
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-limiting aggregation of phospholipid vesicles†
N.
de Lange
 ,
F. A. M.
Leermakers
and
J. M.
Kleijn
*
,
F. A. M.
Leermakers
and
J. M.
Kleijn
*
Physical Chemistry & Soft Matter, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands. E-mail: frans.leermakers@wur.nl
First published on 3rd February 2020
Abstract
Lipid vesicles are widely used as model systems to study biological membranes. The self-assembly of such vesicles into vesicle pairs provides further opportunity to study interactions between membranes. However, formation of vesicle pairs, while subsequently keeping their colloidal stability intact, is challenging. Here, we report on three strategies that lead to stable finite-sized aggregates of phospholipid vesicles: (i) vesicles containing biotinylated lipids are coupled together with streptavidin, (ii) bridging attraction is exploited by adding cationic polymers (polylysine) to negatively charged vesicles, and (iii) temperature as a control parameter is used for the aggregation of vesicles mixed with a thermo-sensitive surfactant. While each strategy has its own advantages and disadvantages for vesicle pair formation, the latter strategy additionally shows reversible limited aggregation: above the LCST of pNIPAm, vesicle pairs are formed, while below the LCST, single vesicles prevail. Mixing protocols were assessed by dynamic and static light scattering as well as fluorescence correlation spectroscopy to determine under which conditions vesicle pairs dominate the aggregate size distribution. We have strong indications that without subsequent perturbation, the individual vesicles remain intact and no fusion or leakage between vesicles occurs after vesicle pairs have formed.
The biological membrane is a cornerstone of life. Not only are cells surrounded by such a double leaflet of lipids, but their interior is also packed with it. Where the primary function of the outer cell membrane is that of a barrier, membranes, in particular intracellular membranes, have a wide variety of different tasks. It is well known that the composition of membranes is tactically adjusted to suit these tasks.1–3 This is evidenced by an extraordinary variety of shapes, structures and even topologies of the membrane present in the cell, each of which is accompanied by a distinct, complex and dynamic composition of the membrane.
Changes in the shape or topology of the membrane, like membrane fusion or the formation of handles between membranes, occur often within cells.4 This allows for content exchange across membranes, which is vital for the cell that needs to transport materials to and from organelles or in and out of the cell. However, the physics behind these membrane interactions remains poorly understood.
Model membrane systems like lipid vesicles, or liposomes, are widely used to study the membrane. They consist of some liquid enclosed by a lipid bilayer and they naturally occur within the cell for transport of material across the cell. Lipid vesicles have been used for various applications like drug delivery5,6 and bioreactors7,8 or to study chemical reactions under confinement and biologically relevant conditions.9 Liposomes can easily be generated in vitro when carefully selected lipids are dispersed in an aqueous solution. This ‘bottom-up’ approach allows for control over the membrane composition. Liposomes, therefore, are ideally suited for studying membranes including various inter-membrane interactions like adhesion10 or (hemi-)fusion as was shown using lipid-anchored DNA11 and SNARE proteins.12
A natural starting point for studying membrane (hemi-)fusion are small aggregates of liposomes, ideally limited to vesicle pairs. To obtain such systems, one has to first start the self-assembly of liposomes and stop further self-assembly at a very early stage. We will refer to such arrested aggregation as limited aggregation.
Aggregation of colloidal particles, such as lipid vesicles, has been a popular research field since the publication of the DLVO theory.13,14 For charged colloids, the colloidal stability is governed by a balance between electrostatic repulsion and van der Waals attraction. The DLVO theory explains the loss of colloidal stability through its dependence on ionic strength as an ‘all or nothing’ process. To control the aggregation, by limiting the aggregation or directing the assembly into specific shapes and structures, several strategies can be applied. Particles of specific shapes,15,16 with symmetrically arranged patches,17,18 or with functionalized surfaces19,20 have been successfully used. In particular, DNA sticky ends are widely used on numerous colloidal particles such as solid metal nanoparticles,21,22 polystyrene microparticles23 and emulsion droplets.24 The aforementioned strategies have provided a rich variety of advanced colloidal materials with various functions.18,25
For vesicles, aggregation needs to be induced by additives as van der Waals attraction is negligible. Additives that can do this may be referred to as ‘linkers’. This approach is not new, as is evidenced by the amount of literature available. Different types of linkers include the previously mentioned tethered DNA strands with complementary sticky ends,26–28 the protein streptavidin which links to biotinylated lipids29–31 or using charged polymers and oppositely charged vesicles.32,33 Other methods include the use of univalent or divalent counterions.34,35 A linker binds vesicles together, which usually leads to large multivesicular aggregates. While such aggregates are easy to obtain, achieving control of the aggregation to obtain vesicle aggregates of a given size vastly expands possible applications, for example multicompartment drug delivery.6 Several strategies have been applied to control aggregation. These include introducing charges in vesicles to slow down the aggregation rate.29 The electrostatic repulsion between vesicles obtained through the charges creates an energy barrier for aggregation, which reduces the amount of successful vesicle collisions. The range of electrostatic repulsion is governed by the Debye length, which can be controlled using salt or other electrolytes.36 The specific properties of biotin and streptavidin can also be used to control vesicle aggregation. As streptavidin is able to bind 2 biotin on either side of the molecule, it is able to bridge vesicles. A prerequisite for this is that a bound streptavidin is able to find a free biotinylated lipid on another vesicle. If, however, the biotin is already bound to another streptavidin, which happens often if the amount of streptavidin present exceeds the amount of biotin present (biotin-starved regime), bridging between vesicles does not occur. This limitation can be exploited for the formation of small vesicle aggregates.30
An aspect of the liposome that can be exploited for limited aggregation is the lateral mobility of the lipids and subsequent linker molecules inserted in the membrane. Upon binding of two vesicles, an adhesion patch is formed. Through the lateral mobility of other linker molecules to this adhesion patch, the strength of the adhesion between two vesicles increases over time.37 A consequence of the local enrichment of linker molecules in an adhesion patch is that these linkers are no longer available to link to other vesicles and that the parts of the vesicles outside the adhesion zone are depleted of the linkers. This intra-aggregate binding is the hallmark for limited aggregation, as was previously shown.26,38
Our aim is to stop the assembly of vesicles as closely as possible at the pair level. For this, we have to obtain the optimal relation between the time scale for vesicle–vesicle collision and the time scale for the lateral mobility of linkers towards the adhesion patches. In short, the vesicle concentration should be low enough so that the characteristic collision time is large compared to the maximum lateral diffusion time. Of course, the number of linkers per vesicle should remain under a threshold value so that a sufficient number of them can be hidden in the adhesion patches. Ideally, we would like to obtain multiple orthogonal approaches to extend the possibilities of manipulating vesicle associates. With an extended toolbox, one can, for example, bring two bilayers together by one of the linkers and compromise the topological stability of such a bilayer pair by another.
We have tested several types of linkers for their capability of linking vesicles to form small stable aggregates. Aggregation of the vesicles was studied using static and dynamic light scattering, with fluorescence correlation spectroscopy and with electrophoretic mobility measurements. We found three scenarios in which the self-limiting association could be established: (1) biotinylated lipids act as receptors on the vesicles with streptavidin as the ligand coupling the vesicles together. This method was shown before to give finite sized aggregates.30 (2) Cationic polymers (polylysine or PLL) are added to a solution with anionic vesicles. Because of the strong cooperative binding of the polymer onto the vesicle, we can induce bridging flocculation. When we limit the number of polymers per vesicle to (close to) unity, the assembly is naturally limited. (3) Thermo-sensitive surfactants containing a C18 tail and a poly-NIPAm head group (C18-pNIPAm) allow the formation of stable small liposome aggregates by a temperature switch: pNIPAm exhibits LCST behaviour and thus the aggregation is triggered by bringing the vesicles to a temperature above T ≈ 32 °C. While this method has not been used for vesicle aggregation before, it has been shown that this thermosensitive surfactant can initiate aggregation of polystyrene particles into small crystallites (order of 100 particles) in a matter of minutes or to destabilize droplet suspensions triggering phase separation using a simple temperature switch.39 For each system, we explore the effects of the relevant physical conditions on the self-assembly of vesicles and discuss in which way vesicle pair formation can be further optimized.
Our working hypothesis is that the ability to fabricate small clusters of vesicles, or more specifically, vesicle pairs, is a useful step to start studying the physical properties of membrane pairs. Combining different attraction strategies systematically with variations in the lipid composition of vesicles may prove to be crucial to alter the topological stability of bilayer pairs and induce membrane (hemi-)fusion or the formation of handles between the vesicles, which could be analyzed by lipid mixing assays or content mixing assays.40,41
1 Materials and methods
1.1 Materials
All chemicals were of analytical grade and were used without further purification. Chloroform solutions of the phospholipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (DOPE-biotin), 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-PE) were purchased from Avanti Polar Lipids Inc. Streptavidin was purchased from Jackson ImmunoResearch. N-Isopropylacrylamide (NIPAm) was obtained from TCI Europe N.V. Other analytical grade chemicals were acquired from Sigma-Aldrich. These include 1-octadecanethiol, 2,2′-azobis(2-methylpropionitrile) (AIBN), 0.1% (w/v) poly-L-lysine solution in H2O (Mw ≈ 1.5 × 105–3.0 × 105 g mol−1), poly-L-lysine hydrobromide (Mw ≈ 1000–5000 g mol−1) and all chemicals used to prepare the buffer solutions.Buffer solutions were prepared using ultrapure water (resistivity >18 MΩ cm). All buffer solutions contained 10 mM tris(2-amino-2-(hydroxymethyl)propane-1,3-diol) and 50 mM NaCl and the pH was adjusted to 7.5 unless otherwise mentioned. Adjusting the pH was done by adding 1 M HCl or 1 M NaOH. All buffers were filtered through 0.2 μm pores before use.
1.2 Vesicle preparation
Vesicle preparation was performed similarly to previous work.42,43 In short, we mixed phospholipids together in the desired ratio in a round bottom flask, evaporated the chloroform under a stream of nitrogen and dried the lipid film under vacuum for at least 2 h. After drying, the lipids were resuspended in the appropriate buffer to a final lipid concentration (Cl ≈ 2.0 × 10−2 M) and hydrated for about 1 h in a rotary evaporator (no vacuum, 323 K, 100 rpm). This resulted in multilamellar vesicles. Subsequently, four freeze–thaw steps were applied using liquid nitrogen and a 40 °C water bath to obtain unilamellar vesicles. After this, the vesicles were extruded 21 times using a mini-extruder (Avanti Polar Lipids, Inc.) equipped with a polycarbonate membrane with 0.2 μm pore size, to obtain vesicles of approximately 75 nm in radius. The vesicles were subsequently collected and stored in a refrigerator until further use. The maximum storage time was 5 days. Characterization of the vesicles was performed using dynamic and static light scattering (DLS and SLS).44,45 Cumulant analysis,46 see the ESI,† revealed the vesicles to be spherical, with hydrodynamic radius Rh ≈ 65–80 nm and polydispersity index (PDI) ≈ 0.1. An overview of all vesicles used can be found in the ESI,† Table S1.1.3 C18-pNIPAm surfactant synthesis
Synthesis of the C18-pNIPAm surfactant was done following the protocol previously described.39 It involves a free radical chain-transfer polymerization of 1-octadecanethiol and NIPAm using AIBN as the initiator. 1.03 g of 1-octadecanethiol (∼3.5 mmol), 29.36 g of NIPAm (∼262.5 mmol) and 1.14 g of AIBN (∼7.0 mmol) were dissolved in a 250 ml round bottom flask with THF (100 ml). This corresponds to a molar ratio of thiol/NIPAm/AIBN of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75:2. The solution was bubbled with nitrogen for 30 minutes after which the reaction was run overnight at 55 °C. Purification was done by precipitation in cold hexane, with subsequent centrifugation. The precipitate was dried under vacuum, collected and stored for further use. The molecular weight of the surfactant was characterized by gel permeation chromatography (GPC) with HFIP (hexafluoro-2-propanol) as the running solvent; 〈Mn〉 = 5708, PDI = 2.8, and degree of polymerization is approximately 50.
:75:2. The solution was bubbled with nitrogen for 30 minutes after which the reaction was run overnight at 55 °C. Purification was done by precipitation in cold hexane, with subsequent centrifugation. The precipitate was dried under vacuum, collected and stored for further use. The molecular weight of the surfactant was characterized by gel permeation chromatography (GPC) with HFIP (hexafluoro-2-propanol) as the running solvent; 〈Mn〉 = 5708, PDI = 2.8, and degree of polymerization is approximately 50.
1.4 Aggregation experiments
For aggregation experiments, vesicles were diluted in buffer to a final lipid concentration Cl ≈ 1.0 × 10−4 M, unless specified otherwise. After measuring the size, mass or electrophoretic mobility of the vesicles, a small amount of linkers, i.e. streptavidin, polylysine (PLL) or C18-pNIPAm surfactants, was added to the sample. Streptavidin was added to vesicles containing biotin (B vesicles or NBD vesicles), PLL was added to vesicles containing DOPG (PG vesicles) and C18-pNIPAm was added to pure DOPC vesicles (S-0 vesicles); see Table S1 (ESI†). After adding the linkers, the sample was mixed gently and the new size and mass were measured. Subsequently, the amount of linkers in the sample was doubled, mixed and measured once again. This cycle was repeated until the molar streptavidin/biotin ratio (Ns/Nb) was 2.4, the molar C18-pNIPAm/DOPC ratio (NC18/NDOPC) was 0.32, or when uncontrolled aggregation of the vesicles was observed. The latter happened at high PLL amounts and could be recognized by an indefinite increase in hydrodynamic radius (Rh) over time.For DOPG vesicles with PLL, reverse aggregation experiments were carried out as well. For this, stable aggregates of vesicles at a fixed PLL per vesicle ratio were subjected to an increasing amount of salt (NaCl) in the solution.
The size and mass of the vesicles and vesicle aggregates were measured using dynamic and static light scattering (DLS and SLS). For NBD vesicles, fluorescence correlation spectroscopy (FCS) was applied.
1.5 Light scattering
All light scattering experiments were performed using an ALV instrument equipped with an ALV5000/60 × 0 external correlator and a 300 mW Cobolt Samba-300 DPSS laser operating at a wavelength λ = 532 nm.For aggregation with streptavidin and biotin or with PLL, standard DLS experiments involved 45–100 measurements of 10 s recorded at an angle of 90°. The diffusion coefficient D and subsequently the Rh were calculated similarly to previous work.47 Once D was steady over time, the average D was determined and this value was used to obtain a normalised diffusion coefficient D0/D or hydrodynamic radius R/R0, with D0 and R0 being the diffusion coefficient and the hydrodynamic radius of single vesicles. SLS experiments were only carried out after DLS experiments showed a steady D over time. Standard SLS experiments consist of measurements performed at a scattering angle (θ) ranging from 30° to 55° with steps of 1°. For each θ, three measurements were recorded of 10 s. To calculate the absolute Rayleigh scattering (Rθ), a similar SLS experiment was performed on buffer solution (10 mM Tris–HCl, 50 mM NaCl, pH 7.5) and on toluene as the reference. The molar mass of the aggregates (Mv) and the radius of gyration (Rg) were estimated using Guinier analysis.48 From Mv, we can calculate the mean aggregate number (M), which is defined as the average number of single vesicles per aggregate. See the ESI† for a detailed explanation on the data analysis of DLS and SLS.
For samples in which C18-pNIPAm surfactants were added to the vesicles, a standard DLS measurement consists of 1000 measurements of 10 s recorded at an angle of 90°. During the recording, the vesicles were subjected to a temperature program. This involved 10 minutes at 25 °C, 30 minutes at 40 °C and the remainder of the experiment back at 25 °C. In repeat experiments, the vesicles were subjected to an extensive temperature program. Here, 30 minute cycles at various temperatures (27.5 °C, 30 °C, 31 °C, 32 °C, 33 °C, 34 °C, 35 °C, 37.5 °C and 40 °C) were alternated with 60 min cycles at 25 °C. SLS experiments were performed similarly to those for the other vesicles, both at 25 °C and at 40 °C.
1.6 Fluorescence correlation spectroscopy
FCS measurements were performed as an extra check for the size and aggregation number of the vesicles containing biotin, before and after self-assembly with streptavidin. For these experiments, vesicles were fluorescently labelled with NBD (NBD vesicles).Comparable to DLS, FCS uses the intensity fluctuations of the detected fluorescent light caused by the diffusion of fluorescent particles in and out of the confocal volume to determine the diffusion coefficient of the fluorescent particles.47,49,50 For this, the dimensions of the confocal spot have to be known. Therefore, Rhodamine 110 (R110), of which the diffusion coefficient is known (D = 4.3 × 10−10 m2 s−1), was used to calibrate the setup. We found a diffusion time of 18 μs and a structural parameter a (= ωz/ωxy, i.e. the ratio of the equatorial and axial radii of the detection volume) between 5 and 10. From this, the confocal volume was found to be approximately 0.2 fL.
To perform FCS experiments, a Leica TCS SP8 X microscope equipped with a 63 × 1.20 NA water immersion objective and a super continuum laser or white light laser (SLL) selecting the 488 nm laser line was used. NBD was excited at 488 nm wavelength with a pulse frequency of 40 MHz. The fluorescence signal was detected through a pinhole, set at 90 μm, and filtered using a 495–550 nm spectral filter. The signal was recorded using an internal hybrid detector, coupled to a PicoHarp 300 TCSPC module (PicoQuant). Measurements were performed in 8-well chamber slides from Nunc™ Lab-Tek™ (Thermo Fischer Scientific). For each sample that contained vesicles, 15 or more measurements were recorded of 4 minutes. For the R110 samples, 20 measurements of 30 seconds each were recorded.
For FCS data analysis, FFS-data processor version 2.3 (Scientific Software Technologies Software Centre) was used.50 Using the program, the average number of particles in the confocal volume (〈N〉), their diffusion coefficient (Df) and the corresponding Rh were calculated. See the ESI† for a detailed explanation of the data analysis.
1.7 Electrophoretic mobility
The electrophoretic mobility of DOPG vesicles with added PLL was measured by laser microelectrophoresis using a zetasizer Nano ZS with a dip cell (Malvern Instruments). See Section 1.4 for the procedure of sample preparation.1.8 Simple model of vesicle pairs
We model the translational diffusion behaviour of the vesicle pairs as if they are prolate ellipsoids. For such objects, the diffusion coefficient is given by Kuipers et al.51 In DLS and FCS, the diffusion coefficient is experimentally determined, and using the Stokes–Einstein equation, an effective hydrodynamic radius, Rh, is calculated. It can be shown (see the ESI†) that for pairs of monodisperse vesicles, Rh is expected to be between 1.32R0 and 1.15R0: the first value refers to the case that the vesicles have a very small contact area (≪R02), and the second value is for an extensive contact area causing the vesicle pair to be perfectly spherical.2 Results and discussion
Using the extrusion method as described in the materials and methods section with 21 push-throughs, we produce monodisperse unilamellar vesicles with an average radius of 65–80 nm, as determined using SLS and DLS (see the ESI,† Table S1).2.1 Vesicle aggregation using biotin and streptavidin
Streptavidin has four binding sites for biotin, two on opposite sides of the protein molecule. If streptavidin is added to vesicles that contain biotin, it acts as a cross-linker between the vesicles, inducing vesicle aggregation. For all measurements reported here, after a short incubation period, the aggregate size, or diffusion coefficient, proved to be stable over time and the aggregation was regarded as limited. The size and aggregate number (M) were subsequently determined from DLS and SLS measurements. Fig. 1 shows D0/D and M as a function of the streptavidin/biotin molar ratio (Ns/Nb) for vesicles with different fractions of biotinylated lipids. | ||
| Fig. 1 (A) Normalized diffusion coefficient (D0/D) as measured using DLS and (B) the aggregation number (M), as a function of the logarithm of the streptavidin/biotin ratio (Ns/Nb). Experiments were carried out for different fractions of biotinylated DOPE phospholipids (fb). The results are shown for vesicles containing fb = 0.0004 (blue), fb = 0.002 (red), fb = 0.004 (orange) and fb = 0.006 (purple). All experiments were performed with a lipid concentration Cl = 0.1 mM. | ||
In Fig. 1, two regimes can be distinguished. In the first regime, at relatively small amounts of streptavidin (Ns/Nb < 0.1), D0/D increases logarithmically (D0/D ∝ log(Ns/Nb)). A transition to the second regime occurs between 0.1 < Ns/Nb < 0.2. Here, Rh/R0 increases more or less sharply depending on the experimental conditions. In the second regime, for Ns/Nb > 0.2, D0/D is practically constant. We find that at the end of the first region M ≈ 2, suggesting a population of mainly vesicle pairs. In the second regime, M > 2 for the samples containing a fraction of biotinylated DOPE phospholipids fb > 0.0004, implying aggregation beyond vesicle pairs.
To further verify the formation of vesicle pairs, FCS measurements were performed. Similar to DLS, from the intensity fluctuations due to the movement of the fluorescent particles in and out of the confocal volume, the hydrodynamic radius is obtained. In addition, the average number of particles (〈N〉) in the (known) confocal volume can be determined. Comparing the concentration of fluorescent particles in the confocal volume with the known total (original) concentration of single vesicles allows distinguishing between the cases of aggregation in mainly vesicle pairs or having a few larger aggregates with many single vesicles. In the first case, the concentration of fluorescent particles is half the total concentration of single vesicles, and in the latter, the concentration of fluorescent particles is only slightly lower compared to that of the single vesicles. Based on the 〈N〉 values, we calculated the aggregation number M as well. The FCS results compared with findings from DLS/SLS can be found in Table 1.
| N s/Nb [−] | D 0/D DLS [−] ± st. dev. | D 0/D FCS [−] ± st. dev. | M SLS [−] ± st. dev. | M FCS [−] ± st. dev. |
|---|---|---|---|---|
| 0 | 1 ± 0.02 | 1 ± 0.08 | 1 ± 0.03 | 1 ± 0.07 |
| 1/25.6 | 1.47 ± 0.03 | 1.56 ± 0.17 | 2.47 ± 0.23 | 2.11 ± 0.35 |
According to our data, at the end of the first regime, D0/D obtained through FCS is equal to D/D0 as measured by DLS. In addition, FCS also gives M ≈ 2, verifying the formation of vesicle pairs (on average). In comparison, M obtained by SLS is slightly higher, which we attribute to the dominant contribution of larger particles to the scattering signal.
The vesicle preparation methods we perform yield polydisperse samples (PDI > 0.1) and the distributions of sizes and aggregate numbers are expected to widen upon aggregation. This is reflected by the somewhat higher standard deviations for the vesicle pairs compared to that of single vesicles, see Table 1. In addition, we performed CONTIN analysis52–54 on the DLS results to follow the changes in the particle distribution during the aggregation experiments (ESI,† Fig. S4). This reveals for the streptavidin–biotin linker system that broadening of the size distributions is quite limited. We will come back to this later in the discussion.
Streptavidin is a protein that can bind a maximum of four biotin moieties. Assuming that half of the biotinylated lipids in a vesicle are on the inner leaflet of the membrane, all the available biotin can bind to streptavidin at 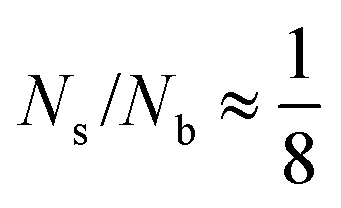 . Below this fraction, we have more biotin than biotin-binding sites. In other words, the amount of streptavidin limits the amount of connections that can be formed. We will therefore refer to regime 1 as the streptavidin-limited regime. At
. Below this fraction, we have more biotin than biotin-binding sites. In other words, the amount of streptavidin limits the amount of connections that can be formed. We will therefore refer to regime 1 as the streptavidin-limited regime. At 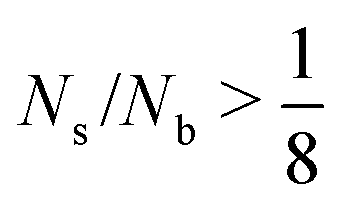 , we have less biotin than biotin binding sites, and then the amount of biotin limits the amount of connections that can be formed. The second regime will therefore be referred to as the biotin-limited regime.
, we have less biotin than biotin binding sites, and then the amount of biotin limits the amount of connections that can be formed. The second regime will therefore be referred to as the biotin-limited regime.
Intra-aggregate binding happens especially if the time required for a bound streptavidin to diffuse to the vesicle–vesicle contact zone (tdiff) is very low compared to the vesicle collision time (τ). We calculated that for a standard aggregation experiment with Cl = 1.0 × 10−4 M and Rv ∼ 65–80 nm, assuming that each collision is successful and that a bound streptavidin has to diffuse the maximum distance, tdiff ∼ 1–2 ms and τ ∼ 80–130 ms (see the ESI† for the full calculation). So, for our standard experiments, τ/tdiff ≈ 70. That we are in the regime where τ ≪ tdiff was confirmed by the observation that the concentration of vesicles has no effect on the results (see ESI†). This heavily favors intra-aggregate binding over coupling to new vesicles, not even taking into account steric hindrance and alignment effects during vesicle collision. As a consequence, although the total amount of links between vesicles increases with increasing amount of streptavidin, aggregation remains limited to mainly vesicle pairs.
At the end of the streptavidin-limited regime, the SLS and FCS measurements confirm a vesicle population of mainly vesicle pairs, as M ≈ 2, see Table 1. At this point, for vesicles with biotin fractions of 0.002, 0.004 and 0.006, the average hydrodynamic radius has increased to about 1.4 times the hydrodynamic radius of a single vesicle (Rvp/R0 = 1.4, corresponding to D0/D = 1.4). Since we are dealing with mainly vesicle pairs, based on the model used for their diffusional behaviour (see the ESI†), we conclude that the contact zones have to be small (i.e., patch radius significantly smaller than R0).
The size of the contact zone is dependent on the balance between the adhesion strength and energy cost associated with the increased membrane curvature. The stronger the adhesion strength, the larger the contact zone, but the membrane curvature at the edges of the contact zone increases as well. Biotin and streptavidin have a very high bond strength, on the order of 30kBT,55 which suggests the formation of large contact areas. This is, however, not the case. As the energy required to pull a phospholipid out of a bilayer is smaller, 16kBT,55 we can imagine that streptavidin attached to a single vesicle is able to pull out other biotinylated and more lipids to form micellar structures on the non-vesicle bound side of streptavidin. Other structural conformations might be possible as well. While bridge formation between two vesicles is the preferred conformation, the adhesion strength could be of much lower strength in comparison to the biotin–streptavidin bond strength. In addition, the energy required for deforming already highly curved membranes, which is the case for our small unilamellar vesicles (SUVs), is high as well. Finding the balance of these two forces as a function of vesicle size and adhesion strength is a highly interesting topic, for which further experiments and theoretical calculations are required, something we aspire to do ourselves.
The sample with fb = 0.0004 hardly shows aggregation beyond vesicle pairs. Apparently, at this low fraction of biotin, practically all biotin is already involved in the contact zones of vesicle pairs, leaving no biotin for further aggregation. We also performed the aggregation experiment for vesicles with relatively high biotin fractions: fb = 0.02 and higher (data not shown). In the streptavidin-limited region, this resulted in similar results as obtained for the other biotin fractions. However, adding more streptavidin resulted in a continuous, unlimited aggregation.
Interestingly, for biotin fractions of 0.002, 0.004 and 0.006, the transition to the streptavidin-limited regime seems to occur around the same Ns/Nb ratio (≈0.075). This suggests that saturation of the contact zones between vesicles occurs at a fixed Ns/Nb ratio, for vesicles of similar size. We, however, expect the transition to occur at higher Ns/Nb ratios for lower biotin fractions. For fb in the range of 0.002–0.006, this is probably not visible because of the limited experimental accuracy. However, as expected, for the sample with fb = 0.0004, a much higher Ns/Nb fraction is necessary to obtain vesicle pairs.
 | ||
| Fig. 2 Normalized hydrodynamic radius of the vesicles determined by DLS as a function of the Ns/Nb ratio in lin-log coordinates. The concentration of streptavidin was increased by subsequently adding an amount of streptavidin to a fixed concentration of vesicles (Cl = 0.1 mM) with biotin fraction fb = 0.004. Each coloured series represents a new experiment in which the initial amount of streptavidin added to the sample is different. This amount corresponds to Ns/Nb of the first (most left) data point of each series. | ||
When the starting amount of streptavidin is low (red curve), the maximum hydrodynamic radius obtained is about twice the radius of a single vesicle. For all the cases with initial ratio Ns/Nb higher than 1/8, the final hydrodynamic radius is lower and drops down to about only 1.3R0 for an initial value Ns/Nb ≈ 2.5 (red dot). This effect can contribute to an overloading of streptavidin onto the biotin. If the initial amount of streptavidin is high, many of the biotinylated lipids in the vesicles immediately bind to a streptavidin molecule and the formation of links with free biotin on other vesicles is hampered, so that less aggregation will occur. The higher the initial amount of streptavidin, the stronger this effect. With proper tuning, vesicle aggregates of specific aggregate sizes could be achieved, including vesicle pairs (green curve, Fig. 2).
2.2 Vesicle aggregation using polycations
Vesicle aggregation experiments using cationic polymers were performed in a similar way to those with biotin and streptavidin: we started with single vesicles and stepwise added linker molecules to induce aggregation. The linker molecule here is the polycation poly-L-lysine (PLL). We continued the addition of PLL until unlimited aggregation was observed. In most cases, this happened close to the point where the amount of positive charges on the added PLL equals the amount of negative charges of the DOPG in the vesicle (Z = 1). The results are shown in Fig. 3; for all measured points, D was constant on the time scale of the experiments (≈0.5 h). | ||
| Fig. 3 DLS results showing the normalized diffusion coefficient D0/D (A and C) and mean aggregation number M (B and D) of PG vesicles as a function of the charge ratio between PLL and vesicles Z (A and B), or the number of PLL molecules per vesicle NPLL/Nv (C and D). Blue: vesicles with a DOPG lipid fraction fDOPG = 0.025. Red: fDOPG = 0.1. Measurements were performed at three NaCl concentrations: 10 mM (circles), 50 mM (triangles) and 100 mM (squares). | ||
All these aggregation experiments have in common that, no matter the DOPG content of the vesicles or the salt concentration, the finite aggregate size increases with the addition of PLL, which is consistent with previous studies involving aggregation of charged vesicles and oppositely charged polymers.32,56 The extent of the exponential increase varies with the salt concentration and the vesicle charge density (σ), as will be discussed in the next paragraph. This is fundamentally different compared to aggregation with biotin and streptavidin, where we found a logarithmic relationship between the size and amount of linkers. Apparently, in this case, newly added linkers form bridges with other vesicles rather than diffusing to the adhesion zone. We attribute this effect to the repulsion between PLL molecules, which prevents accumulation of the polymer in an adhesion patch. As a consequence, when a second PLL molecule attaches to a vesicle, it will tend to attract another vesicle. As such, establishing (mainly) vesicle pairs is only possible if a very limited amount of linker molecules is added. This is reflected in Fig. 3D in which vesicle pairs (M ≈ 2) are obtained at a PLL/vesicle ratio (NPLL/Nv) of 1.5 and below.
Another key difference with the biotin–streptavidin type of linkage is that all positive charges on PLL can interact with the negative charges on one single vesicle, while streptavidin only has four binding sites. As the bond between streptavidin and biotin is very strong,57 we expect that the lipid membrane of a vesicle will adjust its conformation to cover all four binding sites. However, as the binding sites are at opposite sides of the molecule, the conformation in which streptavidin bridges two vesicles is most stable. Furthermore, the bond between streptavidin and biotin is notoriously resilient and independent of experimental conditions.58 In contrast, the strength of the interaction of PLL with the vesicles strongly depends on salt concentration and charge density σ. At low σ in combination with a high salt concentration, limited aggregation was obtained at Z = 1, whereas continuous aggregation was observed at lower Z for all other conditions, see Fig. 3.
When comparing the extent of vesicle aggregation with the NPLL/Nv ratio (Fig. 3), we observe that at low salt concentrations (10 mM), vesicle aggregates with M ≈ 2 are obtained at lower NPLL/Nv for low σ (red circles) compared to high σ (red circles). We anticipate this because at low σ, the charge of PLL is not easily compensated by binding to a single vesicle, and linkage with another vesicle therefore happens more frequently than for vesicles with a high charge density. Interestingly, for Z ≤ 1, the electrophoretic mobility of the stable vesicle aggregates remains constant with increasing PLL concentration and increasing aggregate size. This supports the idea that once PLL binds to a vesicle surface, it initiates aggregation with other vesicles, completely enclosing itself in the contact area.
 | ||
| Fig. 4 Normalized hydrodynamic radius of DOPC/DOPG vesicles measured by DLS as a function of the NaCl concentration obtained by stepwise adding NaCl to the solution. The results are shown for vesicles with fDOPG = 0.025 (blue), fDOPG = 0.05 (red) and fDOPG = 0.1 (orange). | ||
The effect of increasing the salt concentration is strongest for vesicles with a low charge density (fDOPG = 0.025), where small vesicle aggregates break up completely into single vesicles. Also, at higher fDOPG, the average hydrodynamic radius could be reduced by increasing the salt concentration and possibly full reversal to single vesicles occurs at salt concentrations higher than those explored in this experiment.
Reversible aggregation of vesicles is an important feature that may be useful in further research on lipid bilayer interactions and for application in controlled drug delivery strategies, where release may be triggered by aggregation and inhibited when reversed. In practice, however, varying the salt concentration to (repeatedly) induce aggregation and de-aggregation is not possible or at least complicated, since in many systems, the salt concentration cannot be varied, and otherwise decreasing the salt concentration while maintaining other conditions, such as the vesicle concentration, requires dialysis or other labour intensive techniques.
Above a threshold concentration, however, the PLL molecules start to adsorb. Once adsorbed, the chains were also able to form bridges between vesicles, linking vesicles to each other. However, at these high PLL concentrations, the adsorption is such that the equilibrium concentration of PLL in the bulk is hardly affected and therefore uncontrolled aggregation occurs.
2.3 Vesicle aggregation using thermoresponsive poly-NIPAm containing surfactants
The principle behind aggregation using C18-pNIPAm is that its carbon tail is inserted into the vesicle membrane while the pNIPAm part can link the vesicles together depending on the temperature. Above its characteristic LCST (lower critical solution temperature), pNIPAm expels its hydration water, effectively becoming hydrophobic. It tries to form a condensed pNIPAm phase and it can do this better when pNIPAm of an opposite vesicle is also used. Hence, the LCST-behaviour starts vesicle aggregation. This effect is reversible as once the temperature goes below the LCST, the pNIPAm polymers take up water again and swell, and as a result, de-aggregation occurs.To perform an aggregation experiment with C18-pNIPAm, we make a DOPC vesicle solution to which we add a certain amount of the surfactants. After mixing, light scattering measurements were performed while the sample was subjected to a temperature program. This involves a 10 minute step at 25 °C, then half an hour at 40 °C and lastly another 10 minutes at 25 °C. The LCST of pNIPAm lies around 32 °C. In Fig. 5, a typical DLS result is displayed for vesicles with a C18-pNIPAm/DOPC ratio of 0.08. Control measurements on vesicles without surfactants are shown as well.
 | ||
| Fig. 5 Hydrodynamic radius Rh over time for DOPC vesicles undergoing a standard temperature cycle of 10 min at 25 °C, 30 min at 40 °C and another 10 min at 25 °C. The red curve represents the temperature at a given time. The hydrodynamic radius (Rh) is shown for vesicles without C18-pNIPAm (red) and for vesicles with a C18-pNIPAm/DOPC fraction of 0.08 (orange). | ||
As can be seen from the figure, in the presence of C18-pNIPAm, aggregation occurs above the LCST. Without vesicles, the surfactants aggregate very quickly and uncontrollably while the vesicles without surfactants show no aggregation. For the mixture after increasing the temperature to 40 °C, a quick increase in size is observed after which the aggregation size stabilizes, indicating that the aggregation is limited. This process is fully reversible, as once the temperature drops below the LCST, the aggregates quickly disassemble into single vesicles again. As for the streptavidin–biotin system, stabilizing of the aggregation size above the LCST is due to the intra-aggregate binding of the linkers. Basically, C18-pNIPAm surfactants in the membrane of the vesicles diffuse towards the contact areas as they are attracted to the other C18-pNIPAm surfactants already lumped together there. As a result, within a short time, there are no remaining surfactants on the outer surface of the vesicles and therefore binding to other vesicles does not happen anymore.
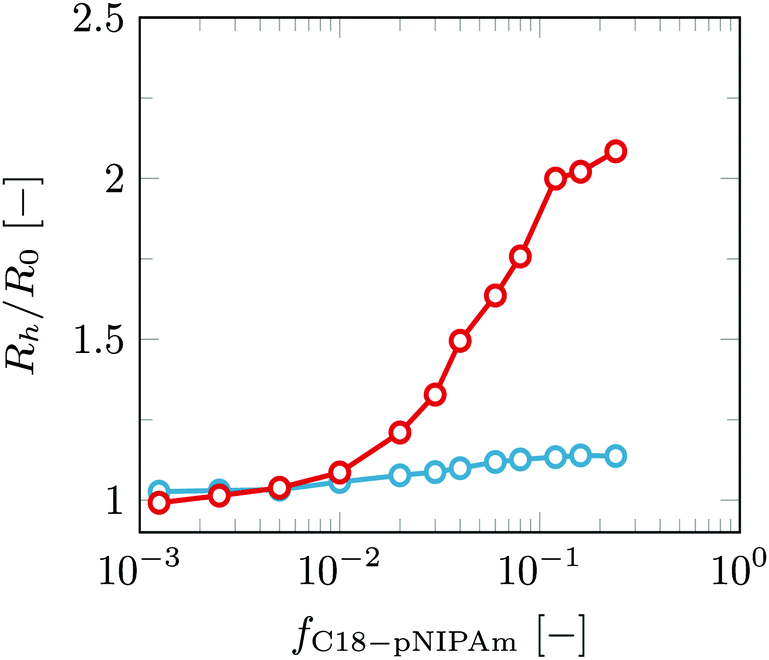 | ||
| Fig. 6 Normalized hydrodynamic radius of the vesicles as a result of stepwise addition of the C18-pNIPAm surfactant to the sample. The average size is shown for each sample at 25 °C (blue) and at 40 °C (red). Lipid concentration Cl = 0.1 mM in 10 mM Tris–HCl buffer. | ||
Each dot represents a standard aggregation experiment as explained earlier. The average size is calculated for both 25 °C and 40 °C. After each experiment, instead of making a new sample, more C18-pNIPAm surfactants are added to the initial sample, mixed and measured using DLS with the standard temperature program. The results clearly show a dependence of the aggregation size on C18-pNIPAm/DOPC ratio. A threshold ratio of around 0.01 has to be reached before aggregation occurs at all. Above this threshold ratio, the aggregation size scales logarithmically with the ratio, confirming accumulation of collapsed pNIPAm in the contact zones (intra-aggregate binding, similar to what happens in the biotin–streptavidin system). Above the LCST and around a C18-pNIPAm/DOPC ratio of 0.04–0.08, the vesicles are mainly present in pairs. Our SLS results show M = 2.62 at 40 degrees at a C18-pNIPAm/DOPC ratio of 0.08. The presence of a threshold value is characteristic of phase separation. This can only set in when the concentration increases above the binodal. In this case, the local pNIPAm concentration needs to be above a binodal value for the C18-pNIPAm anchored onto the membrane. The binodal value appears at relatively high C18-pNIPAm/DOPC ratios, which we attribute to C18-pNIPAm being able to partition partly in the bilayer, near the interface between the lipid head groups and tails, thus already avoiding contact with water to some extent. We anticipate that this binodal/threshold value further decreases with temperature, that is, when the PNIPAm–water demixing is quenched deeper in the two-phase state, we expect the threshold concentration to decrease. This threshold concentration may further be a function of the size of the PNIPAm block.
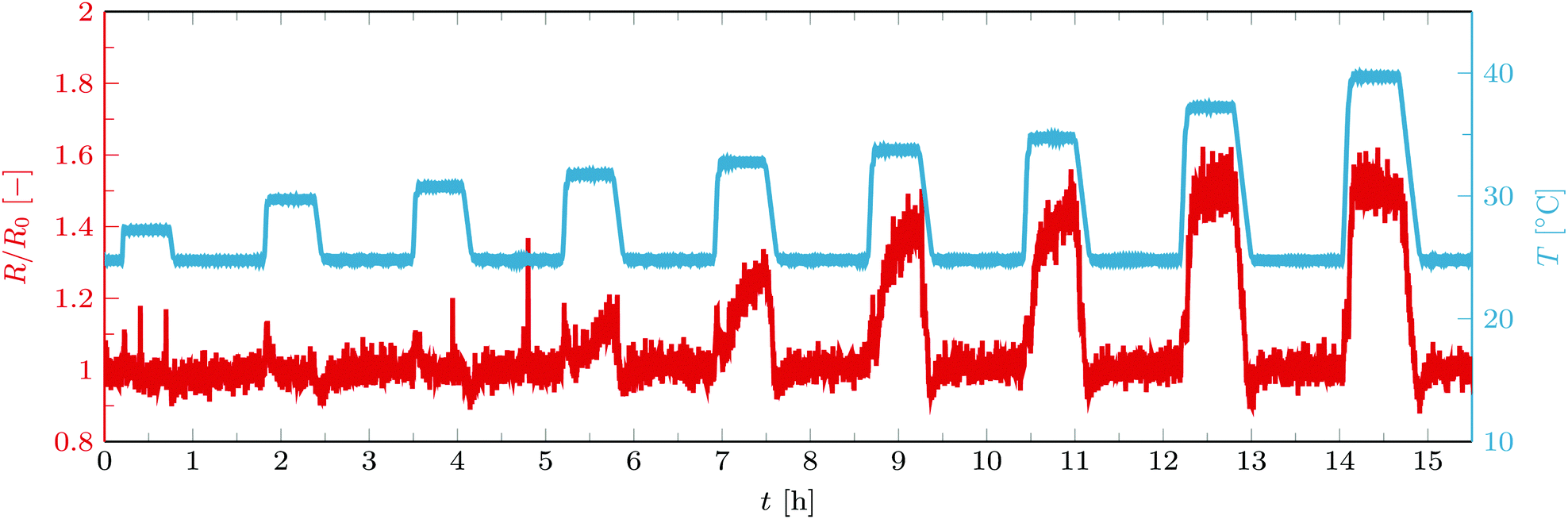 | ||
| Fig. 7 Normalized radius of vesicles containing C18-pNIPAM as they are subjected to a temperature program (red curve). The blue curve shows the temperature. C18-pNIPAM/DOPC ratio = 0.08. | ||
3 General discussion
Limited aggregation, i.e. the formation of stable, small aggregates, is a special feature in the assembly of vesicles triggered by additives. This is emphasized by the fact that not all approaches that we attempted to form stable vesicle pairs were successful. Apart from the strategies reported here, we tried, for example, to induce limited aggregation using telechelic (triblock) copolymers, which combine a hydrophilic middle block with two short aliphatic end blocks. This type of molecule is used as an associative thickener in paints.59,60 We used a variant with C18 tails in combination with a pEO middle block of nominal weight Mw ≈ 35000 g mol−1 (∼800 monomers).61 Already in dilute solutions, these molecules self-assemble and form flower-like micelles. In combination with phospholipid vesicles, we found that the telechelics partition between being assembled in such micelles and being inserted with their hydrophobic blocks in the vesicle membranes. Above a threshold concentration of the telechelics, aggregation of vesicles could be induced as expected. However, the aggregates kept growing over time. We assume that the cause of this unlimited association behaviour stems from the buffering capacity of the flower-like micelle population. Telechelics that were ‘lost’ in adhesion patches could be replenished by disintegrating of the micelles. At ultra-low concentrations of the telechelics, one could imagine that they would be all vesicle-bound and no flowerlike micelles would exist. However, we found no association of vesicles below the CMC of the telechelics. Maybe the length of the PEO and hydrophobic blocks of the telechelics could be tuned such that limited aggregation is possible, but we failed to hit on such an ideal species. As shown in this paper, a similar failure of limited aggregation was observed when a low molecular weight cationic polymer was used to link negatively charged vesicles together. Arguably, the failures to achieve limited aggregation are as instructive as the successes.
Based on both our successful and unsuccessful strategies, two routes can be distinguished that result in limited aggregation of vesicles. The most simple one is just limiting the amount of linkers. In principle, if the amount of linker molecules per vesicle is close to unity, aggregation will be limited and most of the vesicles will aggregate into stable vesicle pairs. For this to happen, the interaction between the linker and the vesicles and the tendency to bridge vesicles need to be strong. We observe this type of limited aggregation for negatively charged vesicles bridged by long PLL molecules. An additional feature is that the cationic polymers repel each other, resulting in only one polymer per adhesion patch. Optimization of the vesicle surface charge and salt concentration is a necessary requirement here.
The second route is to use membrane-bound linkers that attract each other. Limited aggregation then can be obtained provided that the timescale in which linkers diffuse along the membrane to the first formed adhesion patch between two vesicles (tdiff) is small compared to the vesicle collision time (τ). This implies that the vesicle concentration should be relatively low. Following this route, we were able to arrest the aggregation of vesicles at the pair level using biotinylated lipids that can couple by adding streptavidin to the vesicle solution, and by using C18-pNIPAm surfactants that couple above their LCST through hydrophobic interactions.
Each of the three suitable linker types that we used has its own advantages and drawbacks. Limited aggregation by adding an appropriate amount of long polyelectrolytes to charged vesicles has the advantage that it is relatively cheap, simple and straightforward and that aggregation can be reversed by the addition of salt. However, in practice, repeatedly inducing aggregation and de-aggregation is not possible or is complicated. In addition, even if the average aggregation number is two, a fairly broad distribution of single vesicles, vesicles pairs, trimers, etc. is obtained. The strategies using biotin–streptavidin or C18-pNIPAm as linkers however can lead to a sharp distribution of aggregate sizes, with mainly vesicle pairs. This is supported by the development of the particle size distributions during the aggregation experiments with the various linkers, obtained from CONTIN analysis of the DLS results (Fig. S4, ESI†). The links between the vesicles are strong and very insensitive to experimental conditions, which may be advantageous in some cases, but can also be a drawback if reversibility is an issue. C18-pNIPAm surfactant molecules also robustly enable limited aggregation, similar to the streptavidin–biotin linkers. In addition, complete reversibility can be achieved, which can be triggered multiple times by increasing and decreasing the temperature with respect to the LCST.
Possible applications for small vesicle aggregates or vesicle pairs require research into inter-membrane interactions such as membrane (hemi-)fusion. The perfect example of a specific linker molecule that is able not only to connect vesicles but also to induce fusion is DNA.11,26 We aspire to find alternative linkers, such as the ones used in this work, that under certain conditions could achieve similar results. Further experiments are necessary though as there are strong indications that the strategies used to induce limited aggregation of the vesicles occurred without subsequent fusion of the membranes. The reversibility of the aggregation of both vesicles attached by C18-pNIPAm (decreasing temperature) and vesicles attached by PLL (increasing salt concentration) already strongly points to this. A strong indication that fusion did also not occur in the vesicle pairs formed by biotin and streptavidin was found by fitting the SLS data using the form factor of a spherical shell (ESI,† Fig. S5). The presence of an adhesion patch in the vesicle pairs influenced the form factor enough to give a significantly worse fit than for single vesicles. For more direct proof of fusion or the lack of it, we are planning to perform lipid mixing or content mixing experiments using fluorescent molecules in follow up experiments.
4 Conclusions
We have presented three successful strategies that lead to stable finite-sized vesicle aggregates. We showed that by tuning the experimental parameters, it is possible to obtain an average aggregation number of 2. In addition, we analysed vesicle aggregation at this average pair level under various relevant physico-chemical conditions. We distinguish two routes that lead to limited aggregation of vesicles: limiting the amount of linkers, or using membrane-bound linkers that upon contact with another vesicle are able to diffuse to the contact area, depleting the rest of the membrane of linker molecules to bind with other vesicles. It is expected that under well-chosen experimental conditions, the latter route can provide a dominant population of vesicle pairs.Amongst the successful strategies, the one involving C18-pNIPAm as a linker molecule shows an additional feature: the limited aggregation of vesicles is completely and repeatedly reversible using temperature as the trigger. We therefore anticipate that this system can serve as a base to study targeted and triggered interactions between membranes.
We envision that the ability to create stable vesicle pairs is an effective and useful way to study interactions between lipid membranes and the physical properties of membrane pairs. Combining different linker strategies with variations in the lipid composition of the vesicles could be used to unravel the principles underlying the topological stability of bilayer pairs. Furthermore, it enables studies into the nature and time scales of lipid exchange between bilayers and the content exchange of vesicles with or without full scale fusion of the vesicles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Remco Fokkink (Physical Chemistry and Soft Matter, Wageningen University) for assistance with the light scattering experiments, and Jan Willem Borst and Adrie H. Westphal (Laboratory of Biochemistry, Wageningen University) for their help with the FCS experiments and data analysis.Notes and references
- C. Sohlenkamp and O. Geiger, FEMS Microbiol. Rev., 2016, 40, 133–159 CrossRef CAS PubMed.
- L. Vigh, P. V. Escribá, A. Sonnleitner, M. Sonnleitner, S. Piotto, B. Maresca, I. Horváth and J. L. Harwood, Prog. Lipid Res., 2005, 44, 303–344 CrossRef CAS PubMed.
- I. Simidjiev, V. Barzda, L. Mustárdy and G. Garab, Biochemistry, 1998, 37, 4169–4173 CrossRef CAS PubMed.
- B. R. Lentz, V. Malinin, M. E. Haque and K. Evans, Curr. Opin. Struct. Biol., 2000, 10, 607–615 CrossRef CAS PubMed.
- G. V. Betageri and M. B. Yatvin, Liposome drug delivery, US Pat., 6761901, 2004 Search PubMed.
- E. Kisak, B. Coldren, C. Evans, C. Boyer and J. Zasadzinski, Curr. Med. Chem., 2004, 11, 199–219 CrossRef CAS PubMed.
- M. Michel, M. Winterhalter, L. Darbois, J. Hemmerle, J. C. Voegel, P. Schaaf and V. Ball, Langmuir, 2004, 20, 6127–6133 CrossRef CAS PubMed.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS PubMed.
- D. T. Chiu, C. F. Wilson, F. Ryttsén, A. Strömberg, C. Farre, A. Karlsson, S. Nordholm, A. Gaggar, B. P. Modi and A. Moscho, et al. , Science, 1999, 283, 1892–1895 CrossRef CAS PubMed.
- S. F. Fenz, A.-S. Smith, R. Merkel and K. Sengupta, Soft Matter, 2011, 7, 952–962 RSC.
- B. van Lengerich, R. J. Rawle, P. M. Bendix and S. G. Boxer, Biophys. J., 2013, 105, 409–419 CrossRef CAS PubMed.
- T. C. Südhof and J. E. Rothman, Science, 2009, 323, 474–477 CrossRef PubMed.
- B. Derjaguin and L. Landau, Acta Physicochim. URSS, 1941, 14, 633–662 Search PubMed.
- E. J. W. Verwey, J. Phys. Chem., 1947, 51, 631–636 CrossRef CAS PubMed.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381 CrossRef CAS PubMed.
- S. Sacanna, W. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51 CrossRef CAS PubMed.
- D. J. Kraft, R. Ni, F. Smallenburg, M. Hermes, K. Yoon, D. A. Weitz, A. van Blaaderen, J. Groenewold, M. Dijkstra and W. K. Kegel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10787–10792 CrossRef CAS PubMed.
- K. M. Buettner, C. I. Rinciog and S. E. Mylon, Colloids Surf., A, 2010, 366, 74–79 CrossRef CAS.
- F. von der Kammer, S. Ottofuelling and T. Hofmann, Environ. Pollut., 2010, 158, 3472–3481 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- K. Aslan, C. C. Luhrs and V. H. Pérez-Luna, J. Phys. Chem. B, 2004, 108, 15631–15639 CrossRef CAS.
- M.-P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225–4229 CrossRef CAS PubMed.
- Y. Zhang, A. McMullen, L.-L. Pontani, X. He, R. Sha, N. C. Seeman, J. Brujic and P. M. Chaikin, Nat. Commun., 2017, 8, 21 CrossRef PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- L. Parolini, B. M. Mognetti, J. Kotar, E. Eiser, P. Cicuta and L. Di Michele, Nat. Commun., 2015, 6, 5948 CrossRef CAS PubMed.
- U. Jakobsen, A. C. Simonsen and S. Vogel, J. Am. Chem. Soc., 2008, 130, 10462–10463 CrossRef CAS PubMed.
- M. Hadorn and P. E. Hotz, PLoS One, 2010, 5, e9886 CrossRef PubMed.
- S. A. Walker and J. A. Zasadzinski, Langmuir, 1997, 13, 5076–5081 CrossRef CAS.
- E. Kisak, M. Kennedy, D. Trommeshauser and J. Zasadzinski, Langmuir, 2000, 16, 2825–2831 CrossRef CAS.
- S. Chiruvolu, S. Walker, J. Israelachvili, F.-J. Schmitt, D. Leckband and J. A. Zasadzinski, Science, 1994, 264, 1753–1756 CrossRef CAS PubMed.
- T. P. de Souza, G. V. Bossa, P. Stano, F. Steiniger, S. May, P. L. Luisi and A. Fahr, Phys. Chem. Chem. Phys., 2017, 19, 20082–20092 RSC.
- G. V. Bossa, T. P. de Souza and S. May, Soft Matter, 2018, 14, 3935–3944 RSC.
- H. Minami, T. Inoue and R. Shimozawa, J. Colloid Interface Sci., 1993, 158, 460–465 CrossRef CAS.
- A. Di Biasio, F. Bordi and C. Cametti, Trends in Colloid and Interface Science XVI, Springer, 2004, pp. 78–82 Search PubMed.
- S. Chiruvolu, J. Israelachvili, E. Naranjo, Z. Xu, J. Zasadzinski, E. Kaler and K. Herrington, Langmuir, 1995, 11, 4256–4266 CrossRef CAS.
- D. A. Noppl-Simson and D. Needham, Biophys. J., 1996, 70, 1391–1401 CrossRef CAS PubMed.
- S. A. van der Meulen and M. E. Leunissen, J. Am. Chem. Soc., 2013, 135, 15129–15134 CrossRef CAS PubMed.
- T. E. Kodger and J. Sprakel, Adv. Funct. Mater., 2013, 23, 475–482 CrossRef CAS.
- J. Diao, Y. Ishitsuka, H. Lee, C. Joo, Z. Su, S. Syed, Y.-K. Shin, T.-Y. Yoon and T. Ha, Nat. Protoc., 2012, 7, 921 CrossRef CAS PubMed.
- Y.-H. M. Chan, B. van Lengerich and S. G. Boxer, Biointerphases, 2008, 3, FA17–FA21 CrossRef CAS PubMed.
- H. Pera, T. M. Nolte, F. A. Leermakers and J. M. Kleijn, Langmuir, 2014, 30, 14581–14590 CrossRef CAS PubMed.
- N. G. Júnior, M. H. Cardoso, E. S. Cândido, D. van den Broek, N. de Lange, N. Velikova, J. M. Kleijn, J. M. Wells, T. M. Rezende and O. L. Franco, et al. , Sci. Rep., 2018, 8, 11127 CrossRef PubMed.
- W. Brown, Light scattering: Principles and development, Clarendon Press, Oxford, 1996 Search PubMed.
- B. J. Berne and R. Pecora, Dynamic light scattering: with applications to chemistry, biology, and physics, Courier Corporation, 2000 Search PubMed.
- D. E. Koppel, J. Chem. Phys., 1972, 57, 4814–4820 CrossRef CAS.
- A. Nolles, A. H. Westphal, J. A. de Hoop, R. G. Fokkink, J. M. Kleijn, W. J. van Berkel and J. W. Borst, Biomacromolecules, 2015, 16, 1542–1549 CrossRef CAS PubMed.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-rays, 1955 Search PubMed.
- D. Magde, E. L. Elson and W. W. Webb, Biopolymers, 1974, 13, 29–61 CrossRef CAS PubMed.
- V. V. Skakun, M. A. Hink, A. V. Digris, R. Engel, E. G. Novikov, V. V. Apanasovich and A. J. Visser, Eur. Biophys. J., 2005, 34, 323–334 CrossRef PubMed.
- B. Kuipers, M. Van de Ven, R. Baars and A. Philipse, J. Phys.: Condens. Matter, 2012, 24, 245101 CrossRef CAS PubMed.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 229–242 CrossRef.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 213–227 CrossRef.
- P. Stepanek, Dyn. Light Scattering, 1993, 177–241 CAS.
- J. Wong, A. Chilkoti and V. T. Moy, Biomol. Eng., 1999, 16, 45–55 CrossRef CAS PubMed.
- P. Carrara, P. Stano and P. L. Luisi, ChemBioChem, 2012, 13, 1497–1502 CrossRef CAS PubMed.
- P. S. Stayton, S. Freitag, L. A. Klumb, A. Chilkoti, V. Chu, J. E. Penzotti, R. To, D. Hyre, I. Le Trong and T. P. Lybrand, et al. , Biomol. Eng., 1999, 16, 39–44 CrossRef CAS PubMed.
- C. E. Chivers, A. L. Koner, E. D. Lowe and M. Howarth, Biochem. J., 2011, 435, 55–63 CrossRef CAS PubMed.
- A. Yekta, J. Duhamel, H. Adiwidjaja, P. Brochard and M. Winnik, Langmuir, 1993, 9, 881–883 CrossRef CAS.
- A.-C. Hellgren, P. Weissenborn and K. Holmberg, Prog. Org. Coat., 1999, 35, 79–87 CrossRef CAS.
- J. Sprakel, E. Spruijt, M. C. Stuart, N. Besseling, M. Lettinga and J. van der Gucht, Soft Matter, 2008, 4, 1696–1705 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01692a |
| This journal is © The Royal Society of Chemistry 2020 |