
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of oxygen vacancies in water splitting photoanodes
Roser
Fernández-Climent
,
Sixto
Giménez
* and
Miguel
García-Tecedor
 *
*
Institute of Advanced Materials (INAM), Universitat Jaume I, Avenida de Vicent Sos Baynat, s/n, 12006 Castelló de la Plana, Castellón, Spain. E-mail: sjulia@uji.es; tecedor@uji.es
First published on 28th September 2020
Abstract
Photoelectrochemical water splitting has become one of the most reliable solar-energy conversion technologies for clean hydrogen production. In the race of developing and understanding new semiconducting materials for this application, several studies have been focused on the role of oxygen vacancies, which are known to be defects with a high impact on the final optical and electrical properties of the photoelectrodes. These oxygen defective states can introduce either favorable or detrimental pathways to the overall PEC performance. The present topical review aims to summarize the role of oxygen vacancies in four of the most studied semiconducting thin film oxides (BiVO4, Fe2O3, TiO2 and WO3) as photoanodes for solar water splitting.
Introduction
In the current and worrying global environmental situation, light-driven water splitting stands out as one of the most promising alternatives to the obsolete fossil-fuel-based economy for the production of clean energy sources such as hydrogen.1,2 The required materials for solar water splitting are semiconductors, with a certain bandgap, which can generate an electrical current under illumination. Narrow bandgap semiconductors (ideally Eg ∼ 2 eV) are more suitable for solar energy conversion due to their higher optical absorption from the solar spectrum, since all the photons with energies higher than the bandgap can be absorbed. Specifically, n-type semiconductors are required for the Oxygen Evolution Reaction (OER) in order to oxidize water (2H2O → O2 + 4H+ + 4e−) with the photogenerated holes from the valence band (VB) under illumination conditions. Among the most studied materials for the OER, n-type metal oxide semiconductors such as BiVO4,3,4 Fe2O3,5,6 TiO2 (ref. 7 and 8) and WO3 (ref. 9 and 10) stand out due to their promising properties. Furthermore, other promising systems such as TaON,11,12 CN,13etc. have also been reported. Usually, a thin film, around few hundreds of nm, of the active material is deposited onto a transparent oxide conducting layer (back contact, usually FTO or ITO) with a thickness high enough to present a high optical absorption but lower than the carrier diffusion length to allow efficient charge extraction.14,15 Specifically, a given semiconductor must have a thickness of 3/α, being α the absorption coefficient, in order to absorb 95% of the light at that wavelength, as derived from the Beer–Lambert law.16Oxygen vacancies (OVs) constitute one of the most studied defects in oxide semiconductors. These defects, usually intrinsic, are generated by the removal of an oxygen atom from the crystalline structure, which remains charged with two extra electrons.17,18 Oxygen vacancies could be formed by different mechanisms, including: (i) the formation during the growth of a crystal, (ii) the formation to compensate for the charge introduced by a dopant (ionic compensation), or (iii) pre-existing OVs which diffuse and gather at the interfaces, leading to the formation of an interlayer of a different crystal phase due to their influence on the phase stability.19 Oxygen vacancies are the most common defects in metal oxides. Due to this fact, any treatment altering the chemical environment (temperature annealing, treatments under different atmospheres during the growth, laser irradiation, etc.) of the metal oxide will lead to the formation of these oxygen defective states. However, there is a maximum concentration of OVs where the phase stability of the crystal lattice could be compromised.20 Oxygen vacancies are responsible in many cases for the exotic electrical and optical properties of different oxide semiconductors.21–23 Additionally, in recent years, the possibility to tune the population and energy distribution of these defects, through different physical and chemical treatments or doping processes, for specific goals has been extensively exploited.24–27 However, the detection of structural defects in condensed matter physics has always been a challenge due to their diluted and elusive nature. Several efforts have been undertaken to find suitable strategies to address this issue. Currently, there is a wide range of available techniques, some of them indirect,28 for the detection of OVs,29 based on different energy sources such as: (i) electrons, (ii) X-rays or (iii) photons. (i) Among the detection techniques using electrons as the energy source, the most relevant ones are: High-Resolution Transmission Electron Microscopy (HRTEM), corrected by spherical aberration, which can lead to the direct visualization of OVs,30 Scanning Tunneling Microscopy (STM)31 and Cathodoluminescence (CL).32 (ii) X-ray based tools include X-ray Photoelectron Spectroscopy (XPS),33 X-ray Absorption Spectroscopy (XAS)34 and X-ray Diffraction (XRD).25 (iii) Using photons as the energy source, the most usual techniques are Photoluminescence (PL),35 Raman Spectroscopy,36 and Fourier Transform Infrared Spectroscopy (FTIR).37 In the specific case of solar water splitting, the effect of oxygen vacancies on the performance of different photoanodes has been widely reported in different systems.25,38–40 Oxygen vacancies can act as doping elements introducing defect levels inside the bandgap of the semiconductor, and thus, modifying the different pathways for the electronic processes such as recombination, charge extraction and injection.41 Specifically, OVs can induce favorable effects on the overall PEC performance by increasing the carrier concentration, being responsible in many cases for the intrinsic n-type conductivity,42 which favors the formation of a space-charge layer trough doping,43 or enhancing light absorption,44 but can also induce detrimental effects such as activation of recombination channels, acting as charge-carrier traps or recombination centers.45,46
The present review aims to summarize the main contributions about the role of oxygen vacancies in four of the most studied semiconducting metal oxides as photoanodes for the OER (BiVO4, Fe2O3, TiO2 and WO3).
Bismuth vanadate, BiVO4
During the last few years, BiVO4 has emerged as one of the most promising candidates to fabricate reliable photoanodes for photoelectrochemical (PEC) solar water splitting. BiVO4 presents three possible phases: tetragonal scheelite, tetragonal zircon and monoclinic scheelite.47 Among these structures, the monoclinic scheelite structure (Fig. 1), showing a layered nature and belonging to the C2/c spatial group, is the most active for PEC applications.48 BiVO4 is an n-type semiconductor, with a 2.4 eV bandgap and consequently a theoretical solar to hydrogen (STH) efficiency of 9.2% with a maximum photocurrent of 7.5 mA cm−2 under AM 1.5 G illumination, low overpotential and favorable band-edge positions.49 However, BiVO4 also presents poor electron transport, high surface recombination and slow water oxidation kinetics. Enormous efforts have been made in the past few years in order to mitigate these drawbacks through different approaches such as nanostructuring,50,51 doping,52,53 heterostructuring4,54 and the use of efficient co-catalysts.55,56 | ||
| Fig. 1 Monoclinic structure of BiVO4 obtained with VESTA software. | ||
An additional strategy to improve the performance of BiVO4 photoanodes relies on the intentional creation of oxygen vacancies.57,58 Oxygen vacancies in BiVO4, usually associated with V4+ species, are structural defects, which can introduce shallow levels inside the bandgap. These shallow levels can have favorable effects on the final performance, reducing the recombination processes between photo-generated carriers, and thus, increasing the overall PEC activity. Alternatively, these defects can act as recombination centers and then activate recombination channels, which decrease the final performance. Specifically, the charge separation efficiency can be improved in BiVO4 photoanodes by the intentional introduction of a high density of oxygen vacancies, as previously reported.36,59,60 In these studies, the enhanced performance was systematically correlated with a higher density of oxygen deficiencies, detected mainly by XPS. Frequently, the samples showing a higher density of OVs also showed a higher Incident Photon to Current Efficiency (IPCE), which is a direct measurement of the External Quantum Efficiency (EQE), related to a higher density of extracted photogenerated carriers for the same light irradiation, mainly due to a more efficient optical absorption. However, as explained above, there are other studies, where the defect levels inside the bandgap introduced by oxygen vacancies show a detrimental behavior. As an example, a recent study showed the suppression of the formation of oxygen vacancies leading to a higher PEC performance due to an increase of the conductivity as a consequence of the release of bound polarons.61
The synthesis of black BiVO4 has been reported, as a new black material.63 Herein, nanosized black BiVO4 colloids were prepared by laser irradiation of bulk particles. By using Raman Spectroscopy and XPS, the black color was associated with the presence of a high concentration of oxygen vacancies in the nanostructure after laser radiation. The new black BiVO4 showed a clear improvement in the performance as an anode material for Sodium Ion Batteries (SIBs), compared to pristine BiVO4. The enhanced performance was related not only to the decreased particle size of the black BiVO4, but also to the presence of oxygen vacancies, which can alter the electronic structure improving the conductivity.
Additionally, the presence of oxygen vacancies clearly increases the optical absorption in the visible range, which is favorable for photocatalytic applications such as PEC water splitting. However, an excess density of oxygen vacancies could induce a metal-like behavior (degenerated semiconductor), leading to the deactivation of the photoactivity of the material and consequently, decreasing the overall performance towards the OER. Although the application explored in this study is out of the scope of the present review, this work opens a new possibility for the design of black BiVO4 as a photoanode for water splitting with enhanced performance towards the OER.
Several theoretical studies have also tried to shed light on the role of oxygen vacancies in BiVO4 photoanodes.64,65 Oxygen vacancies have been found to present two possible geometries of a V–O–V bridge, allowing excess electrons to be delocalized in a two-dimensional charge state or localized in different V centers.62 These V–O–V bonds present low transition energies, which allow their consecutive formation and destruction, leading to the migration of oxygen vacancies in the bulk of BiVO4 and increasing both electronic and ionic conductivities.66,67 Due to the complexity of BiVO4, all these possible electronic configurations (Fig. 2) have a direct impact on its final performance towards solar water splitting. Another recent theoretical study reported that the location of oxygen vacancies, in the bulk or at the surface, influences the overall performance of BiVO4 photoanodes in a completely different direction.64 Specifically, this study claimed that both oxygen vacancies in the bulk and at the surface introduce donor levels inside the band gap. However, the formation of bulk polarons, due to the migration of electrons associated with oxygen vacancies, contributes to the conductivity while surface polarons likely do not, acting as possible recombination centers.
 | ||
| Fig. 2 Intra-gap states introduced by the different electronic configurations associated with oxygen vacancies in BiVO4. This figure has been reproduced from ref. 62 with permission from AIP, copyright 2020. | ||
The occupancy of the electronic states associated with OVs can also be modulated in order to move one step forward in the modification of BiVO4 photoanodes. In a recent study,68 the population of the electronic states associated with OVs in BiVO4 photoanodes were modulated by using several spectroscopic techniques. This study concluded that oxygen vacancies participate in the trapping of photogenerated carriers in addition to their well-known role enhancing the n-doping and the formation of the space-charge layer. Photogenerated holes are trapped by VOV4+ states in the bulk, leading to a loss pathway, since these states are above the water oxidation level and thus are not able to participate in the OER. However, photogenerated electrons are trapped in VOV5+ states in the space-charge layer with an activation energy of around 0.2 eV. Thus, these electrons can be extracted with some thermal activation, improving the PEC performance of the BiVO4 photoanodes. The role of oxygen vacancies in trapping and transport processes of BiVO4 photoanodes is depicted in the scheme shown in Fig. 3.
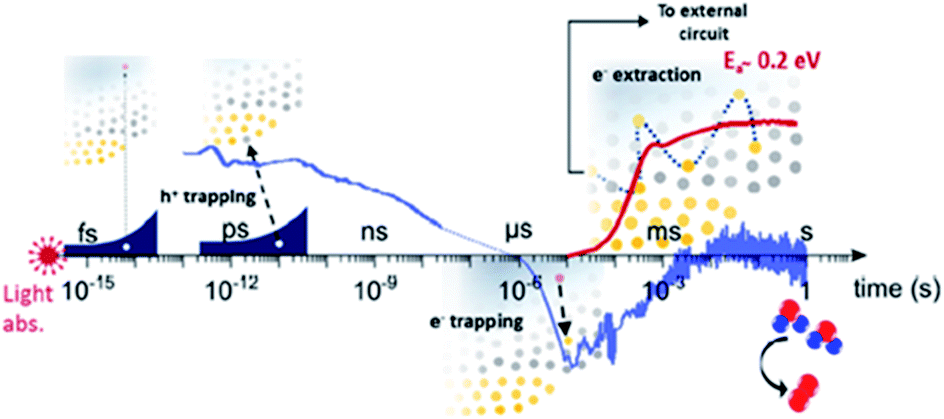 | ||
| Fig. 3 Scheme of the role of oxygen vacancies in trapping and transport processes in BiVO4 photoanodes. This figure has been reproduced from ref. 68 with permission from ACS, copyright 2019. | ||
A recent study has reported the suppression of OVs in the bulk with the concomitant increase in the generation of these defects at the surface of BiVO4, by a photoetching process.69 This study also claims that the formation of oxygen vacancies at the surface leads to a noticeable improvement in the final PEC activity towards water oxidation due to an increase in the carrier density, enhancing the band-bending, and then improving the charge separation efficiency by a factor 2.3 compared to pristine BiVO4.
An additional approach to improve the performance of BiVO4 photoanodes consists of the deposition of a certain electrocatalyst with a high concentration of oxygen vacancies. It has been already reported that oxygen vacancies lead to an increase of the density of catalytic sites improving then the overall performance of the BiVO4-catalyst system.70 The same strategy was followed to improve the PEC activity of α-Fe2O3 photoanodes with a high oxygen-defective CoOx electrocatalytic layer.71 However, this approach is out of the scope of this review.
Hematite, Fe2O3
Hematite has been widely studied as a solar water splitting photoanode due its abundance, low-cost, non-toxicity, and favorable physical properties, such as its narrow bandgap (2.1 eV) and high chemical stability.5,72–74 Fe2O3 has a theoretical maximum photocurrent of 12.6 mA cm−2, which corresponds to a STH efficiency of 16.8%.75 Among the different existing phases, α-Fe2O3, with an orthorhombic–trigonal structure (Fig. 4) belonging to the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c spatial group, is the most active phase for solar water splitting. Nevertheless, hematite also shows severe limiting factors as poor electrical conductivity, a short hole diffusion length (2–4 nm),76 poor OER kinetics,77 and non-favorable band positions for water splitting.78 Several approaches have been reported to overcome these drawbacks, such as extrinsic doping,79,80 nanostructuring,81 the use of electrocatalysts82,83 and complex architectures.84 An additional possibility consists of the intrinsic doping of hematite photoanodes with oxygen vacancies. The role of these defects in α-Fe2O3 photoanodes for solar water oxidation has been extensively reported as a beneficial factor to their final performance.
c spatial group, is the most active phase for solar water splitting. Nevertheless, hematite also shows severe limiting factors as poor electrical conductivity, a short hole diffusion length (2–4 nm),76 poor OER kinetics,77 and non-favorable band positions for water splitting.78 Several approaches have been reported to overcome these drawbacks, such as extrinsic doping,79,80 nanostructuring,81 the use of electrocatalysts82,83 and complex architectures.84 An additional possibility consists of the intrinsic doping of hematite photoanodes with oxygen vacancies. The role of these defects in α-Fe2O3 photoanodes for solar water oxidation has been extensively reported as a beneficial factor to their final performance.
 | ||
| Fig. 4 Orthorhombic–trigonal structure of Fe2O3 obtained with VESTA software. | ||
Several studies reported fabrication methods to tune the concentration of oxygen vacancies for improved PEC performance: Li et al.85 reported a fabrication method of α-Fe2O3 photoanodes trough thermal decomposition of β-FeOOH in an N2-rich atmosphere. The resulting hematite showed enhanced photoactivity due an increase in the electrical conductivity through a polaron hopping mechanism86 as a result of the formation of oxygen vacancies, and thereby Fe2+ sites. Other studies combined the generation of oxygen vacancies with extrinsic doping (with Ti, Sn, etc.) to improve the activity25 by increasing the electrical conductivity. Specifically, Sun et al.87 claimed that oxygen vacancies increase the donor density with a lower oxidation state of iron (Fe2+), while Ti-doping improves the catalytic response by the introduction of active sites, increasing the final performance of the fabricated photoanodes. The final activity of hematite photoanodes can also be tuned through thermal annealing in different environments.88 In this study, the effect of thermal annealing under different atmosphere on Nb- and Si-doped Fe2O3 photoanodes was analyzed. When the samples were annealed in air, oxygen can be incorporated as O2− in the hematite lattice (OO). This excess oxygen is then compensated for by the creation of cation vacancies  following the equation:
following the equation:
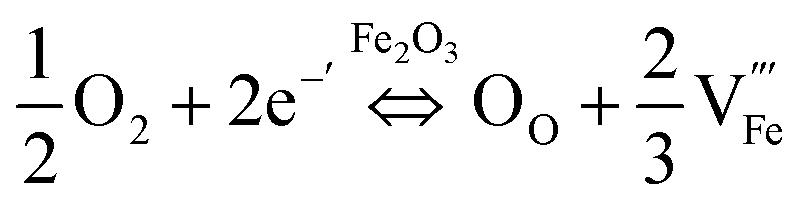 | (1) |
These cation vacancies are surrounded by anions and act as critical recombination centers for holes, decreasing the photocurrent. On the other hand, annealing under an oxygen-free atmosphere (Ar) leads to the formation of oxygen vacancies, due to the reduced oxygen partial pressure, increasing the carrier concentration present in the photoanodes and thus, the photocurrent. In the same direction, a recent study reported the enhanced performance of Fe2O3 photoanodes after thermal annealing in a low oxygen atmosphere, showing that oxygen vacancies improved the transport of photogenerated holes.89
Theoretical Density Functional Theory (DFT) studies have also been focused on the effect of oxygen vacancies on the overpotential and final performance of Fe2O3 photoanodes. It has been reported that the most efficient way to reduce the overpotential in hematite photoanodes relies on the control of the population of oxygen vacancies.90 Specifically, the authors calculated an overpotential as low as 470 mV for an optimal concentration of oxygen vacancies of 12.6 vacancies per nm2, which is one of the lowest densities among theoretical studies,91,92 as shown in Fig. 5. The enhanced performance of hematite photoanodes after N2 treatments has also been reported.93 The experimental results showed that the enhanced performance induced by the presence of OVs was related to an increase of both charge separation and charge transfer efficiencies. Combining these experimental results with DFT calculations, it was found that the enhanced charge separation was achieved by the increased electron density combined with a decrease in the surface potential which improves the hole transport from the bulk to the (110) surface. Additionally, oxygen vacancies expose Fe atoms on the surface leading to an increase of the population of active sites.
 | ||
| Fig. 5 Calculated overpotentials as a function of the concentration of oxygen vacancies. This figure has been reproduced from ref. 90 with permission from ACS, copyright 2016. | ||
A recent study has reported the effect of the location of oxygen vacancies on the final behavior of the Fe2O3 photoanode.94 Bulk oxygen vacancies were reported to have a negative impact on the performance of the hematite photoanodes for water oxidation. However, the surface oxygen vacancies were able to narrow the space charge region width, boosting the charge separation and charge transport of the photoanodes (Fig. 6a and b). Additionally, the water adsorption and hydrophilicity of α-Fe2O3 were modified by the surface oxygen vacancies, improving the kinetics of water oxidation.
 | ||
| Fig. 6 (a) Charge separation (ηsep) and (b) charge injection (ηcat) efficiencies of pristine and oxygen vacancy-rich α-Fe2O3 (OV) photoanodes. This figure has been reproduced from ref. 94 with permission from ACS, copyright 2020. | ||
Titanium dioxide, TiO2
Since the pioneering study of Fujishima and Honda in 1972,95 many researchers focused their studies on TiO2 materials for photocatalytic applications due to its earth abundance, low cost, chemical and thermal stability, high refractive index, non-toxicity, and high photocatalytic activity under adequate radiation.96–98 Specifically, TiO2 is an n-type semiconductor with three different polymorphs: rutile (Eg = 3.0 eV), anatase (Eg = 3.2 eV), and brookite (Eg = 1.9 eV), rutile being thermodynamically stable whereas the other two phases are metastable.99,100 Rutile and anatase have been the most studied phases, anatase (Fig. 7) being the most efficient one for photocatalytic applications due to the lower charge carrier recombination and higher surface adsorption for hydroxyl groups.101 Despite all the positive properties mentioned above, the large bandgap of anatase and rutile TiO2 limits their optical absorption to ultraviolet (UV) light, leading to the efficient utilization of only a small part of the solar spectrum (∼5%), giving a theoretical maximum photocurrent of 1.1 mA cm−2 and a STH efficiency of 1.3% for anatase and 1.8 mA cm−2 and 2.2% STH for rutile.49 Moreover, due to its wide bandgap, TiO2 also suffers from rapid electron–hole recombination, which decreases its overall conversion efficiency.102,103 Different groups tried to overcome the aforementioned limitations to enhance the photocatalytic performance by extending the absorption edge of the solar spectrum to the visible region. Some strategies have been proposed to enhance the light harvesting efficiency based on bandgap engineering of TiO2 by dye104 or quantum dot sensitization,105 doping with N, C, and S atoms7,106–108 or heterostructuring.109–111 | ||
| Fig. 7 Crystalline structure of the tetragonal anatase TiO2, belonging to the spatial group I41/amd, obtained with VESTA software. | ||
However, the most efficient and widely employed strategy for enhancing the photocatalytic activity of TiO2 photoanodes is by tuning the density of OVs and Ti3+ interstitials, which are the dominant intrinsic defects in this material.112–115 Several studies have demonstrated that OVs and Ti3+ play a critical role in determining the surface and electric properties of TiO2 since they are capable of creating localized mid gap states. Ti3+ surface defects are generated by the reduction of Ti4+ ions due to the loss of oxygen atoms in a reductive treatment or due to electron trapping under UV radiation.24,112,116 The presence of OVs in TiO2 could increase the donor density and effectively promote the separation and transport of photogenerated electron–hole pairs.117,118 OVs are easily induced by the reduction of TiO2 (TiO2−x). Many synthetic routes have been reported to reduce TiO2, such as metal reduction,119 ionothermal processes,120 NaBH4 reduction,121 vacuum annealing122,123 and hydrogenation.124
The synthesis of OV-rich TiO2 resulted in a black material with enhanced solar light harvesting and photocatalytic performance. Although in 2001 Lu et al.125 firstly reported an opaque black color of TiO2 after a reducing treatment due to some induced defects, the term “Black TiO2” did not appear until 2011, when Chen et al.126 synthesized black TiO2 by introducing surface disorder into the TiO2 nanocrystals through a hydrogenation process. The term “Black” was employed due to the visual aspect of this material, in contrast to the usual whitish color of pristine TiO2. Such a significant enhancement in the photocatalytic performance of black TiO2 photoanodes is due to the presence of OVs and/or Ti3+ defects. The electron transition from the valence band (VB) to the OVs mid-levels and/or from the OVs to the VB results in visible and infrared light absorption. This TiO2 material with a high concentration of structural defects showed a higher performance towards water oxidation under full illumination (all over the solar electromagnetic spectrum), as shown in Fig. 8a.119 However, photogenerated holes from the energy levels of OVs cannot contribute to PEC performance because they are located above the H2O/O2 oxidation potential, and consequently did not contribute to enhance the PEC performance of black TiO2 under visible light illumination (Fig. 8b), although these oxygen defective states can oxidize other scavengers127 and reduce recombination.128 The enhanced PEC activity of black TiO2 under full solar light could be mainly ascribed to the enhanced charge transport and charge separation together with visible light photoresponse.129
 | ||
| Fig. 8 Current density for pristine TiO2 nanotubes (TNTs) and black TiO2 nanotubes (B-TNTs) in the dark and under solar illumination of the (a) full spectrum or only (b) visible light. This figure has been reproduced from ref. 119 with permission from RSC, copyright 2014. | ||
Some studies reveal an anodic shift of the VB position of TiO2 due to the presence of OVs119,130 that could be followed by a band tail121 resulting in a narrowed band gap which extends its optical absorption to the visible region, as shown in Fig. 9a. In a recent study, vacuum annealing and Zr doping were explored to improve the performance of nanostructured TiO2 photoanodes.123 Both doping and vacuum treatment led to a slightly stressed surface and consequently to a catalytically more active TiO2 surface, concomitant with the creation of oxygen vacancies, which enhanced carrier transport. Vacuum annealing induced an anodic band shift, thermodynamically beneficial for the accumulation of photogenerated holes needed for water oxidation. Moreover, both Zr doping and vacuum annealing induced optical scattering, enhancing the optical pathway of incoming light and slightly increasing the light harvesting efficiency of the TiO2 nanostructured films. However, this topic is controversial among the scientific community since other studies reported no displacement in the VB location,129,131 as shown in Fig. 9b. Maybe these differences could be due to the employed synthetic route, since it has been claimed that hydrogen treatment has a negligible effect on the valence band position at the TiO2 surface.118
 | ||
| Fig. 9 Energy diagrams for pristine and black TiO2 (a) with and (b) without VB shift. These figures have been reproduced from ref. 121 and 129 with permission from RSC, copyright 2013 and ACS, copyright 2017, respectively. | ||
Other studies have been focused on modifying the synthetic conditions of black TiO2 to find an optimum material and simultaneously gaining mechanistic insights into the behavior of defects. Hou et al.131 calcined a TiO2 sample at different temperatures in air and a vacuum to generate different OV concentrations and studied its photocatalytic performance. The experimental results indicated that samples calcined in air had only bulk OVs while those calcined in a vacuum possessed bulk and surface OVs. The bulk OVs mainly act as recombination centers for the photogenerated charges under total illumination, but extended the light absorption under visible light irradiation. However, surface OVs improve the visible light response and capture photogenerated electrons, inhibiting recombination. Then, the visible light photocatalytic performance was noticeable improved under the combined influence of the bulk and surface oxygen vacancies. Park et al.132 extended the optical absorption to the visible range (300–420 nm) of black TiO2 photoanodes by introducing a “crystal-deficient” TiO2 overlayer. In addition, this “crystal-deficient” overlayer induced a 50-fold enhancement of the electrical conductivity, which noticeably increased the electron diffusion length to ∼20 μm, ensuring electron collection, since the employed TiO2 nanostructures were around 2 μm long, leading to enhanced PEC activity.
Tungsten oxide, WO3
WO3 is a widely studied metal oxide semiconductor for PEC water splitting. Among the different crystalline polymorphs, the monoclinic γ-WO3, belonging to the P21/c space group, is the most common phase (Fig. 10).133 WO3 is an n-type semiconductor with a bandgap of 2.6 eV and a thermodynamically favorable valence band (VB) position for water oxidation.134 This semiconducting oxide has a theoretical maximum photocurrent of 3.9 mA cm−2, which corresponds to a STH efficiency of 4.8%.135 Additionally, WO3 also shows a good photocurrent onset for water oxidation, particularly in an acidic environment.136 However, WO3 presents a limited performance towards the OER due to a high electron–hole recombination.137 Analogous approaches, applied to other water splitting photoanodes to improve their performance, such as extrinsic doping138 and the use of hetero-architectures139,140 have also been reported for WO3. | ||
| Fig. 10 Monoclinic structure of γ-WO3 obtained with VESTA software. | ||
Oxygen vacancies are intrinsic dopants which have a highly relevant role in WO3 photoanodes by introducing donor levels inside the bandgap and being responsible for the n-doping.141 The formation of OVs in WO3 (in chemical equilibrium) can be expressed as:
 | (2) |
 represents the neutral oxygen atom and
represents the neutral oxygen atom and  represents the neutral oxygen vacancy with two trapped electrons, introducing a donor level in the gap. Upon increasing the temperature, the donor levels are successively ionized, with an activation energy Ea, generating free electrons, e, in the conduction band. This process can be expressed as:
represents the neutral oxygen vacancy with two trapped electrons, introducing a donor level in the gap. Upon increasing the temperature, the donor levels are successively ionized, with an activation energy Ea, generating free electrons, e, in the conduction band. This process can be expressed as: | (3) |
 | (4) |
The electrical conductivity of WO3 has been reported to be a consequence of the carrier generation originating from the formation of oxygen vacancies at the surface.141 This result was also confirmed by theoretical studies.142 In this last work, the authors also reported that the localized character of the polaron depended on the concentration of oxygen vacancies in WO3 photoanodes. The polaron state was a highly localized state for concentrations of OVs ∼0.8%, whereas it has a 2D character for concentrations of ∼0.4% or smaller. In the case of concentrations of ∼0.1%, the extension of the polaron was calculated to be approximately 1 nm, in agreement with experimental results. This 2D polaron induces a delocalized state at the surface enhancing surface reactivity towards water oxidation intermediates and thus, improving the performance. In good agreement with the latter study, another theoretical study also reported that the excess of charge generated by OVs was partially delocalized at the surface.143 Additionally, the authors analyzed the interaction between H2O and WO3 on pristine and defective surfaces and reported that OVs did not strongly influence the H2O adsorption, the process being, in both cases, non-dissociative.
Additionally, the density of oxygen vacancies present in the WO3 lattice can be tuned to improve its performance. By combining thermal treatments under a H2 atmosphere and the deposition of plasmonic nanoparticles (Au and Ag) on WO3 photoanodes, Mathur et al.144 were able to increase the population of OVs, leading to a higher optical absorption (the samples change from transparent to a greyish color), higher stability in acidic environments and an increase in the carrier density and hence, improving the PEC performance. Lately, S.-P. Chai et al.137 tuned the OV concentration by adjusting the concentration of HCl employed during the synthesis. Moreover, it was also observed that a higher amount of this acid leads to a more completed formation of pristine WO3, while a lower amount caused bulk OVs to form instead. The surface oxygen vacancies narrow the bandgap, as observed in other oxide semiconductors,145 and downshift both the VB and CB, leading to a better utilization of the solar spectrum and thermodynamically more favorable energetics towards water oxidation, decreasing the overpotential associated with this reaction. Besides, OVs induced sub-band states which improve the charge separation, as schemed in Fig. 11.
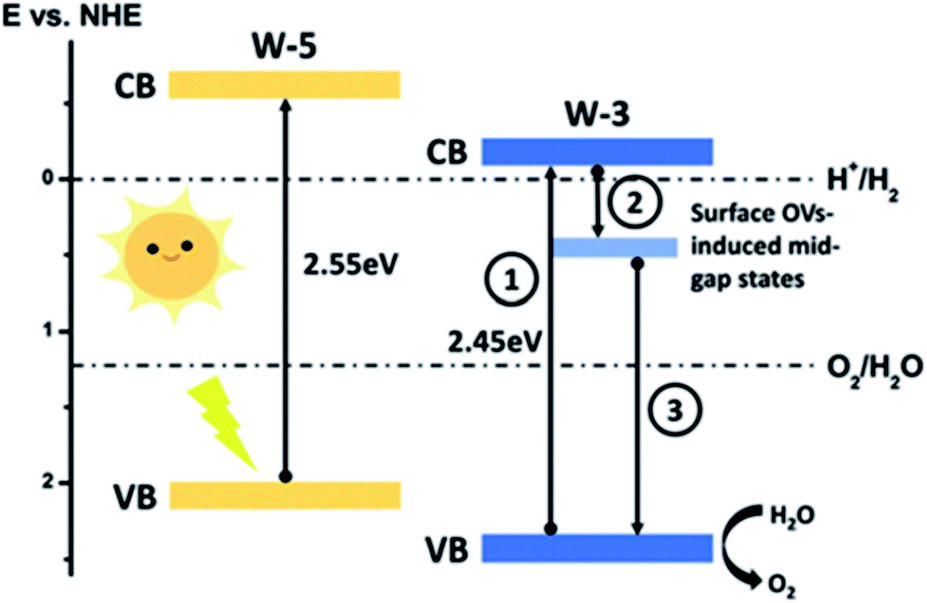 | ||
| Fig. 11 Energy band diagram of pristine (W-5) and oxygen-vacancies rich (W-3) WO3. This figure has been reproduced from ref. 137 with permission from RSC, copyright 2019. | ||
However, other groups reported experimental methods to suppress the excessive formation of OVs at the surface, which could enhance the recombination kinetics and thus, reduce the PEC performance.146 Specifically, an ozone treatment was employed to reoxidize the W5+ ions on the surface, reducing the concentration of excess OVs and shifting the overpotential cathodically by around 150 mV. There are several studies where the population of oxygen vacancies is optimized to improve the final PEC performance of WO3 photoanodes.147,148 In a very recent study,149 the performance of WO3 photoanodes was analyzed as a function of the concentration of bulk OVs from a kinetic perspective, since apart from all the thermodynamic effects detailed above, such as narrowing the bandgap, introducing defect levels and shifting the VB maximum, oxygen vacancies in WO3 photoanodes also induce kinetic effects. In this work, four different concentrations of OVs were studied, “very high” (∼5.8% oxygen atoms), “high” (∼2.3% oxygen atoms), “medium” (∼2% oxygen atoms) and “low” (∼0.5% oxygen atoms), using transient absorption spectroscopy (TAS) to analyze their impact on the charge carrier recombination kinetics over different time scales, from picoseconds to seconds. The sample with an excess concentration of OVs showed the lowest photocurrent due to high hole trapping kinetics taking place faster than the instrument detection. The sample with a “high” concentration of OVs has a higher bulk trap recombination rate (from hole trapping) limiting the concomitant carrier collection. In the sample with a low concentration of OVs, charge separation is limited, since there are too few defect states, increasing the bimolecular recombination. However, the sample with the balanced concentration of OVs delivered the highest photocurrent due to a beneficial equilibrium between charge separation and trap-mediated recombination. The performance of this four different samples is summarized in Fig. 12.
 | ||
| Fig. 12 The role of oxygen vacancies in charge separation and electron transport in WO3, from femtoseconds to seconds. This figure has been reproduced from ref. 149 with permission from RSC, copyright 2020. | ||
Another kinetically focused study150 revealed that WO3 containing oxygen vacancies is more prone to follow the four-hole pathway, instead of the more likely two-hole pathway followed by pristine WO3.
This result explained the better performance of WO3 containing OVs in terms of faster kinetics, higher stability and higher faradaic efficiency towards oxygen compared to stoichiometric WO3. The four-hole pathway is schematized in Fig. 13 for both pristine WO3 and OV-rich WO3 photoanodes, where the route for the latter is energetically more favorable. In contrast, the oxidation process from H2O to H2O2, a two-hole pathway, was found to be energetically more favorable in pristine WO3.
 | ||
| Fig. 13 Potential energy profiles for water dehydrogenation to O2. This figure has been reproduced from ref. 150 with permission from ACS, copyright 2019. | ||
Summary and outlook
The present topical review is focused on the role of oxygen vacancies in four highlighted water splitting photoanode materials, BiVO4, Fe2O3, TiO2 and WO3. Oxygen vacancies are spontaneously present, as intrinsic dopants, in all these materials inducing their n-type behavior and introducing defect levels at different depths inside the bandgap. The particular location of these oxygen-defective states within the bandgap of the semiconductor leads to different consequences: specifically, surface oxygen vacancies are beneficial for the final performance of water splitting photoanodes, since they improve the charge separation by narrowing the space charge layer. However, in general, bulk oxygen vacancies are detrimental since they enhance recombination dynamics and thus, activate loss channels, with a concomitant decrease of the photocurrent. The concentration of these defects also has a significant effect on the final performance, and the optimization of defect density is crucial for the development of competitive photoanodes for PEC water splitting. But then, a key question arises: are oxygen vacancies beneficial or detrimental to the final PEC performance of water splitting photoanodes? Most likely, the answer to this question is “depending on the concentration and its location”. Since the presence of these defects, often intrinsic, as well as their concentration and their location within the bandgap, induces severe modifications, the smart tuning of these parameters can lead to significantly enhanced water splitting photoanodes and other solar-related applications. As it has been acutely detailed herein, the population of OVs can be easily tuned by several techniques, leading to a dramatically modified performance towards the OER. Consequently, we believe that the modulation of the population of OVs, and other structural defects, will be greatly exploited in the race for improving solar water splitting photoanodes in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the EU FET programme (A-LEAF 732840) and from the Ministerio de Ciencia, Innovación y Universidades of Spain (project ENE2017-85087-C3-1-R).References
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS.
- D. Huang, S. Chen, G. Zeng, X. Gong, C. Zhou, M. Cheng, W. Xue, X. Yan and J. Li, Coord. Chem. Rev., 2019, 385, 44–80 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 1245026 Search PubMed.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928–1933 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS.
- I. Cesar, A. Kay, J. A. Gonzalez Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS.
- S. U. Khan, M. Al-Shahry and W. B. Ingler, science, 2002, 297, 2243–2245 CrossRef CAS.
- M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- F. Wang, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2012, 116, 8901–8909 CrossRef CAS.
- V. Cristino, S. Caramori, R. Argazzi, L. Meda, G. L. Marra and C. A. Bignozzi, Langmuir, 2011, 27, 7276–7284 CrossRef CAS.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 RSC.
- S.-T. Bae, H. Shin, J. Y. Kim, H. S. Jung and K. S. Hong, J. Phys. Chem. C, 2008, 112, 9937–9942 CrossRef CAS.
- T. W. Hamann, Dalton Trans., 2012, 41, 7830–7834 RSC.
- S. Wang, P. Chen, J. H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS.
- Y. Zhang, Y. Guo, H. Duan, H. Li, C. Sun and H. Liu, Phys. Chem. Chem. Phys., 2014, 16, 24519–24526 RSC.
- C. Künneth, R. Batra, G. A. Rossetti Jr, R. Ramprasad and A. Kersch, in Ferroelectricity in Doped Hafnium Oxide: Materials, Properties and Devices, Elsevier, 2019, pp. 245–289 Search PubMed.
- S. Švarcová, K. Wiik, J. Tolchard, H. J. Bouwmeester and T. Grande, Solid State Ionics, 2008, 178, 1787–1791 CrossRef.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 026103 CrossRef.
- B. M. Rajbongshi, A. Ramchiary and S. Samdarshi, Mater. Lett., 2014, 134, 111–114 CrossRef CAS.
- L. Liu, Z. Mei, A. Tang, A. Azarov, A. Kuznetsov, Q.-K. Xue and X. Du, Phys. Rev. B, 2016, 93, 235305 CrossRef.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- T.-Y. Yang, H.-Y. Kang, U. Sim, Y.-J. Lee, J.-H. Lee, B. Koo, K. T. Nam and Y.-C. Joo, Phys. Chem. Chem. Phys., 2013, 15, 2117–2124 RSC.
- M. García-Tecedor, D. Maestre, A. Cremades and J. Piqueras, J. Phys. D: Appl. Phys., 2017, 50, 415104 CrossRef.
- S. Carrettin, Y. Hao, V. Aguilar-Guerrero, B. C. Gates, S. Trasobares, J. J. Calvino and A. Corma, Chem. - Eur. J., 2007, 13, 7771–7779 CrossRef CAS.
- G. Pacchioni, ChemPhysChem, 2003, 4, 1041–1047 CrossRef CAS.
- A. Sarkar and G. G. Khan, Nanoscale, 2019, 11, 3414–3444 RSC.
- K. Yoshida, T. Kawai, T. Nambara, S. Tanemura, K. Saitoh and N. Tanaka, Nanotechnology, 2006, 17, 3944 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- M. García-Tecedor, J. Bartolomé, D. Maestre, A. Trampert and A. Cremades, Nano Res., 2019, 12, 441–448 CrossRef.
- M. W. Gaultois and A. P. Grosvenor, J. Mater. Chem., 2011, 21, 1829–1836 RSC.
- Q. Wu, Q. Zheng and R. van de Krol, J. Phys. Chem. C, 2012, 116, 7219–7226 CrossRef CAS.
- D. Li, H. Haneda, N. K. Labhsetwar, S. Hishita and N. Ohashi, Chem. Phys. Lett., 2005, 401, 579–584 CrossRef CAS.
- J.-M. Wu, Y. Chen, L. Pan, P. Wang, Y. Cui, D. Kong, L. Wang, X. Zhang and J.-J. Zou, Appl. Catal., B, 2018, 221, 187–195 CrossRef CAS.
- B. Choudhury and A. Choudhury, J. Lumin., 2012, 132, 178–184 CrossRef CAS.
- T. W. Kim, Y. Ping, G. A. Galli and K.-S. Choi, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- Q. Liu, D. Ding, C. Ning and X. Wang, Int. J. Hydrogen Energy, 2015, 40, 2107–2114 CrossRef.
- W. Li, J. Li, X. Wang and Q. Chen, Appl. Surf. Sci., 2012, 263, 157–162 CrossRef CAS.
- G. Wang, Y. Ling, X. Lu, F. Qian, Y. Tong, J. Z. Zhang, V. Lordi, C. Rocha Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957–10964 CrossRef CAS.
- R. Van de Krol and M. Grätzel, Photoelectrochemical Hydrogen Production, Springer, 2012 Search PubMed.
- D.-D. Qin, T. Wang, Y.-M. Song and C.-L. Tao, Dalton Trans., 2014, 43, 7691–7694 RSC.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- M. Sachs, J.-S. Park, E. Pastor, A. Kafizas, A. A. Wilson, L. Francàs, S. Gul, M. Ling, C. Blackman and J. Yano, Chem. Sci., 2019, 10, 5667–5677 RSC.
- Y. K. Kho, W. Y. Teoh, A. Iwase, L. Mädler, A. Kudo and R. Amal, ACS Appl. Mater. Interfaces, 2011, 3, 1997–2004 CrossRef CAS.
- L. Zhou, W. Wang, L. Zhang, H. Xu and W. Zhu, J. Phys. Chem. C, 2007, 111, 13659–13664 CrossRef CAS.
- S. Giménez and J. Bisquert, Photoelectrochemical Solar Fuel Production, Springer, 2016 Search PubMed.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- S. P. Berglund, D. W. Flaherty, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Phys. Chem. C, 2011, 115, 3794–3802 CrossRef CAS.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- S. K. Pilli, T. E. Furtak, L. D. Brown, T. G. Deutsch, J. A. Turner and A. M. Herring, Energy Environ. Sci., 2011, 4, 5028–5034 RSC.
- W. Luo, Z. Yang, Z. Li, J. Zhang, J. Liu, Z. Zhao, Z. Wang, S. Yan, T. Yu and Z. Zou, Energy Environ. Sci., 2011, 4, 4046–4051 RSC.
- S. Selim, L. Francas, M. García-Tecedor, S. Corby, C. Blackman, S. Gimenez, J. R. Durrant and A. Kafizas, Chem. Sci., 2019, 10, 2643–2652 RSC.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- D. Cardenas-Morcoso, R. Ifraemov, M. García-Tecedor, I. Liberman, S. Gimenez and I. Hod, J. Mater. Chem. A, 2019, 7, 11143–11149 RSC.
- D. Kong, J. Qi, D. Liu, X. Zhang, L. Pan and J. Zou, Trans. Tianjin Univ., 2019, 25, 340–347 CrossRef CAS.
- Q. Pan, K. Yang, G. Wang, D. Li, J. Sun, B. Yang, Z. Zou, W. Hu, K. Wen and H. Yang, Chem. Eng. J., 2019, 372, 399–407 CrossRef CAS.
- S. Wang, P. Chen, Y. Bai, J. H. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- Y. Chen, M. Yang, J. Du, G. Ke, X. Zhong, Y. Zhou, F. Dong, L. Bian and H. He, J. Mater. Sci., 2019, 54, 671–682 CrossRef CAS.
- W. Qiu, S. Xiao, J. Ke, Z. Wang, S. Tang, K. Zhang, W. Qian, Y. Huang, D. Huang and Y. Tong, Angew. Chem., 2019, 131, 19263–19271 CrossRef.
- N. Daelman, F. S. Hegner, M. Rellán-Piñeiro, M. Capdevila-Cortada, R. García-Muelas and N. López, J. Chem. Phys., 2020, 152, 050901 CrossRef CAS.
- X. Xu, Y. Xu, F. Xu, G. Jiang, J. Jian, H. Yu, E. Zhang, D. Shchukin, S. Kaskel and H. Wang, J. Mater. Chem. A, 2020, 8(4), 1636–1645 RSC.
- W. Wang, P. J. Strohbeen, D. Lee, C. Zhou, J. K. Kawasaki, K.-S. Choi, M. Liu and G. Galli, Chem. Mater., 2020, 32, 2899–2909 CrossRef CAS.
- J. K. Cooper, S. B. Scott, Y. Ling, J. Yang, S. Hao, Y. Li, F. M. Toma, M. Stutzmann, K. Lakshmi and I. D. Sharp, Chem. Mater., 2016, 28, 5761–5771 CrossRef CAS.
- X. Yang, A. J. Fernández-Carrión, J. Wang, F. Porcher, F. Fayon, M. Allix and X. Kuang, Nat. Commun., 2018, 9, 1–11 CrossRef.
- F. S. Hegner, D. Forrer, J. R. Galán-Mascarós, N. López and A. Selloni, J. Phys. Chem. Lett., 2019, 10, 6672–6678 CrossRef CAS.
- S. Selim, E. Pastor, M. García-Tecedor, M. R. Morris, L. Francas, M. Sachs, B. Moss, S. Corby, C. A. Mesa and S. Gimenez, J. Am. Chem. Soc., 2019, 141, 18791–18798 CrossRef CAS.
- S. Feng, T. Wang, B. Liu, C. Hu, L. Li, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 2044–2048 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed., 2018, 57, 2248–2252 CrossRef CAS.
- S. Zhu, L.-a. Huang, Z. He, K. Wang, J. Guo, S.-e. Pei, H. Shao and J. Wang, J. Electroanal. Chem., 2018, 827, 42–50 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef.
- Y. Lin, S. Zhou, S. W. Sheehan and D. Wang, J. Am. Chem. Soc., 2011, 133, 2398–2401 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS.
- A. Murphy, P. Barnes, L. Randeniya, I. Plumb, I. Grey, M. Horne and J. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and P. R. Trevellick, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2027–2041 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CrossRef CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS.
- D. A. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682–6702 RSC.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS.
- S. Shen, S. A. Lindley, X. Chen and J. Z. Zhang, Energy Environ. Sci., 2016, 9, 2744–2775 RSC.
- Y. Ling, G. Wang, J. Reddy, C. Wang, J. Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 4074–4079 CrossRef CAS.
- M. Zhang, W. Luo, Z. Li, T. Yu and Z. Zou, Appl. Phys. Lett., 2010, 97, 042105 CrossRef.
- A. Pu, J. Deng, M. Li, J. Gao, H. Zhang, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 2491–2497 RSC.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef CAS.
- Y. Makimizu, N. T. Nguyen, J. Tucek, H. J. Ahn, J. Yoo, M. Poornajar, I. Hwang, S. Kment and P. Schmuki, Chem. - Eur. J., 2020, 26, 2685–2692 CrossRef CAS.
- X. Zhang, P. Klaver, R. van Santen, M. Van De Sanden and A. Bieberle-Hütter, J. Phys. Chem. C, 2016, 120, 18201–18208 CrossRef CAS.
- M.-T. Nguyen, S. Piccinin, N. Seriani and R. Gebauer, ACS Catal., 2015, 5, 715–721 CrossRef CAS.
- A. Hellman, B. Iandolo, B. Wickman, H. Grönbeck and J. Baltrusaitis, Surf. Sci., 2015, 640, 45–49 CrossRef CAS.
- J. Hu, X. Zhao, W. Chen and Z. Chen, ACS Omega, 2018, 3, 14973–14980 CrossRef CAS.
- Q. Yang, J. Du, J. Li, Y. Wu, Y. Zhou, Y. Yang, D. Yang and H. He, ACS Appl. Mater. Interfaces, 2020, 12, 11625–11634 CrossRef CAS.
- A. Fujishima and K. Honda, nature, 1972, 238, 37–38 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- F. Fresno, R. Portela, S. Suárez and J. M. Coronado, J. Mater. Chem. A, 2014, 2, 2863–2884 RSC.
- A. J. Haider, R. H. AL–Anbari, G. R. Kadhim and C. T. Salame, Energy Procedia, 2017, 119, 332–345 CrossRef CAS.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- R. Zallen and M. Moret, Solid State Commun., 2006, 137, 154–157 CrossRef CAS.
- D. A. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- C. Byrne, G. Subramanian and S. C. Pillai, J. Environ. Chem. Eng., 2018, 6, 3531–3555 CrossRef CAS.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS.
- X.-F. Gao, H.-B. Li, W.-T. Sun, Q. Chen, F.-Q. Tang and L.-M. Peng, J. Phys. Chem. C, 2009, 113, 7531–7535 CrossRef CAS.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS.
- J. Yan, H. Wu, H. Chen, Y. Zhang, F. Zhang and S. F. Liu, Appl. Catal., B, 2016, 191, 130–137 CrossRef CAS.
- H. M. El-Bery, Y. Matsushita and A. Abdel-moneim, Appl. Surf. Sci., 2017, 423, 185–196 CrossRef CAS.
- C. Hao, W. Wang, R. Zhang, B. Zou and H. Shi, Sol. Energy Mater. Sol. Cells, 2018, 174, 132–139 CrossRef CAS.
- H. Liu, H. Ma, X. Li, W. Li, M. Wu and X. Bao, Chemosphere, 2003, 50, 39–46 CrossRef CAS.
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, 1996 Search PubMed.
- J. Nowotny, T. Bak, M. Nowotny and L. Sheppard, J. Phys. Chem. B, 2006, 110, 18492–18495 CrossRef CAS.
- G. Vásquez, S. Z. Karazhanov, D. Maestre, A. Cremades, J. Piqueras and S. E. Foss, Phys. Rev. B, 2016, 94, 235209 CrossRef.
- T. Berger, M. Sterrer, O. Diwald, E. Knözinger, D. Panayotov, T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2005, 109, 6061–6068 CrossRef CAS.
- D. Cronemeyer, Phys. Rev., 1959, 113, 1222 CrossRef CAS.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS.
- H. Cui, W. Zhao, C. Yang, H. Yin, T. Lin, Y. Shan, Y. Xie, H. Gu and F. Huang, J. Mater. Chem. A, 2014, 2, 8612–8616 RSC.
- G. Li, Z. Lian, X. Li, Y. Xu, W. Wang, D. Zhang, F. Tian and H. Li, J. Mater. Chem. A, 2015, 3, 3748–3756 RSC.
- Q. Kang, J. Cao, Y. Zhang, L. Liu, H. Xu and J. Ye, J. Mater. Chem. A, 2013, 1, 5766–5774 RSC.
- T. Xia, Y. Zhang, J. Murowchick and X. Chen, Catal. Today, 2014, 225, 2–9 CrossRef CAS.
- M. N. Shaddad, D. Cardenas-Morcoso, M. García-Tecedor, F. Fabregat-Santiago, J. Bisquert, A. M. Al-Mayouf and S. Gimenez, ACS Omega, 2019, 4(14), 16095–16102 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS.
- T.-C. Lu, S.-Y. Wu, L.-B. Lin and W.-C. Zheng, Phys. B, 2001, 304, 147–151 CrossRef CAS.
- X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- J. Tang, A. J. Cowan, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2011, 115, 3143–3150 CrossRef CAS.
- F. M. Pesci, G. Wang, D. R. Klug, Y. Li and A. J. Cowan, J. Phys. Chem. C, 2013, 117, 25837–25844 CrossRef CAS.
- H. Song, C. Li, Z. Lou, Z. Ye and L. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 8982–8987 CrossRef CAS.
- S. Li, J. Qiu, M. Ling, F. Peng, B. Wood and S. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 11129–11135 CrossRef CAS.
- L. Hou, M. Zhang, Z. Guan, Q. Li and J. Yang, Appl. Surf. Sci., 2018, 428, 640–647 CrossRef CAS.
- K. Zhang, S. Ravishankar, M. Ma, G. Veerappan, J. Bisquert, F. Fabregat-Santiago and J. H. Park, Adv. Energy Mater., 2017, 7, 1600923 CrossRef.
- X. Liu, F. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7894–7911 RSC.
- G. Liu, J. Han, X. Zhou, L. Huang, F. Zhang, X. Wang, C. Ding, X. Zheng, H. Han and C. Li, J. Catal., 2013, 307, 148–152 CrossRef CAS.
- S. Sfaelou, L.-C. Pop, O. Monfort, V. Dracopoulos and P. Lianos, Int. J. Hydrogen Energy, 2016, 41, 5902–5907 CrossRef CAS.
- Q. Mi, A. Zhanaidarova, B. S. Brunschwig, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 5694–5700 RSC.
- Y.-H. Chew, J.-Y. Tang, L.-J. Tan, B. W. J. Choi, L.-L. Tan and S.-P. Chai, Chem. Commun., 2019, 55, 6265–6268 RSC.
- S. S. Kalanur, J.-Y. Hwang and H. Seo, J. Catal., 2017, 350, 197–202 CrossRef CAS.
- Y. Bu, Z. Chen and C. Sun, Appl. Catal., B, 2015, 179, 363–371 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, Appl. Catal., B, 2012, 111, 288–296 CrossRef.
- M. Gillet, C. Lemire, E. Gillet and K. Aguir, Surf. Sci., 2003, 532, 519–525 CrossRef.
- M. Gerosa, F. Gygi, M. Govoni and G. Galli, Nat. Mater., 2018, 17, 1122–1127 CrossRef CAS.
- E. Albanese, C. Di Valentin and G. Pacchioni, ACS Appl. Mater. Interfaces, 2017, 9, 23212–23221 CrossRef CAS.
- T. Singh, R. Müller, J. Singh and S. Mathur, Appl. Surf. Sci., 2015, 347, 448–453 CrossRef CAS.
- S. A. Ansari, M. M. Khan, S. Kalathil, A. Nisar, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 9238–9246 RSC.
- J. Zhang, X. Chang, C. Li, A. Li, S. Liu, T. Wang and J. Gong, J. Mater. Chem. A, 2018, 6, 3350–3354 RSC.
- T. Soltani, A. Tayyebi, H. Hong, M. H. Mirfasih and B.-K. Lee, Sol. Energy Mater. Sol. Cells, 2019, 191, 39–49 CrossRef CAS.
- J. Cen, Q. Wu, D. Yan, W. Zhang, Y. Zhao, X. Tong, M. Liu and A. Orlov, RSC Adv., 2019, 9, 899–905 RSC.
- S. Corby, L. Francàs, A. Kafizas and J. R. Durrant, Chem. Sci., 2020, 11, 2907–2914 RSC.
- M. Yang, H. He, J. Du, H. Peng, G. Ke and Y. Zhou, J. Phys. Chem. Lett., 2019, 10, 6159–6165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |