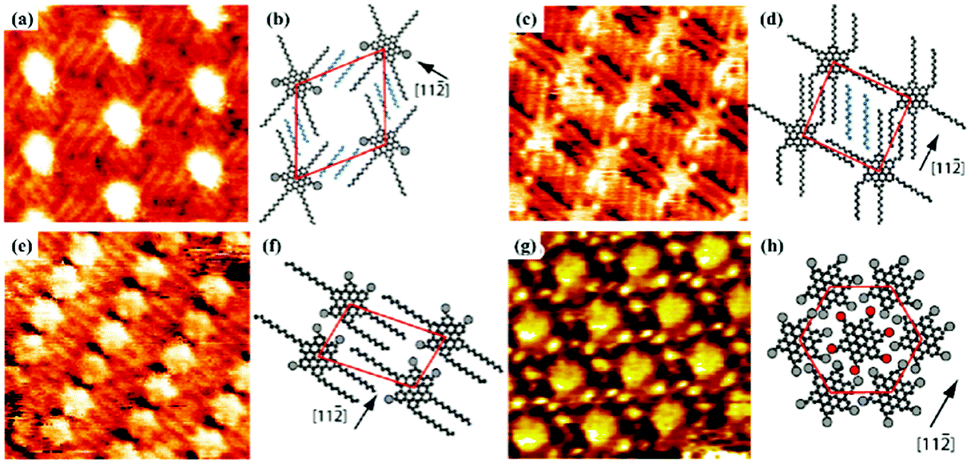
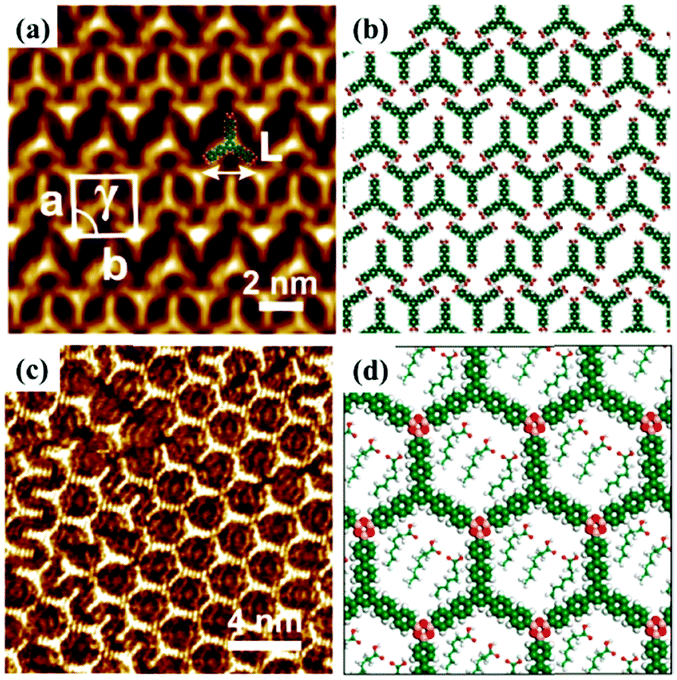
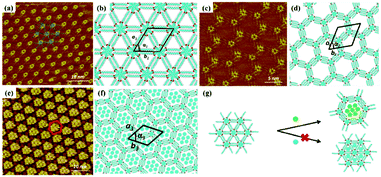
Host–guest supramolecular chemistry is regarded as a crucial approach in 2D supramolecular crystal engineering. The regulation of supramolecular architectures can be fulfilled by introducing guests whose size and shape match well with host networks to co-locate on the surface for the stabilization of the whole system. The sensitivity of the host architectures to the guest molecules can be promising in the trace measurements of guest molecules. Coronene (COR), phthalocyanine and fullerene are three common molecules selected to initiate the structural reconstruction of hosts.25–30 In comparison to rigid host architectures with fixed nanopores, the flexible architecture is more easily modulated for trapping guests. A HPB molecule is composed of a hexaphenylbenzene unit with six alkyl chains and can interact with others through alkyl-chain integrations.31 Snowflake (Fig. 1(a)) and honeycomb (Fig. 1(c)) architectures are fabricated by HPB molecules on HOPG, but the latter is unstable and easily substituted by the former. A snowflake architecture then totally turns into a honeycomb architecture evoked by COR molecules so that the new hexagonal cavities are suitable for accommodating COR heptamers (Fig. 1(e)). Though the size of triangle nanopores in the snowflake architecture is identical to that of COR, the steric hindrance impedes the accommodation of COR in the snowflake architecture (Fig. 1(g)). In addition, the reconstructed host architectures are related with the concentration of guests. As an acid-functionalized benzo phenanthrene derivative, H3TTCA can transform into a honeycomb architecture possessing hexagonal nanopores formed by six H3TTCA molecules through O–H⋯O H-bonds on HOPG (Fig. 2(a)).32 After adding COR at a concentration of 10% or 50% saturation, the original honeycomb architecture is divided into numerous individual hexagonal rings with COR molecules occupying the triangular and even hexagonal cavities resulting from segmental H-bond cleavage (Fig. 2(c) and (e)). When the concentration reaches 100% saturation, the H3TTCA host architecture turns into parallel lines and COR molecules arrange in a linear orientation (Fig. 2(g)). Besides, the supramolecular bilayer of the host on the surface can be successfully fabricated by the introduction of nonplanar C60 molecules,33,34 and the host architecture can be simultaneously tuned by multiple guests.25,35,36
 |
| | Fig. 1 High-resolution STM images of HPB's adlayers physisorbed on HOPG: (a and c) before adding COR, (e) after adding COR. (b), (d), (f) Built molecular packing patterns of the adlayer in (a), (c), (e) respectively. (g) Schematic illustration of the HPB's adlayer capturing seven COR molecules. (Reprinted with permission from ref. 31. Copyright 2015 American Chemical Society). | |
 |
| | Fig. 2 High-resolution STM images of H3TTCA's adlayers physisorbed on HOPG: (a) before adding COR and (c), (e), (g) after adding COR at a concentration of 10%, 50% and 100% saturation respectively. (b), (d), (f), (h) The corresponding built molecular packing models of the adlayers in (a), (c), (e), (g) respectively. (Reprinted with permission from ref. 32. Copyright 2015 The Royal Society of Chemistry). | |
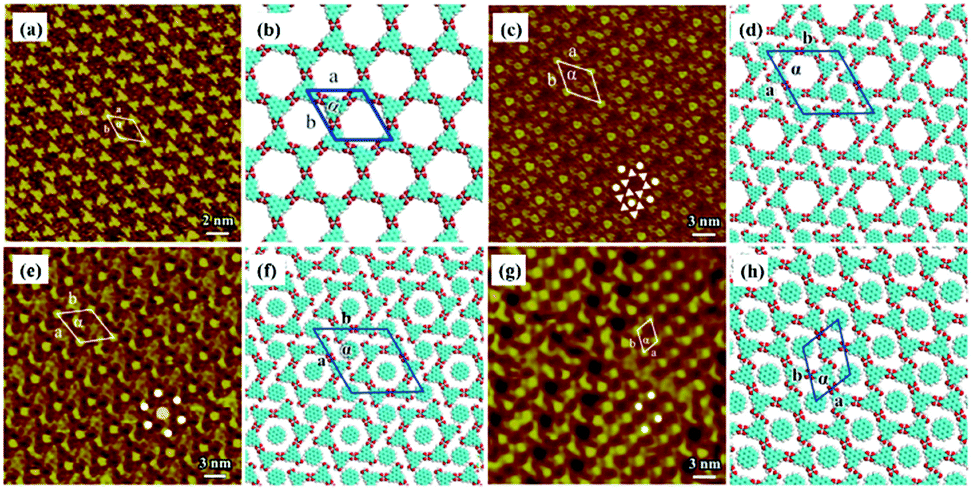
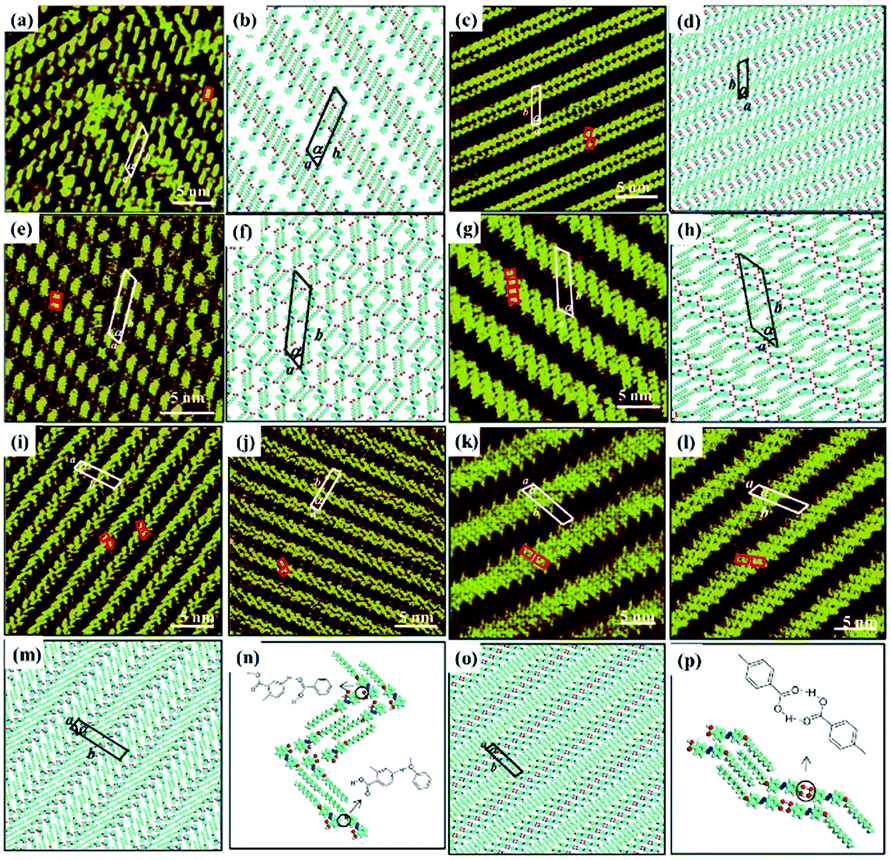
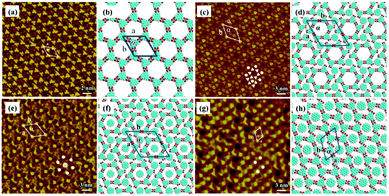
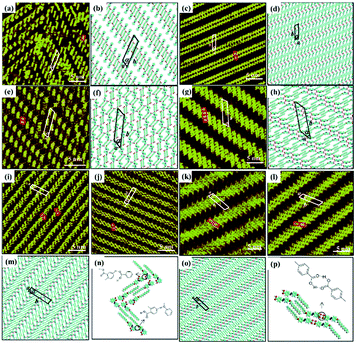
Other types of functionalized guest molecules such as pyridine and melamine derivatives regulate host architecture through host–guest non-covalent interactions and the ultimate outcome is relevant to the concentration and chemical structure of guest molecules.37–41 H4TCPE and H4ETTC are tetraphenylethylene derivatives end-capped with carboxyl groups and aggregated into lamellar and quadrilateral architectures via intermolecular O–H⋯O H-bonds at the solid/liquid interface separately (Fig. 3(a) and (e)).39 Owing to the higher hydrogen density of H4TCPE's individual architecture and the spatial chemical structure of H4TCPE molecules, bipyridine derivatives (DPE, PEBP-C4, and PEBP-C8) almost cannot trigger any phase transition of H4TCPE (Fig. 3(i) and (k)). The stability of the H4TCPE's individual architecture is also demonstrated by DFT results wherein the total energy per unit area of the H4TCPE's individual architecture is lower than that of H4TCPE/pyridine, which indicates the difficulty in mediating the individual architecture of H4TCPE by pyridine derivatives. While regarding H4ETTC with an extra benzene ring on each branch, its architecture can be destroyed by the participation of bipyridine derivatives, and replaced by the acid–pyridine alternating architecture ascribed to the formed H4ETTC–pyridine O–H⋯N H-bonds (Fig. 3(c), (g), (m) and (o)). Simultaneously, increasing the concentration of DPE induces H4ETTC molecules to generate a densely-packed architecture by interacting with more DPE molecules (Fig. 3(c) and (g)), and the steric hindrance originated from alkoxyl chains of pyridine derivatives leads to distinct structural transitions of H4ETTC.
 |
| | Fig. 3 High-resolution STM images and the calculating molecular models of H4TCPE's adlayers physisorbed on HOPG: (a), (b) before adding pyridine derivatives; (i), (j) after the addition of PEBP-C4; (k), (l) after the addition of PEBP-C8. High-resolution STM images and the calculated molecular models of H4ETTC's adlayers physisorbed on HOPG: (e), (f) before adding pyridine derivatives; (c), (d) after the addition of a saturated DPE solution; (g), (h) after the addition of a 33.33% saturated DPE solution; (m), (n) after the addition of PEBP-C4; (o), (p) after the addition of PEBP-C8 (Reprinted with permission from ref. 39. Copyright 2018 Springer Nature). | |
3. Regulatory effect of light irradiation
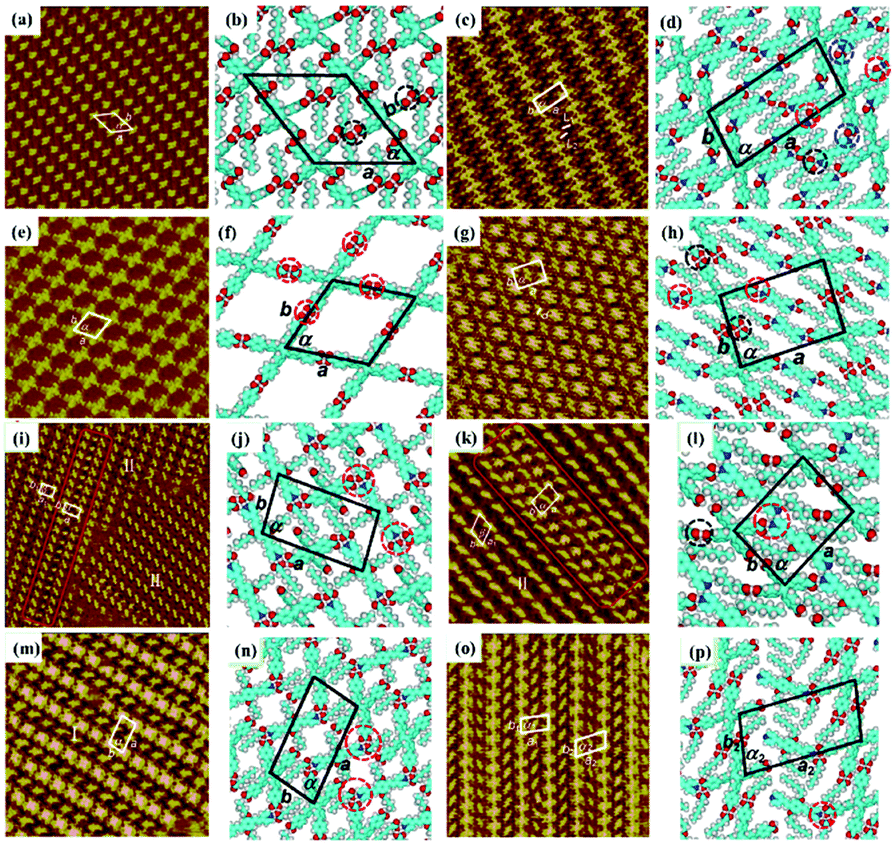
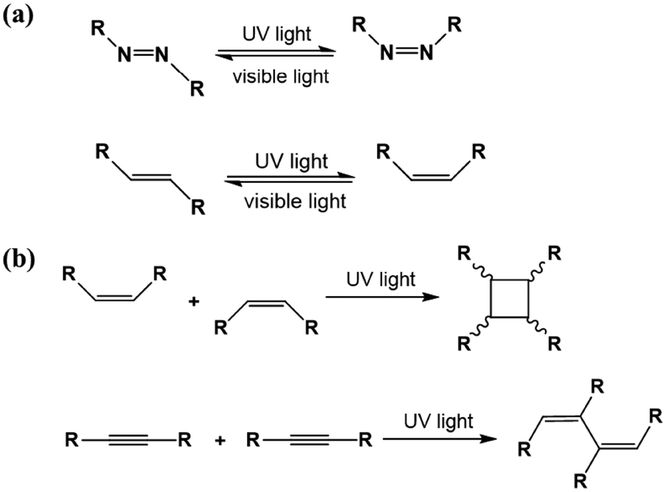
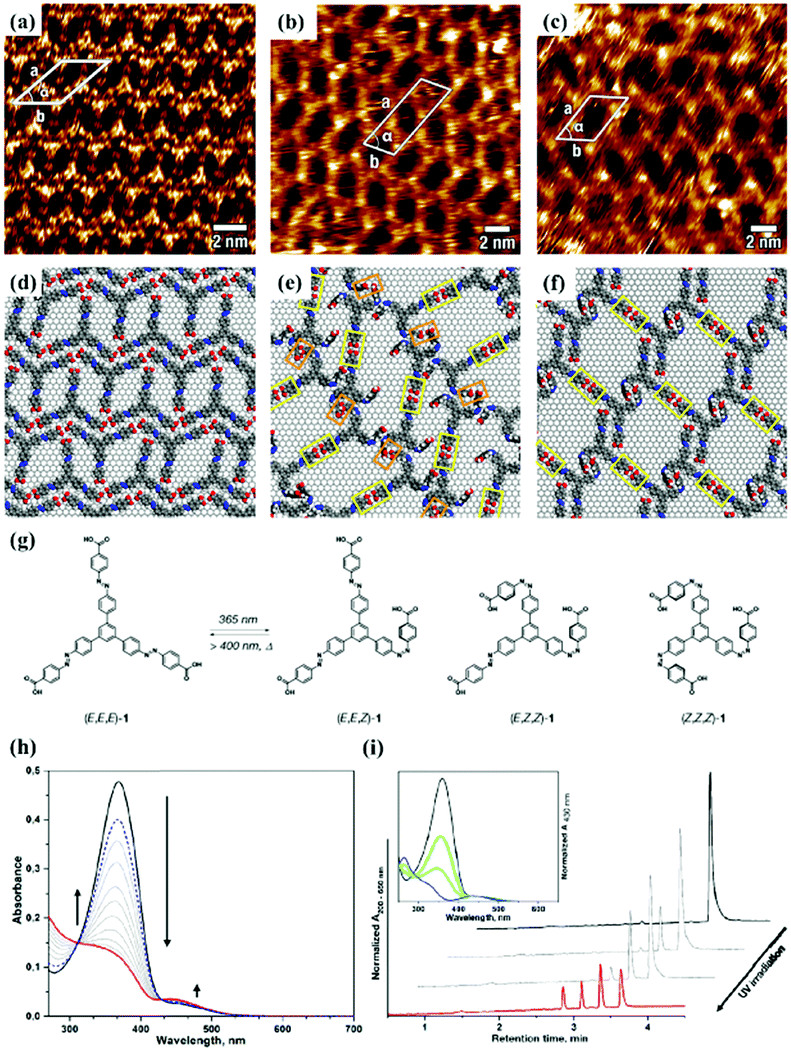
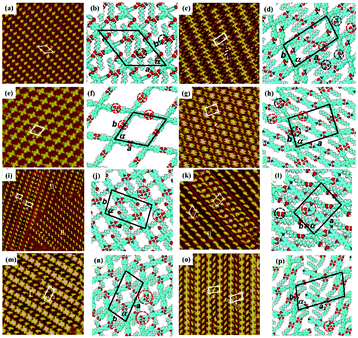
As an effective way to mediate supramolecular architecture, photochemistry should be brought to the forefront. The original bond cleavage and new bond generation can be caused by the adsorption of photons, consequently leading to the phase transition or chemical reactions. Organic molecules containing photo-sensitive units, such as azobenzene moieties, double bonds and diacetylene bonds (Fig. 4), can undergo geometrical rearrangement or chemical reactions after exposure to an appropriate wavelength.42–44 Such photo-responsive characteristics make these organic molecules promising in optoelectronic devices such as molecular switches and optical information collectors. To the best of our knowledge, switchable cis–trans isomerization can occur in the azobenzene or olefin derivatives upon irradiation by UV/visible light. Initially, the photoisomerization processes of 4NN-macrocycle molecules comprising four azobenzene moieties are revealed by STM with the help of a TCDB template.45 Both the cis and trans conformations of 4NN-macrocycle can be distinguished by the shapes of spots in the STM images, and it is found that the conformational transition of 4NN-macrocycle molecules from (t, t, t, t) to (t, c, t, c) and (t, t, t, c) is controlled by UV light at 365 nm. Later, the responsiveness of the individual supramolecular system of the acid-terminated compound 1 that contains three azobenzene moieties located at the 1, 3, 5 positions of a benzene ring to light irradiation is investigated.46 The azobenzene branches decorated with carboxyl groups are utilized to generate intermolecular H-bonds for managing the self-assembly. Nonirradiated (E,E,E)-1 spontaneously constitutes a densely-packed morphology via O–H⋯O H-bonds (Fig. 5(a)), while the subsequent photoirradiation only gives rise to two loosely porous assembly architectures of (E,E,Z)-1 and (E,Z,Z)-1 molecules respectively (Fig. 5(b) and (c)). Molecular mechanics/dynamics simulations indicate that the probable reason for no observation of (Z,Z,Z)-1 molecules on the graphite surface is the weaker molecule–substrate interactions owing to the non-planar characteristics of compound 1's cis geometry compared with its trans geometry. To further confirm the impact of light irradiation on the chemical structure of compound 1 concluded from STM observations, UV-vis absorption spectroscopy (Fig. 5(h)) and HPLC analysis (Fig. 5(i)) are also conducted. In Fig. 5(h), the absorption maxima of (E,E,E)-1 is located at 370 nm for the π–π* band and 455 nm for the n–π* band. After UV irradiation, the absorption maxima for π–π* and n–π* bands decrease and increase, respectively, which implies the E → Z isomerization of compound 1. HPLC (Fig. 5(i)) is utilized to further separate the photogenerated isomers and reveal three additional peaks of compound 1 after UV irradiation. The inset absorption spectra of compound 1's four possible isomers inosculate with the computed absorption spectra, which verifies the transition of compound 1 from (E,E,E)-1 to (E,E,Z)-1, (E,Z,Z)-1 and (Z,Z,Z)-1 triggered by UV irradiation. In some cases, photoirradiation makes no difference to the self-assembly adlayers on the surface. In a latest work, the azobenzene units modified by alkoxyl chains and carboxyl groups can form stable monolayers by themselves on HOPG.47 Importantly, carboxyl groups located at different positions – the end of the alkoxyl chains or the meta/ortho/para positions of azobenzene rings – are found to significantly affect the final assembly architectures on HOPG caused by photoirradiation. As for Azo-1 (Fig. 6(a) and (e)) and meta-Azo molecules (Fig. 6(c) and (g)), the STM results indicate that the molecular shape changes from linear to curving spots triggered by UV illumination, indicating the isomer transition from trans to cis. Afterwards, the isomer transition gives rise to the changes in intermolecular interactions, and accordingly the dramatic realignments of molecules. However, both the pure assembly architectures of ortho-Azo (Fig. 6(i) and (j)) and para-Azo molecules (Fig. 6(k) and (l)) on the surface are insensitive to photoirradiation and still remain unchanged due to their extremely high stability. The photo-induced isomerization effect of light irradiation on the multicomponent system has also been studied. In the rectangle-like trans-NN4A/DPE binary architecture, each trans-NN4A molecule connects with four DPE molecules via N⋯H–O H-bonds before light irradiation.48 UV light can trigger the primary architecture to turn into a cis-NN4A/DPE zigzag architecture attributed to the isomerization of NN4A. Eventually, the visible light gives rise to trans-NN4A accounting for the re-emergence of the original rectangle trans-NN4A/DPE architecture.
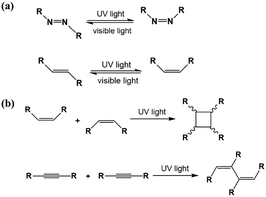 |
| | Fig. 4 (a) trans–cis transformation of the azobenzene moiety and the double bond responding to light irradiation. (b) Photodimerization and polymerization reactions of double and diacetylene bonds induced by light irradiation, respectively. | |
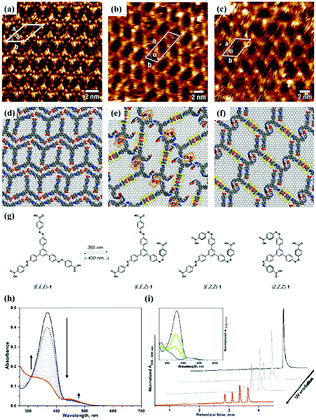 |
| | Fig. 5 High-resolution STM images of compound 1's adlayers physisorbed on HOPG: (a) before UV light irradiation; (b) and (c) after UV light irradiation. (d)–(f) The corresponding built molecular packing models of the adlayers in (a), (b), (c), respectively. (g) Schematic illustration of compound 1's isomerization exposure to UV light. (h) UV-vis absorption spectroscopy of compound 1 in DMSO before (black curves) and after (red curves), UV irradiation and after visible light irradiation (dashed blue curves). (i) HPLC traces recorded for compound 1 solutions before (black curves) and after UV irradiation (red curves). Inset, UV-vis absorption spectra of (E,E,E)-1 (black curves), (E,E,Z)-1 (green curves), (E,Z,Z)-1 (light blue curves) and (Z,Z,Z)-1 (blue curves). (Reprinted with permission from ref. 46. Copyright 2018 American Chemical Society). | |
 |
| | Fig. 6 STM images and the corresponding proposed models of the adlayers of four azobenzene derivatives before UV light irradiation: (a) and (b) Azo-1; (c) and (d) meta-Azo; (i), (m) and (n) ortho-Azo; (k), (o) and (p) para-Azo. High-resolution STM images and the corresponding proposed models of the adlayers of four azobenzene derivatives after UV irradiation: (e) and (f) Azo-1; (g) and (h) meta-Azo; (j) ortho-Azo; (l) para-Azo. (Reprinted with permission from ref. 47. Copyright 2019 The Royal Society of Chemistry). | |
Moreover, [2+2] photodimerization reactions can be efficiently implemented under the condition wherein the reacting photo-sensitive units are parallel to each other and the distance between neighbouring photo-sensitive groups is less than 4.2 Å.49 Molecule 2, a stilbene derivative substituted with alkoxyl chains, can constitute densely packed monolayers with dimers governed by van der Waals forces on HOPG (Fig. 7(a)).50 After exposure to UV irradiation for 5 min, a new architecture slightly different from the initial in domain A is observed in domain B (Fig. 7(b)), where molecule 2 has transformed into cis-conformation, and molecule 2 dimers are arranged in conformation 1/conformation 2 indicated by white/yellow arrows (Fig. 7(c)). With the irradiation time extending to 20 min, photodimerization reactions merely occur in the regions of conformation 1 (Fig. 7(f)) where the double bond is parallel to the other bond in each dimer and the distance is within 4.2 Å. On the other hand, in conformation 2, the distance between units reaches to 6.2 Å so that the photodimerization reactions are suppressed. According to the UV-vis spectra of molecule 2 before (black curves) and after (red curves) UV irradiation for 5 min, and after 20 min (blue curves), as shown in Fig. 7(h), the absorption maxima changed from 323 nm to 274 nm after UV irradiation for 5 min, confirming the transition of molecule 2 from trans-2 to cis-2. With the irradiation time prolonged to 20 min, the absorption maxima (274 nm) decreases, indicating that the photodimerization reaction occurs in molecule 2 except for photoisomerization. Another example is the light-induced polymerization of diacetylene acid derivatives depending on the relative position of the diacetylene bond and the carboxyl group in the molecule.51 DA-17, DA-19 and DA-21 molecules whose diacetylene unit directly combined with the carboxyl group can form a paralleled architecture where alkyl chains interdigitate and the distance between neighbouring parallel diacetylene bonds exceeds 5 Å so that they are not expected to be photopolymerizable. But concerning DA-23, DA-25 with a diacetylene moiety located away from the carboxyl group, lamellar structures with alkyl chains in a tail-to-tail assembly before light irradiation can be detected by STM and the short distance between adjacent diacetylene units satisfies the requirements of photodimerization reactions.
 |
| | Fig. 7 (a) STM image of molecule 2's adlayers physisorbed on HOPG before UV irradiation. Large-scale and high-resolution STM images of molecule 2's self-assembled adlayers after UV irradiation: (b) and (c) for 5 min; (e) and (f) for 20 min. (d) and (g) The corresponding built molecular models of the adlayers in (c) and (f) respectively. (h) UV-vis spectra of molecule 2 in chloroform before (black curves), and after (red curves) UV light irradiation at 365 nm for 5 min and for 20 min (blue curves). (Reprinted with permission from ref. 50. Copyright 2014 American Chemical Society). | |
4. Regulatory effect of temperature
Temperature is an essential factor for regulating self-assembly processes, which could have an obvious effect on thermodynamics and kinetics. In the formation stage of molecular nanoarchitecture at the solution/solid interface, the elevated temperature can intensify the mobility of an adsorbate, change the viscosity of a solvent and even improve the solution concentration as a solvent evaporates, leading to diversified nanoarchitectures possessing various densities by breaking kinetic trapping effects.52–54 An early STM study on CoOEP and NiOEP unambiguously reveals the enhanced mobility of the adsorbate at high temperature and suggests the extreme stability of dynamically controlled supramolecular networks through highly tight binding of adsorbates with the surface below 100 °C.55 Rico Gutzler et al. found that the nanoarchitecture of BTB at the n-nonanoic acid/graphite interface can be converted between two architectures – nanoporous chicken-wire56 and densely-packed strip architectures – provoked by temperature, which is mostly caused by the adsorption and desorption of the solvent on the graphite surface.57 At room temperature, the BTB's chicken-wire architecture instead of the strip architecture is observed at the interface, thanks to the significant contribution of numerous n-nonanoic acid molecules located in the nanopores of the chicken-wire architecture to the lower Gibbs free energy. While at a high temperature, the n-nonanoic acid molecules desorb from the surface and enter into the solution, resulting in the improvement of Gibbs free energy of the chicken-wire architecture without n-nonanoic acid molecules and the transformation from a chicken-wire architecture into a strip architecture. Another similar example is the temperature-dependent research on the self-assembly nanoarrays of HBC-C12 at the n-tetradecane/Au(111) interface.58 As the temperature rises from 20 °C to 50 °C (Fig. 8), the n-tetradecane molecules and the alkyl chains of HBC-C12 desorb gradually from the Au surface, but more HBC-C12 molecules adsorb on the surface, which generates changes in the dominant forces in the HBC-C12 supramolecular system and three phase transformations. At the same time, the phase transition temperature is correlated with the solvent species. As for an aliphatic acid solvent, the binding enthalpies with graphite enhance with the increased length of alkyl chains, leading to difficulties in the desorption of solvent molecules and the increased phase transition temperature.57 The phase transition temperature is also closely associated with the concentration of the adsorbate. Matthew O. Blunt et al. mention that DBA-OC16 molecules at a higher concentration can undergo a structural transformation at a lower temperature, and propose a thermodynamic model to estimate enthalpy and entropy changes related to phase transition.59
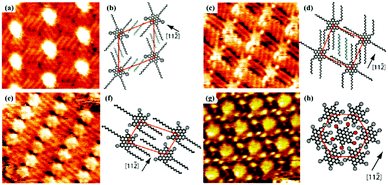 |
| | Fig. 8 High-resolution STM images of HBC-C12's packing adlayers at different temperatures at the n-tetradecane/Au(111) interface: (a) 20 °C, (c) 25 °C, (e) 35 °C, (g) 50 °C. (b), (d), (f) and (h) The corresponding calculating models of adlayers in (a), (c), (e) and (g) respectively. (Reprinted with permission from ref. 58. Copyright 2010 American Chemical Society). | |
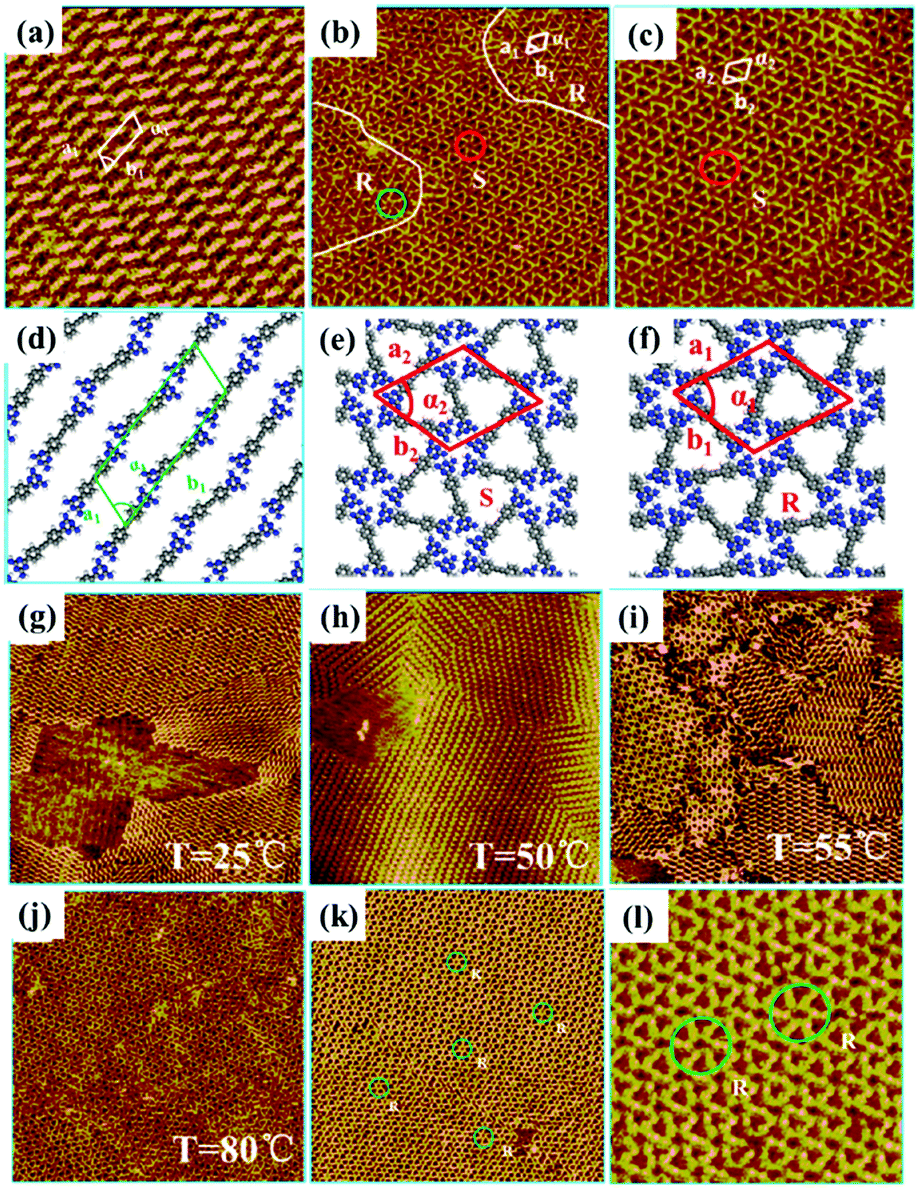
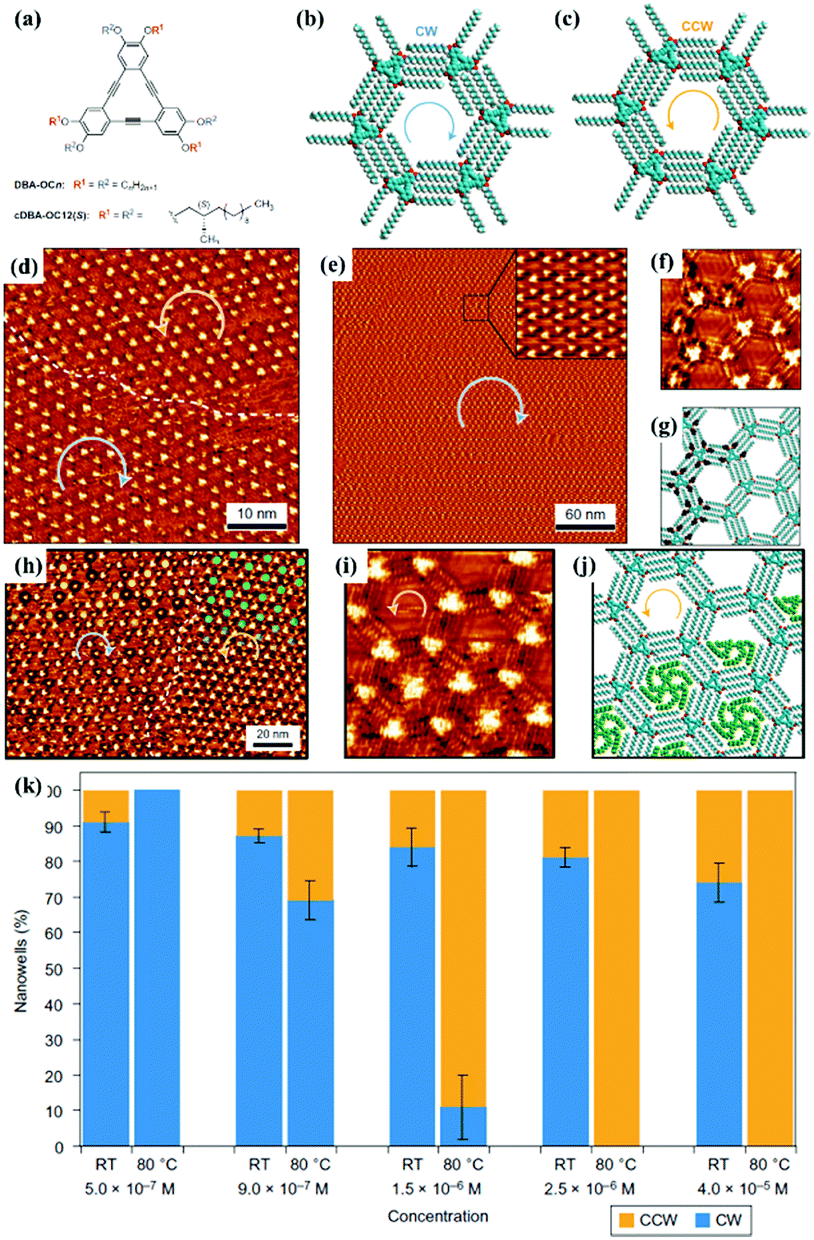
Surface chirality has received much attention due to its promising potential in enantioselective catalysis chiral molecular recognition, etc. Generally, simple annealing can only trigger the formation of racemic chiral architectures composed of achiral molecules on a surface without breaking the surface mirror symmetry. By combining temperature with other experimental parameters, a homochiral architecture can be acquired on a surface. A recent study reports the homochiral architecture of amino-functionalized triazine derivatives controlled by temperature and chiral solvents.60 At room temperature, a well-organized strip self-assembled architecture of compound 1 exists stably at the octanoic acid/HOPG interface, in which every compound 1 connects with two neighbouring molecules (Fig. 9(a)). The strip architecture begins to change to flower racemic chiral architectures at 55 °C (Fig. 9(i)). When the temperature rises to 80 °C (Fig. 9(j)), the more thermodynamically stable racemic chiral architectures demonstrated by theoretical calculations cover the whole surface by overcoming kinetic limitations. In the flower S (Fig. 9(e)) or R architecture (Fig. 9(f)), six compound 1 molecules aggregate into a hexamer through intermolecular N–H⋯N H-bonds with a counter clockwise (S) rotation or clockwise (R) rotation. With the replacement of octanoic acid by (S)-2-octanol solvents, the initial strip architecture of compound 1 totally transformed into a flower R architecture (Fig. 9(k) and (l)). Yuan Fang et al. suggest that temperature can also cooperate with a few chiral cDBA-OC12(S) guest molecules to guide the preparation of 2D homochiral architectures of numerous achiral DBA-OC12 molecules (Fig. 10(a)).61 At room temperature, DBA-OC12 molecules obey the sergeant–soldier principle and are arranged into an abundant clockwise (CW) homochiral network induced by cDBA-OC12(S) molecules at a low concentration (Fig. 10(d) and (g)). The domains of the CW network (Fig. 10(e)) further amplify and almost cover the surface after heating treatment resulting from temperature-assisted ripening,62,63 while the increased concentration of cDBA-OC12(S) leads to an expanded anticlockwise (CCW) homochiral network at the expense of CW regions triggered by annealing (Fig. 10(k)). It is guessed that during the nucleation and free growth period, the assembly behaviour of molecules is governed by the sergeant–soldier principle. Alternatively, during the ripening period, the host–guest pathway is dominantly assisted by temperature, which means that the subsequent annealing can result in the desorption of cDBA-OC12(S) sergeants on a surface and the accommodation of cDBA-OC12(S) in the cavities of the DBA-OC12's CCW network forming very thermodynamically stable host–guest complexes (Fig. 10(i) and (j)). The handedness of the supramolecular network depends on the competition between the two pathways which is determined by temperature and guest concentration.
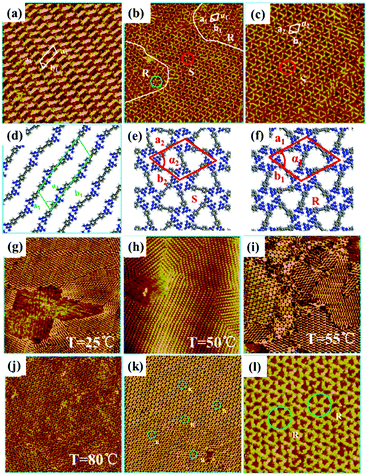 |
| | Fig. 9 STM images of compound 1's adlayers at the octanoic acid/HOPG interface: (a) before annealing; (b) and (c) after annealing. (d)–(f) Calculated molecular packing models of the adlayers in (a)–(c) separately. STM images of various compound 1's adlayers at the octanoic acid/HOPG interface under different temperatures: (g) 25 °C, (h) 50 °C, (i) 55 °C, (j) 80 °C. (k) and (l) Large-scale and high-resolution STM images of compound 1's adlayers at the (S)-2-octanol/HOPG interface after annealing at 80 °C. (Reprinted with permission from ref. 60. Copyright 2016 American Chemical Society). | |
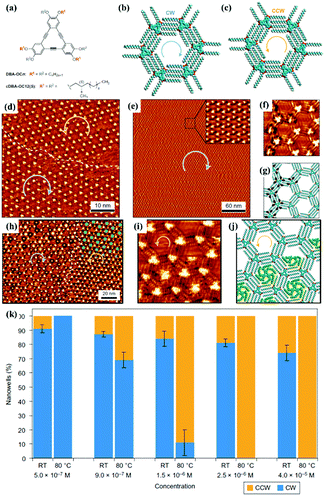 |
| | Fig. 10 (a) Chemical structures of DBA-OC12 and cDBA-OC12(S) molecules. (b) and (c) Schematic illustrations of CW and CCW nanoarrays of DBA-OC12 molecules respectively. STM images of cDBA-OC12's adlayers (10 mol% cDBA-OC12(S), C = 5.0 × 10−7 M): (d) before heating, (e) and (f) after heating at 80 °C. (h) and (i) STM images of cDBA-OC12(S) guests adsorbing in CW and CCW regions of the DBA-OC12 adlayer. (g) and (j) Calculated molecular models of the adlayers in (f) and (i) separately. (k) Histograms depicting the relationships among the DBA-OC12 adlayers, concentration and temperature. (Reprinted with permission from ref. 61. Copyright 2016 Springer Nature). | |
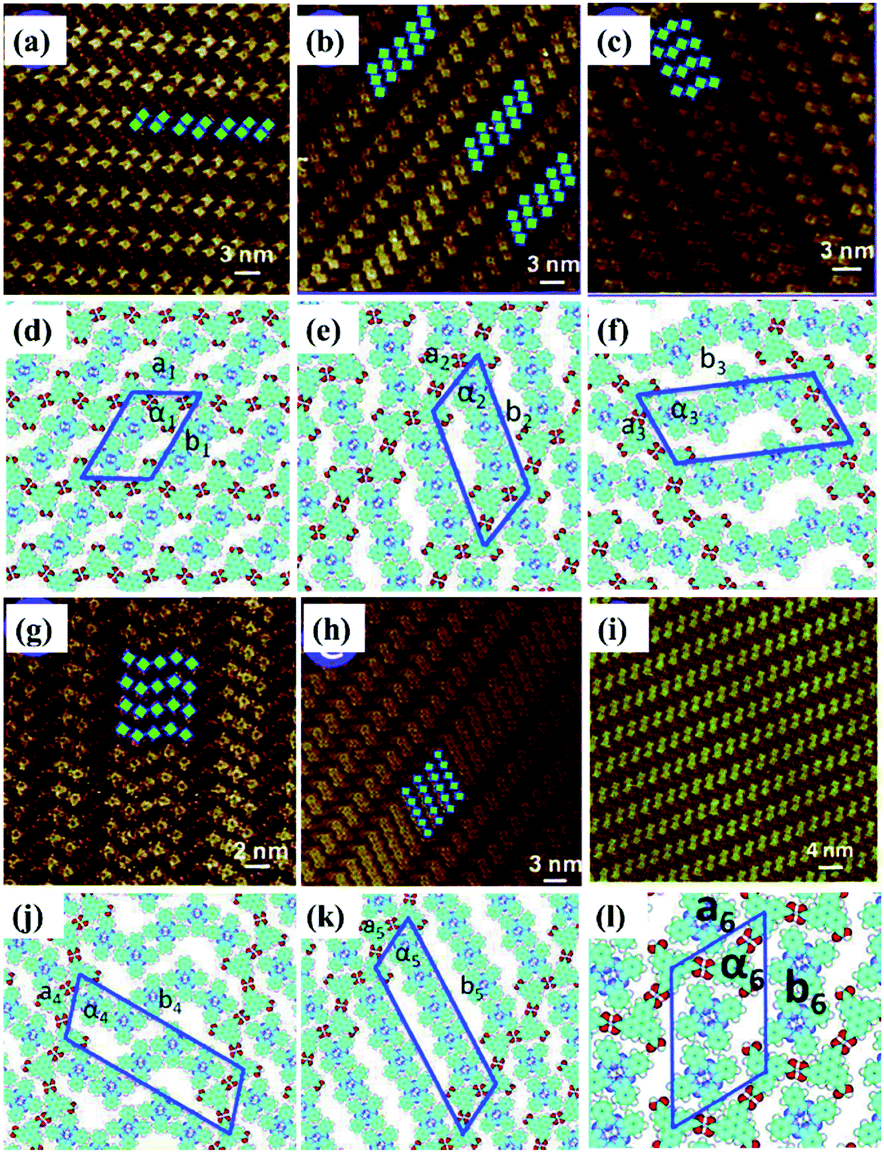
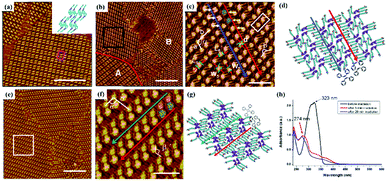
Temperature is of great significance for multi-component supramolecular systems as well as mono-component systems.64 Fangyun Hu et al. even carried out temperature-dependent research on the H3TTCA/H2Pc binary system.65 At room temperature, two, three, four or five H2Pc molecules randomly arrange in the nanopores of the H3TTCA H-bonded template, and five types of H3TTCA/H2Pc co-assembly adlayers are adsorbed on HOPG (Fig. 11). After undergoing heating treatment at 323 K for 10 minutes, only one of the five nanoarrays can exist stably on HOPG (Fig. 11(i)), which is the most thermodynamically stable as demonstrated by DFT. In addition, surface reactions including C–C coupling,66 Schiff base67–69 and boronic anhydridation reactions70 in multi-component supramolecular architectures can be induced by temperature, which is helpful for preparing a 2D covalent organic framework by taking advantage of the surface-confined effect.
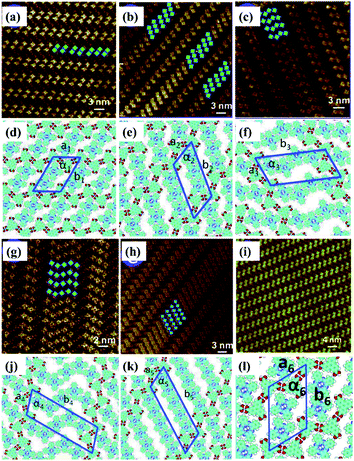 |
| | Fig. 11 (a)–(c), (g) and (h) Five types of H3TTCA/H2Pc co-assembled adlayers on HOPG. (d), (e), (f), (j) and (k) Molecular models of the adlayers in (a), (b), (c), (g) and (h) respectively. (i) and (l) High-resolution STM image and molecular packing model of H3TTCA/H2Pc adlayer after heating at 323 K. (Reprinted with permission from ref. 65. Copyright 2014 The Royal Society of Chemistry). | |
5. Regulatory effect of the electric field
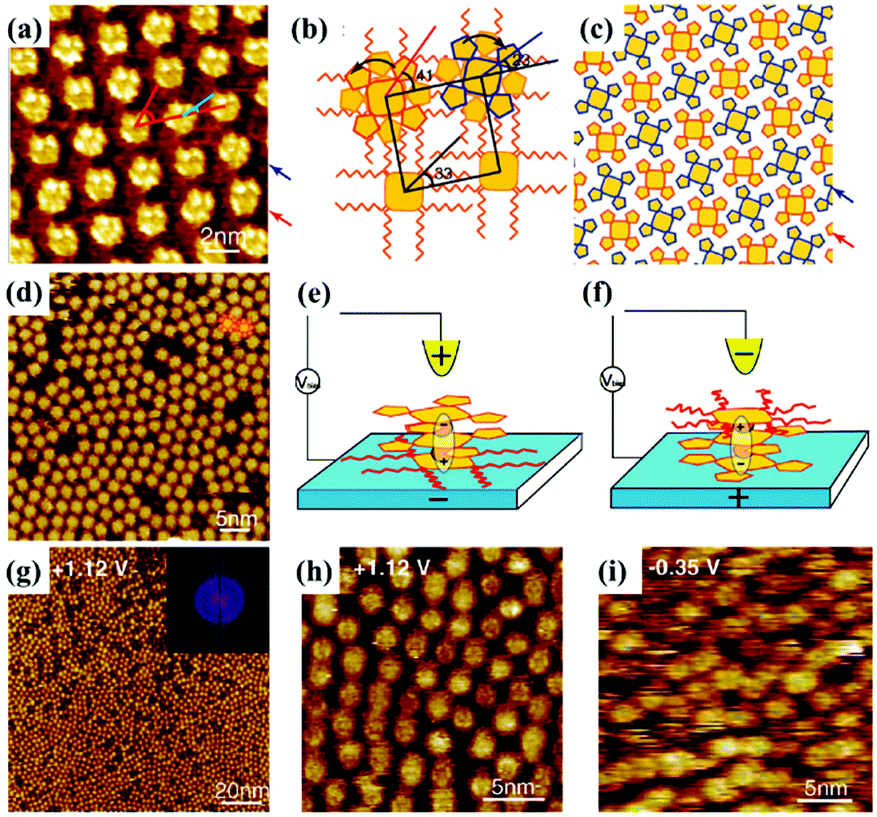
In addition to being a characterization technique, STM is also capable of functioning as an effective stimulus to manipulate molecular configuration, orientation and as a supramolecular adlayer on a surface, thanks to its electric field effect produced by altering the imaging bias voltage or giving pulses to the STM tip. For instance, the trans and cis configurations of TBA molecules can be switched reversibly on the Au(111) substrate by the adjustment of the electric field.71 The interactions between molecular polarity/electronics and the electric field play a crucial role in manipulating molecular switchable orientation and monolayers. Cpd 1, a polar triple-decker sandwich molecule containing two negatively charged Pc(15C5)4 units and a positively charged PcOC8 unit, generates a well-organized architecture with PcOC8 units adsorbed on the surface at a negative substrate bias (Fig. 12(a)).72 The extra stability energy contributed by the electric field can be given by Eenergy = −Pmolecular![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dipole·Eelectricfield, whereas at the positive substrate bias, the well-organized architecture is replaced by a disordered architecture where the orientations of Pc(15C5)4 and PcOC8 units are exchanged arising from the interactions between the molecular dipole and the electric field (Fig. 12(d)). Influenced by the electric field, the absence of intermolecular interactions between Pc(15C5)4 units lying on the surface and without sufficient alkyl–alkyl interdigitation between PcOC8 units weaken the stabilization of adsorption layers of Cpd 1 resulting in the observed disordered architecture. However, the molecular orientation of non-polar Cpd 2 remains unchanged in spite of the varied bias voltage (Fig. 12(g)–(i)). Later, the assembling architecture of another polar triple decker phthalocyanine complex 1 molecule assisted by a TCDB template network also depends on the direction of the electric field.73 The orientation of complex 1 trapped in TCDB cavities on the surface can adjust with the substrate bias, appearing as the conversion between ordered and disordered architectures in STM images. In another research effort, the positive substrate bias may induce the dipole moment of the non-dipolar BTB molecule, leading to BTB's bending orientation toward the HOPG substrate and consequently a tightly-packed linear architecture distinct from the pristine honeycomb structure with BTB molecules paralleling to the surface at a negative substrate bias.74 Kunal S. Mali et al. selected PQPClO4 molecules, a type of positively charged molecule, as the target molecule. This molecule exists in solution in the form of salt, causing the sensitivity of its constituent ions to interfacial polarization and the correlation of its assembly architectures dominated by electrostatic forces on the HOPG surface under an external field.23 When the HOPG substrate is substituted with Au, the reversible morphology transformation between the monolayer and the bilayer of the PQPClO4 molecules can be accomplished by electric field stimuli.75
dipole·Eelectricfield, whereas at the positive substrate bias, the well-organized architecture is replaced by a disordered architecture where the orientations of Pc(15C5)4 and PcOC8 units are exchanged arising from the interactions between the molecular dipole and the electric field (Fig. 12(d)). Influenced by the electric field, the absence of intermolecular interactions between Pc(15C5)4 units lying on the surface and without sufficient alkyl–alkyl interdigitation between PcOC8 units weaken the stabilization of adsorption layers of Cpd 1 resulting in the observed disordered architecture. However, the molecular orientation of non-polar Cpd 2 remains unchanged in spite of the varied bias voltage (Fig. 12(g)–(i)). Later, the assembling architecture of another polar triple decker phthalocyanine complex 1 molecule assisted by a TCDB template network also depends on the direction of the electric field.73 The orientation of complex 1 trapped in TCDB cavities on the surface can adjust with the substrate bias, appearing as the conversion between ordered and disordered architectures in STM images. In another research effort, the positive substrate bias may induce the dipole moment of the non-dipolar BTB molecule, leading to BTB's bending orientation toward the HOPG substrate and consequently a tightly-packed linear architecture distinct from the pristine honeycomb structure with BTB molecules paralleling to the surface at a negative substrate bias.74 Kunal S. Mali et al. selected PQPClO4 molecules, a type of positively charged molecule, as the target molecule. This molecule exists in solution in the form of salt, causing the sensitivity of its constituent ions to interfacial polarization and the correlation of its assembly architectures dominated by electrostatic forces on the HOPG surface under an external field.23 When the HOPG substrate is substituted with Au, the reversible morphology transformation between the monolayer and the bilayer of the PQPClO4 molecules can be accomplished by electric field stimuli.75
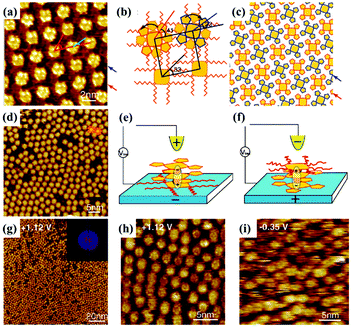 |
| | Fig. 12 STM images of Cpd 1's adlayers on the graphite substrate: (a) under negative bias, (d) under positive bias. (b) and (c) Calculated molecular packing models of the adlayer in (a). (e) and (f) Schematic illustrations of Cpd 1's reversible orientations under different bias. (g) and (h) Large-scale and high-resolution STM images of Cpd 2's adlayers on graphite under positive bias separately. (i) High-resolution STM image of Cpd 2's adlayers on graphite under negative bias. (Reprinted with permission from ref. 72. Copyright 2008 American Chemical Society). | |
As for multi-component systems, the effect of electric field should also be noticeable.76,77 Regarding the TMA/BTB binary system, a nanoporous co-assembly architecture is acquired on HOPG at a negative substrate bias and then the surface is totally occupied by a tightly-packed architecture at a positive substrate bias.77 The change of substrate dipole and the formed strong intermolecular carboxylate–carboxyl H-bonds after the segmental deprotonation of molecules are two possible reasons for the observed densely-packed architecture. Simultaneously, the final outcome triggered by surface dipole is determined by the concentration of TMA and BTB, and further influenced by the addition of the third molecule COR. Importantly, a 3D self-assembly system can be prepared by availing the applied electric field. Qing-Na Zheng et al. successfully obtained a flower bilayer architecture containing TMA and TP molecules at the OA/HOPG interface.78 In the bilayer architecture, the monolayer of TP molecules adsorbs atop the nanoporous H-bonded network of TMA (Fig. 13(c) and (d)). Then it can rapidly transform into a Kagomé monolayer architecture where TP molecules are accommodated in the TMA network by adjusting the bias voltage from positive to negative (Fig. 13(e) and (f)). Both the changes of substrate dipole and the protonation of TP molecules under an acidic environment are responsible for the constructed bilayer architecture. The preparation of the bilayer nanostructure by taking advantage of the electric field is meaningful for achieving crystalline engineering.
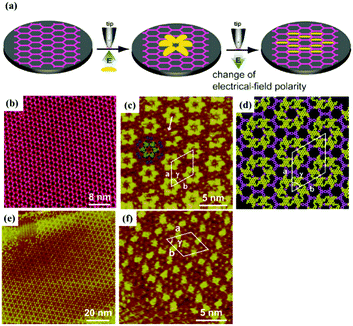 |
| | Fig. 13 (a) Schematic illustration of transformation from flower bilayers to Kagomé monolayers triggered by the electric field. (b) STM image of TMA's adlayer on the HOPG substrate. (c) and (d) STM image and molecular model of TMA/TP flower bilayer under positive bias. (e) and (f) Large-scale and high-resolution STM images of TMA/TP Kagomé monolayers under negative bias. (Reprinted with permission from ref. 78. Copyright 2014 Wiley-VCH). | |
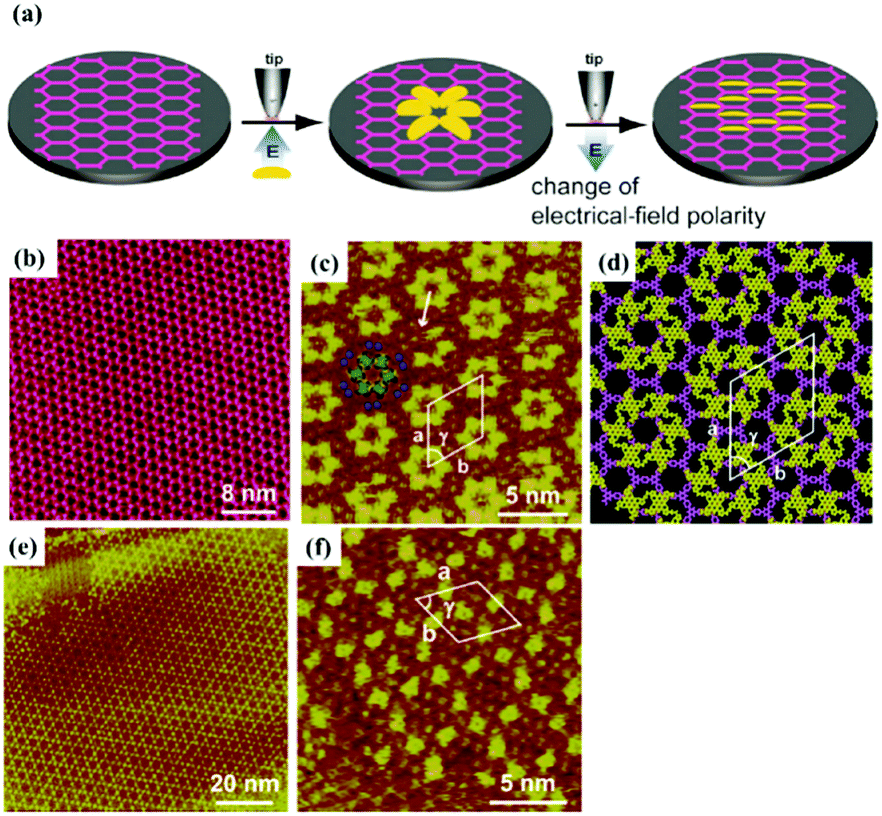
Furthermore, surface chemical reactions can be tuned by the electric field because the effect of the electric field on the stabilities of chemical species can be influenced by stabilizing or destabilizing charge-separated resonance contributors.79 For instance, Min Li et al. prepared the μ-oxo dimers of Mn(III)–PP molecules in the regions near the STM tip with a pulse voltage.21 Recently, Zhen-Feng Cai et al.80 achieved the dehydration reactions of TBPBA with boronic acids at the 1-octanoic acid/HOPG interface and obtain a dynamic covalent-organic framework (COF) monolayer promising in self-healing materials/surfaces under an electric field without heating. The reversible switching between the self-assembly adlayer (Fig. 14(a)) and the covalent-organic framework of TBPBA (Fig. 14(c)) is relevant to the electric field. When 1-octanoic acid is replaced by a 1-phenyloctane solvent, the TBPBA's self-assembly adlayer is insensitive to the electric field. The applied electric field is speculated to have an impact on the adsorption/desorption behaviour of hydrophobic 1-octanoic acid molecules on HOPG, which further influences interfacial water content and the chemical equilibrium of the reactions.
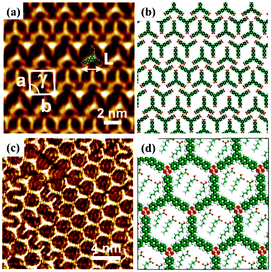 |
| | Fig. 14 (a) and (b) STM image and molecular packing model of TBPBA's adlayers at the OA/HOPG interface. (c) and (d) STM image and molecular model of TBPBA's COF monolayer at the OA/HOPG interface under negative bias. (Reprinted with permission from ref. 80. Copyright 2019 American Chemical Society). | |
6. Conclusions and outlook
In this review, the adsorption of molecules on a surface and the dynamic surface-assisted nanostructures tuned by external stimuli – guest species, light irradiation, varied temperature and electric field – have been revealed by STM. Guest molecules can participate in the assembled nanostructures of target molecules. Light irradiation, temperature and electric field can affect the configuration, mobility and orientation of adsorbates physisorbed on a surface, respectively. All the external stimuli bring about the phase transition of interfacial nanostructures by changing the pairwise interactions among absorbates, solvents and substrates. In some cases, the final regulated nanostructures are the outcome of multiple external stimuli. The preparation of chiral and bilayer architectures, and the conversion from a supramolecular network to a covalent organic network can be achieved by taking full advantage of the external factors. In future, more research studies will focus on quantitatively analysing the self-assembly monolayer at the liquid/solid interface, concluding universal laws from the research findings for precisely regulating nanostructures, constructing 3D multilayered self-assembly nanostructures on a surface, and exploring the effects of diverse supramolecular nanostructures on the properties of nanodevices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Basic Research Program of China (No. 2016YFA0200700), the National Natural Science Foundation of China (No. 21773041 and 21972031) and the Engineering Research Center of Nano Geomaterials of Ministry of Education (No. NGM2018KF001 and NGM2018KF004).
References
- F. Wang and C. L. Feng, Angew. Chem., 2018, 130, 5757–5761 CrossRef.
- R. Zhong, Q. Tang, S. Wang, H. Zhang, F. Zhang, M. Xiao, T. Man, X. Qu, L. Li, W. Zhang and H. Pei, Adv. Mater., 2018, 30, 1706887 CrossRef.
- N. R. Jber, N. H. Karam and A. H. Al-Dujaili, Mol. Cryst. Liq. Cryst., 2018, 675, 29–38 CrossRef CAS.
- G.-Y. Wu, L.-J. Chen, L. Xu, X.-L. Zhao and H.-B. Yang, Coord. Chem. Rev., 2018, 369, 39–75 CrossRef CAS.
- J.-H. Kim, K. Tahara, J. Jung, S. De Feyter, Y. Tobe, Y. Kim and M. Kawai, J. Phys. Chem. C, 2012, 116, 17082–17088 CrossRef CAS.
- Y. Yoneda, K. Mereiter, C. Jaeger, L. Brecker, P. Kosma, T. Rosenau and A. French, J. Am. Chem. Soc., 2008, 130, 16678–16690 CrossRef CAS.
- S. Ahn, C. N. Morrison and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 7946–7947 CrossRef CAS.
- X. Hu, B. Zha, Y. Wu, X. Miao and W. Deng, Phys. Chem. Chem. Phys., 2016, 18, 7208–7215 RSC.
- Y. Wu, J. Li, Y. Yuan, M. Dong, B. Zha, X. Miao, Y. Hu and W. Deng, Phys. Chem. Chem. Phys., 2017, 19, 3143–3150 RSC.
- A. Langner, S. L. Tait, N. Lin, R. Chandrasekar, M. Ruben and K. Kern, Angew. Chem., Int. Ed., 2008, 47, 8835–8838 CrossRef CAS.
- Y.-f. Geng, P. Li, J.-z. Li, X.-m. Zhang, Q.-d. Zeng and C. Wang, Coord. Chem. Rev., 2017, 337, 145–177 CrossRef CAS.
- D. Ecija, J. I. Urgel, A. P. Seitsonen, W. Auwarter and J. V. Barth, Acc. Chem. Res., 2018, 51, 365–375 CrossRef CAS.
- Z. Ma, Y.-Y. Wang, P. Wang, W. Huang, Y.-B. Li, S.-B. Lei, Y.-L. Yang, X.-L. Fan and C. Wang, ACS Nano, 2007, 1, 160–167 CrossRef CAS PubMed.
- L. Cai, L. Wang, S. Kang, Y. Geng, K. Deng, Q. Zheng and Q. Zeng, J. Phys. Chem. C, 2016, 120, 27259–27267 CrossRef CAS.
- K. Xiao, W. Deng, J. K. Keum, M. Yoon, I. V. Vlassiouk, K. W. Clark, A.-P. Li, I. I. Kravchenko, G. Gu, E. A. Payzant, B. G. Sumpter, S. C. Smith, J. F. Browning and D. B. Geohegan, J. Am. Chem. Soc., 2013, 135, 3680–3687 CrossRef CAS.
- T. Balandina, K. Tahara, N. Saendig, M. O. Blunt, J. Adisoejoso, S. Lei, F. Zerbetto, Y. Tobe and S. De Feyter, ACS Nano, 2012, 6, 8381–8389 CrossRef CAS.
- K. Tahara, S. Furukawa, H. Uji-i, T. Uchino, T. Ichikawa, J. Zhang, W. Mamdouh, M. Sonoda, F. C. De Schryver, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2006, 128, 16613–16625 CrossRef CAS.
- Y. Yang and C. Wang, Curr. Opin. Colloid Interface Sci., 2009, 14, 135–147 CrossRef CAS.
- M. Shen, Z. Luo, S. Zhang, S. Wang, L. Cao, Y. Geng, K. Deng, D. Zhao, W. Duan and Q. Zeng, Nanoscale, 2016, 8, 11962–11968 RSC.
- D. Rohde, C.-J. Yan, H.-J. Yan and L.-J. Wan, Angew. Chem., Int. Ed., 2006, 45, 3996–4000 CrossRef CAS.
- M. Li, D. den Boer, P. Iavicoli, J. Adisoejoso, H. Uji-i, M. Van der Auweraer, D. B. Amabilino, J. A. A. W. Elemans and S. De Feyter, J. Am. Chem. Soc., 2014, 136, 17418–17421 CrossRef CAS.
- D. Wang, Q. Chen and L.-J. Wan, Phys. Chem. Chem. Phys., 2008, 10, 6467–6478 RSC.
- K. S. Mali, D. Wu, X. Feng, K. Muellen, M. Van der Auweraer and S. De Feyter, J. Am. Chem. Soc., 2011, 133, 5686–5688 CrossRef CAS.
- L. Piot, C. Marie, X. Feng, K. Muellen and D. Fichou, Adv. Mater., 2008, 20, 3854–3858 CrossRef CAS.
- Y.-T. Shen, L. Guan, X.-M. Zhang, S. Wang, L.-H. Gan, Q.-D. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2013, 15, 12475–12479 RSC.
- S. Yoshimoto, Y. Honda, O. Ito and K. Itaya, J. Am. Chem. Soc., 2008, 130, 1085–1092 CrossRef CAS.
- X. Miao, L. Xu, Y. Li, Z. Li, J. Zhou and W. Deng, Chem. Commun., 2010, 46, 8830–8832 RSC.
- J. Lu, S. B. Lei, Q. D. Zeng, S. Z. Kang, C. Wang, L. J. Wan and C. L. Bai, J. Phys. Chem. B, 2004, 108, 5161–5165 CrossRef CAS.
- Y. Li, C. Liu, Y. Xie, X. Li, X. Li, X. Fan, K. Deng, Q. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2013, 15, 125–128 RSC.
- L. Piot, F. Silly, L. Tortech, Y. Nicolas, P. Blanchard, J. Roncali and D. Fichou, J. Am. Chem. Soc., 2009, 131, 12864–12865 CrossRef CAS.
- S. Chang, R. Liu, L. Wang, M. Li, K. Deng, Q. Zheng and Q. Zeng, ACS Nano, 2015, 10, 342–348 CrossRef PubMed.
- S. Zhang, J. Zhang, K. Deng, J. Xie, W. Duan and Q. Zeng, Phys. Chem. Chem. Phys., 2015, 17, 24462–24467 RSC.
- J. D. C. González, M. Iyoda and J. P. Rabe, Nat. Commun., 2017, 8, 14717 CrossRef.
- M. O. Blunt, J. C. Russell, M. del Carmen Gimenez-Lopez, N. Taleb, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Nat. Chem., 2011, 3, 74 CrossRef CAS.
- J. Adisoejoso, K. Tahara, S. Okuhata, S. Lei, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7353–7357 CrossRef CAS PubMed.
- S.-L. Lee, C.-H. Lin, K.-Y. Cheng, Y.-C. Chen and C.-h. Chen, J. Phys. Chem. C, 2016, 120, 25505–25510 CrossRef CAS.
- M. Li, Y.-L. Yang, K.-Q. Zhao, Q.-D. Zeng and C. Wang, J. Phys. Chem. C, 2008, 112, 10141–10144 CrossRef CAS.
- C.-A. Palma, J. Bjork, M. Bonini, M. S. Dyer, A. Llanes-Pallas, D. Bonifazi, M. Persson and P. Samori, J. Am. Chem. Soc., 2009, 131, 13062–13071 CrossRef CAS PubMed.
- X. Peng, L. Cheng, X. Zhu, Y. Geng, F. Zhao, K. Hu, X. Guo, K. Deng and Q. Zeng, Nano Res., 2018, 11, 5823–5834 CrossRef CAS.
- Y. Qian, Y. Gu, J. Feng, W. Jiang, Y. Geng, W. Duan, J. Ma, Q. Zeng and Z. Wang, J. Phys. Chem. C, 2018, 122, 24158–24163 CrossRef CAS.
- H. Liang, W. Sun, X. Jin, H. Li, J. Li, X. Hu, B. K. Teo and K. Wu, Angew. Chem., Int. Ed., 2011, 50, 7562–7566 CrossRef CAS.
- K. Tahara, K. Inukai, J. Adisoejoso, H. Yamaga, T. Balandina, M. O. Blunt, S. De Feyter and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 8373–8376 CrossRef CAS.
- M. M. S. Abdel-Mottaleb, S. De Feyter, A. Gesquiere, M. Sieffert, M. Klapper, K. Mullen and F. C. De Schryver, Nano Lett., 2001, 1, 353–359 CrossRef CAS.
- A. Deshpande, C.-H. Sham, J. M. P. Alaboson, J. M. Mullin, G. C. Schatz and M. C. Hersam, J. Am. Chem. Soc., 2012, 134, 16759–16764 CrossRef CAS.
- Y.-T. Shen, L. Guan, X.-Y. Zhu, Q.-D. Zeng and C. Wang, J. Am. Chem. Soc., 2009, 131, 6174–6180 CrossRef CAS.
- A. Galanti, V. Diez-Cabanes, J. Santoro, M. Valášek, A. Minoia, M. Mayor, J. Cornil and P. Samorì, J. Am. Chem. Soc., 2018, 140, 16062–16070 CrossRef CAS.
- X. Zhu, Y. Geng, X. Zhu, P. Duan, F. Li, Q. Zeng and J. Qi, New J. Chem., 2019, 43, 6262–6266 RSC.
- X. Zhang, S. Wang, Y. Shen, Y. Guo, Q. Zeng and C. Wang, Nanoscale, 2012, 4, 5039–5042 RSC.
- Y.-F. Han, G.-X. Jin and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 9263–9266 CrossRef CAS.
- L.-y. Liao, Y.-b. Li, X.-m. Zhang, Y.-f. Geng, J.-y. Zhang, J.-l. Xie, Q.-d. Zeng and C. Wang, J. Phys. Chem. C, 2014, 118, 15963–15969 CrossRef CAS.
- C. Guo, J. Xue, L. Cheng, R. Liu, S. Kang, Q. Zeng and M. Li, Phys. Chem. Chem. Phys., 2017, 19, 16213–16218 RSC.
- Y. Li, C. Liu, Y. Xie, X. Li, X. Li, X. Fan, K. Deng, Q. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2013, 15, 125–128 RSC.
- D. C. Y. Nguyen, L. Smykalla, T. N. H. Nguyen, T. Rüffer and M. Hietschold, J. Phys. Chem. C, 2016, 120, 11027–11036 CrossRef CAS.
- Z. Mu, O. Rubner, M. Bamler, T. Bloemker, G. Kehr, G. Erker, A. Heuer, H. Fuchs and L. Chi, Langmuir, 2013, 29, 10737–10743 CrossRef CAS.
- A. Bhattarai, U. Mazur and K. Hipps, J. Am. Chem. Soc., 2014, 136, 2142–2148 CrossRef CAS.
- L. Kampschulte, M. Lackinger, A.-K. Maier, R. S. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829–10836 CrossRef CAS.
- R. Gutzler, T. Sirtl, J. r. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084–5090 CrossRef CAS.
- C. Marie, F. Silly, L. Tortech, K. Mullen and D. Fichou, ACS Nano, 2010, 4, 1288–1292 CrossRef CAS.
- M. O. Blunt, J. Adisoejoso, K. Tahara, K. Katayama, M. Van der Auweraer, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2013, 135, 12068–12075 CrossRef CAS.
- L. Cheng, Y. Li, C.-Y. Zhang, Z.-L. Gong, Q. Fang, Y.-W. Zhong, B. Tu, Q. Zeng and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 32004–32010 CrossRef CAS.
- Y. Fang, E. Ghijsens, O. Ivasenko, H. Cao, A. Noguchi, K. S. Mali, K. Tahara, Y. Tobe and S. De Feyter, Nat. Chem., 2016, 8, 711 CrossRef CAS.
- M. Lackinger, S. Griessl, L. Kampschulte, F. Jamitzky and W. M. Heckl, Small, 2005, 1, 532–539 CrossRef CAS.
- A. Bellec, C. Arrigoni, G. Schull, L. Douillard, C. Fiorini-Debuisschert, F. Mathevet, D. Kreher, A.-J. Attias and F. Charra, J. Chem. Phys., 2011, 134, 124702 CrossRef PubMed.
- H. Dai, W. Yi, K. Deng, H. Wang and Q. Zeng, ACS Appl. Mater. Interfaces, 2016, 8, 21095–21100 CrossRef CAS.
- F. Hu, Y. Gong, X. Zhang, J. Xue, B. Liu, T. Lu, K. Deng, W. Duan, Q. Zeng and C. Wang, Nanoscale, 2014, 6, 4243–4249 RSC.
- A. C. Marele, R. Mas-Balleste, L. Terracciano, J. Rodriguez-Fernandez, I. Berlanga, S. S. Alexandre, R. Otero, J. M. Gallego, F. Zamora and J. M. Gomez-Rodriguez, Chem. Commun., 2012, 48, 6779–6781 RSC.
- H. Shi, Y. Liu, J. Song, X. Lu, Y. Geng, J. Zhang, J. Xie and Q. Zeng, Langmuir, 2017, 33, 4216–4223 CrossRef CAS.
- F.-Y. Hu, X.-M. Zhang, X.-C. Wang, S. Wang, H.-Q. Wang, W.-B. Duan, Q.-D. Zeng and C. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1583–1587 CrossRef CAS.
- X.-H. Liu, C.-Z. Guan, S.-Y. Ding, W. Wang, H.-J. Yan, D. Wang and L.-J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS.
- C.-Z. Guan, D. Wang and L.-J. Wan, Chem. Commun., 2012, 48, 2943–2945 RSC.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446–14447 CrossRef CAS.
- S.-B. Lei, K. Deng, Y.-L. Yang, Q.-D. Zeng, C. Wang and J.-Z. Jiang, Nano Lett., 2008, 8, 1836–1843 CrossRef CAS.
- F. Gao, X.-M. Zhang, L. Cui, K. Deng, Q.-D. Zeng and J.-L. Zuo, Sci. Rep., 2014, 4, 5928 CrossRef CAS.
- F. P. Cometto, K. Kern and M. Lingenfelder, ACS Nano, 2015, 9, 5544–5550 CrossRef CAS.
- K. Cui, K. S. Mali, O. Ivasenko, D. Wu, X. Feng, M. Walter, K. Muellen, S. De Feyter and S. F. L. Mertens, Angew. Chem., Int. Ed., 2014, 53, 12951–12954 CrossRef CAS.
- S.-L. Lee, Y. Fang, G. Velpula, F. R. Cometto, M. Lingenfelder, K. Muellen, K. S. Mali and S. De Feyter, ACS Nano, 2015, 9, 11608–11617 CrossRef CAS.
- G. Velpula, J. Teyssandier, S. De Feyter and K. S. Mali, ACS Nano, 2017, 11, 10903–10913 CrossRef CAS.
- Q.-N. Zheng, X.-H. Liu, X.-R. Liu, T. Chen, H.-J. Yan, Y.-W. Zhong, D. Wang and L.-J. Wan, Angew. Chem., Int. Ed., 2014, 53, 13395–13399 CrossRef CAS.
- A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Nature, 2016, 531, 88 CrossRef.
- Z.-F. Cai, G. Zhan, L. Daukiya, S. Eyley, W. Thielemans, K. Severin and S. De Feyter, J. Am. Chem. Soc., 2019, 141, 11404–11408 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab