
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild thermolytic solvolysis of technical lignins in polar organic solvents to a crude lignin oil
Panos D.
Kouris
a,
Dannie J. G. P.
van Osch
a,
Geert J. W.
Cremers
a,
Michael D.
Boot
ab and
Emiel J. M.
Hensen
 *a
*a
aLaboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bEnergy Technology, Department of Mechanical Engineering, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 19th October 2020
Abstract
A mild thermal solvolysis process using alcohols for the valorization of technical Protobind soda lignin into crude lignin oil (CLO) is presented. The solubilization process results in lower molecular weight lignin fragments (1250–1550 g mol−1cf. 2500 g mol−1 of parent lignin), while rejecting heavy compounds and other solid impurities. The influence of the reaction temperature (100–350 °C), residence time (0.5–4 h), lignin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :solvent ratio (1:15–1:2 w/v) and alcohol solvent (methanol, ethanol, 1-propanol, 1-butanol, and 1-octanol) on the amount and type of products is investigated. At a high lignin loading (ratio < 1:5 w/v) and under optimum conditions for lignin solubilization (T = 200 °C, t = 0.5 h), the condensation reactions and solvent consumption are minimized. Methanol exhibits the highest solvolytic efficacy resulting in an overall lignin solubilization degree of 61 wt%, which includes some heavier lignin fractions originating from condensation reactions. The other alcohols resulted in a lignin solubilization degree of 57 wt% for ethanol, 53 wt% for 1-propanol, 51 wt% for 1-butanol and 38 wt% for 1-octanol. The solvent losses based on GC-MS analysis of the obtained CLOs were 1.1 wt% for methanol, 1.4 wt% for ethanol and 2.2 wt% for 1-butanol. Hansen solubility parameters are employed to discuss the effect of solvent on the solubilization process. Gel permeation chromatography and heteronuclear single quantum coherence NMR of solubilized fractions revealed cleavage of β-O-4 bonds during thermal solvolysis, explaining the molecular weight reduction. Methanol is the most favourable solvent and is utilized in solubilization of 5 different biorefinery lignins. In all cases, this led to CLO with a lower molecular weight of the lignin fragments, a lower polydispersity and an increased hydroxyl group content.
:solvent ratio (1:15–1:2 w/v) and alcohol solvent (methanol, ethanol, 1-propanol, 1-butanol, and 1-octanol) on the amount and type of products is investigated. At a high lignin loading (ratio < 1:5 w/v) and under optimum conditions for lignin solubilization (T = 200 °C, t = 0.5 h), the condensation reactions and solvent consumption are minimized. Methanol exhibits the highest solvolytic efficacy resulting in an overall lignin solubilization degree of 61 wt%, which includes some heavier lignin fractions originating from condensation reactions. The other alcohols resulted in a lignin solubilization degree of 57 wt% for ethanol, 53 wt% for 1-propanol, 51 wt% for 1-butanol and 38 wt% for 1-octanol. The solvent losses based on GC-MS analysis of the obtained CLOs were 1.1 wt% for methanol, 1.4 wt% for ethanol and 2.2 wt% for 1-butanol. Hansen solubility parameters are employed to discuss the effect of solvent on the solubilization process. Gel permeation chromatography and heteronuclear single quantum coherence NMR of solubilized fractions revealed cleavage of β-O-4 bonds during thermal solvolysis, explaining the molecular weight reduction. Methanol is the most favourable solvent and is utilized in solubilization of 5 different biorefinery lignins. In all cases, this led to CLO with a lower molecular weight of the lignin fragments, a lower polydispersity and an increased hydroxyl group content.
Introduction
The transition to a sustainable economy is one of the great challenges of the 21st century. The primary source of fuels and chemicals is currently based on crude oil, while natural gas and coal also contribute significantly to cover the global energy demand.1 Concerns about climate change call for the replacement of fossil carbon by renewable alternatives. Lignocellulosic biomass is the largest resource of renewable carbon, stored in a variety of biopolymers formed by photosynthesis. Unfortunately, upgrading second-generation lignocellulosic biomass into fuels and chemicals is still not competitive with conventional fossil alternatives. Lignocellulosic biomass is predominantly made up of cellulose, hemicellulose and lignin. The first steps in lignocellulosic biomass valorization date back nearly two millennia to ancient China around 105 A.D., when the art of paper manufacturing was first recorded.2 In papermaking, and more recently also in cellulosic ethanol production, the lignocellulosic matrix is separated into (hemi)cellulose and lignin. The former compound is valorized into valuable products such as paper and bio-ethanol, while the latter is typically burned on-site for energy. This concept of sacrificing lignin in favor of extracting value from cellulose has thus been the norm since antiquity. Today, over 98% of all lignin produced is burnt to serve plant energy needs. To improve overall profitability of lignocellulose conversion, it is therefore important to valorize lignin, which may constitute up to 30 wt% of the total mass, into valuable products as well. The absence of industrial processes that add value to lignin can be largely attributed to its chemical recalcitrance and structural complexity.Lignin is part of the cell walls found in almost all terrestrial biomass and is the second most abundant natural polymer in the world after cellulose. The total amount of lignin present in the biosphere exceeds 300 billion tonnes and increases annually by around 20 billion tonnes.3 Lignin is an amorphous three-dimensional polymer network consisting of methoxylated phenylpropane structures, cross-linked by C–O–C (β-O-4′, α-O-4′, and 4-O-5′) and C–C (β-1′ and β-β, 5–5′) bonds.4 It confers strength and rigidity to plants and protects the cellulose and hemicellulose from microbial attack.5 Therefore, it is broadly recognized that depolymerizing lignin into useful compounds for various applications, ranging from polyurethane foams and epoxy resins to additives for concrete or rubber, is a viable strategy.6–8
Considering its structure, lignin is also the largest renewable source of aromatic building blocks in nature and has significant potential to serve as starting material for the production of bulk aromatic compounds and offer suitable alternatives to the large volumes of BTX (benzene, toluene, and xylene) and phenol derived from petroleum oil.9 However, depolymerization technologies are required to obtain these products. The past decade has witnessed strong growth in scientific research in this direction. Lignin depolymerization is a challenging task because of the already mentioned structural complexity and recalcitrance of lignin, which is derived from the random C–C and C–O interlinkages between the primary constituents.9
A wide variety of chemical treatment methods aimed at breaking down lignin into fragments has been explored,4,5,10–12 and can be categorized into thermochemical, hydrolytic, reductive and oxidative approaches. Catalytic reductive depolymerization is a promising method for obtaining fuel additives and aromatic chemicals, because radical coupling reactions of the intermediate fragments can be avoided in the presence of hydrogen. However, such processes often require solvents, metal catalysts and hydrogen for efficient deoxygenation. Solvents including methanol,13–15 ethanol,15–19 ethanol/water,20,21 methanol/water,9 propanol, or dioxane15,22 have been investigated for tandem solvolysis and hydrogenolysis reactions. The latter reactions can be catalyzed by precious group metals (such as Pt, Pd, Ru, or Rh) and the more abundant base metals (such as Cu or Ni). Importantly, for effective bond cleavage and high yields of aromatic compounds, relatively severe conditions are typically applied.
In earlier work,19 we investigated important capital expenditure (CAPEX) and operational expenditure (OPEX) indicators for commercialization of such catalytic depolymerization technologies using the supercritical ethanolysis process catalyzed by a Cu–Mg–Al mixed oxide catalyst as a case study.17,18,23 It was concluded that a trade-off exists between depolymerization efficacy and overall product cost. The lignin content of the solvolytic step was found to be the most critical parameter. Although the use of more solvents led to higher monomer yield owing to less catalyst fouling, the cost price became progressively higher. On the CAPEX side, the reason is that reactor cost scales with the mass rates. Regarding OPEX, relative solvent losses and energy consumption increase with increasing lignin dilution. This intrinsic trade-off led to the suggestion to separate lignin depolymerization process into two distinct steps: (i) thermal solvolysis into lignin fragments and (ii) heterogeneous catalytic upgrading of these intermediates into desired products. The present work explores in detail the first step.
Supercritical alcohols have attracted attention as a useful medium for the solvolysis of lignin to biofuels and value-added chemicals because of their unique physicochemical properties. Fluids approaching supercritical points have solvent powers comparable to those of liquids and they are much more compressible than dilute gases.24 Nielsen et al. have reported a non-catalytic solvolysis process of biorefinery lignin in supercritical ethanol that can produce a heptane-soluble bio-oil without the need to add a catalyst or a reducing agent such as hydrogen.25 Due to the high price of alcohols, solvent consumption severely impacts the commercial viability of this process. The same group later demonstrated that solvent decomposition occurs in three primary ways: (i) decomposition of the alcohol to gases through decarbonylation, (ii) the formation of light condensation products through condensation and dehydration reactions, and (iii) formation of ethers or esters through alcohol condensation with carboxyl and hydroxyl groups present in lignin.26 Solvent consumption, together with char fouling, can be controlled to some extent by lowering the reaction temperature and reaction time. Choi et al. investigated the effect of various reaction parameters on solvolysis of lignin in both sub- and supercritical ethanol to low molecular weight phenols.12 Similar to the other studies, this work explored high solvent dilution of the feedstock and required severe supercritical reaction conditions to partially deoxygenate and crack the lignin feed. The first common characteristic of these studies is the application of high solvent dilution to suppress char formation. The problem of char formation at high lignin loadings and temperatures in the range of 250–450 °C was reported extensively by Nielsen et al.25,26 A second similarity relates to the primary objective, which is in all cases to (partially) deoxygenate and crack the lignin, thereby necessarily requiring severe (i.e., supercritical) process conditions.
The heterogeneous and solid nature of lignin renders its valorization very challenging. We consider that for the purpose of downstream processing it would be beneficial to present lignin in a form that can be easily processed and from which the unconvertible parts have already been removed. Based on the above, a thermal solvolysis step will be explored which yields a solubilized from of lignin in a solvent. This mixture is called crude lignin oil (CLO). An advantage of this approach is the compatibility with the crude oil value chain, which deals primarily with (viscous) liquids at a very large scale. A process that can convert lignin locally into crude lignin oil at a smaller scale can obviate scale mismatches with respect to the downstream processing, which will include deoxygenation and cracking steps utilizing most likely heterogeneous catalysts. This work is motivated by earlier investigations on lignin solvolysis in alcohol solvents and explores lower process severity (reaction time, temperature), targeting oligomeric and solvent-soluble forms of lignin, whilst maximizing lignin loadings together with final product yields.
Materials and methods
Chemicals and materials
Isolated lignin L1 was obtained from a commercial-scale lignin recovery plant in Canada using lignin from a pulp mill processing forestall wood. Technical lignin L2 was purchased from a paper and pulp company in the south of the Netherlands processing miscanthus. Isolated lignin L3 was obtained from a process that extracts (hemi)cellulose and lignin using acid pretreatment on hardwood. Protobind 1000 alkali lignin L4 was purchased from GreenValue, which is obtained by soda pulping of wheat straw. Biorefinery lignin L5 was purchased from Renmatix, which uses a supercritical-water pre-treatment technology for deconstructing hardwood into useable industrial sugars. The detailed composition analysis of all 5 technical lignins was carried out according to the NREL method described in the literature,27 and is presented in Table 1. All commercial chemicals were analytical reagents and were used without further purification.| Sample | Klason lignin (wt%) | Residual carbohydrates (wt%) | Other organics (wt%) | Ash content (wt%) |
|---|---|---|---|---|
| a All data are presented as wt% of total dry matter. | ||||
| L1 | 96 | 0 | 4 | 0 |
| L2 | 70 | <2 | 15 | 15 |
| L3 | 68 | 11 | 20 | 1 |
| L4 | 94 | 4 | 0 | 2 |
| L5 | 79 | 12 | 9 | 0 |
Experimental procedures
A 100 mL stirred batch autoclave (Amar) was used for all solvolysis experiments. A series of experiments was conducted in the temperature range of 100–350 °C using reaction times between 0.5 h and 4 h. An amount of lignin-containing starting material (2.66–20 g) was suspended in 40 mL of alcohol solvent (methanol, ethanol, 1-propanol, 1-butanol, or 1-octanol). The starting lignin material was ground until a fine powder was obtained followed by sieving to remove particles larger than 100 μm. The reactor was then sealed and purged several times with nitrogen to remove oxygen. The reactor was then leak-tested at a pressure of 10 bar. The reaction mixture was heated to the desired temperature under continuous stirring at 500 rpm. Under these hydrodynamic conditions, we could exclude an effect of the stirring speed. The reaction time was considered from the moment the reactor reached the final reaction temperature. After the reaction, the heating oven was removed, and the reactor was allowed to cool to room temperature in an ice bath. The reaction mixture was collected, and the autoclave was washed with the solvent used in the experiment. The reaction mixture and the washing solution were combined and filtered over a filter crucible (porosity 4). The filter cake was washed with the same solvent several times and dried at 80 °C until constant weight. For the experiments carried out at a 1:5 w/v lignin:solvent ratio, the filter cake was washed with THF (tetrahydrofuran) to distinguish the unconverted lignin from the collected char fractions. The reactor walls and the stirrer were cleaned carefully with a spatula to recover all the fouling char fractions. The filtrate was rotary evaporated (45–70 °C, 5 mbar) for 1 h. The intermediate product fraction represents the solubilized lignin oligomers and carbohydrates of the parent technical lignin. The product fraction was dried at 80 °C until constant weight. To remove the carbohydrate fractions a liquid–liquid extraction with ethyl acetate–water was applied. The lignin oligomers will be extracted in the organic ethyl acetate layer, while the sugars remain in the aqueous phase. Therefore, a mixture of 60 mL ethyl acetate and 40 mL H2O was added into the dried product fraction (oligomers and carbohydrates). After rigorous mixing, the total mixture was left for 24 h in order to achieve phase separation. The resulting two phases were separated from each other using a separatory funnel. The ethyl acetate phase comprising the lignin fraction was subjected to rotary evaporation (45 °C, 5 mbar) and yielded the final lignin oligomeric fraction.
Analytical procedures
:1, v/v) under stirring. This was followed by the addition 100 μL of cyclohexanol (10.85 mg mL−1) as an internal standard and 100 μL of chromium(III) acetylacetonate solution (5 mg mL−1 in anhydrous pyridine and deuterated chloroform 1.6:1, v/v) as a relaxation reagent. Finally, the mixture was reacted with 100 μL of phosphorylating reagent (2-chloro-1,3,2-dioxaphospholane) for about 10 min and transferred into a 5 mm NMR tube for NMR analysis.
Hansen solubility parameter
Hansen solubility parameters have found extensive use to understand issues of solubility, diffusion and dispersion. These aspects can be characterized for solvents, polymers and particles by three parameters for dispersion, polarity and hydrogen bonding. The term solubility parameter was first introduced by Hildebrand in 1949.29 Solubility parameters are cohesion energy parameters as they derive from the energy required to convert a liquid to a gas state. All types of bonds holding molecules together are broken in the vaporization process. Thus, the energy of vaporization is a direct measure of the total cohesive energy holding the liquid molecules together.30 The Hildebrand solubility parameter δ = (E/V)1/2 depends on the molar volume of the pure solvent (V) and the energy of vaporization (E) and can be used for molecules that have no significant polar or hydrogen bonding possibilities. Hansen proposed a new theory overcoming the two aforementioned intrinsic limitations. The basis of Hansen solubility parameters (HSPs) is that the total cohesive energy (E) of a liquid consists of three major intermolecular interactions: (nonpolar) dispersion forces, (polar) permanent dipole–permanent dipole forces, and (polar) hydrogen bonding.30 The most general is the nonpolar cohesive energy (ED), derived from induced dipole forces. All molecules contain these types of attractive forces. The second type is the polar cohesion energy (EP), which results from inherent molecular interactions and is essentially found in polar molecules. The third major cohesive energy source (EH) comes from hydrogen bonds which, according to a modern definition,31 are “attractive interactions between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H and an atom or a group of atoms in the same or a different molecule in which there is evidence of bond formation”. Therefore, the basic equation governing the assignment of Hansen parameters is that E must be the sum of the individual energies that make it up, as shown in eqn (1):| E = ED + EP + EH | (1) |
Dividing this by the molar volume gives the square of the total solubility parameter as the sum of the squares of Hansen components:
| E/V = (ED/V) + (EP/V) + (EH/V) | (2) |
| δ2 = δD2 + δP2 + δH2 | (3) |
According to Hansen, any molecular substance can be represented as a point in a three-dimensional space with coordinates as the three different types of intermolecular interactions δD, δP and δH.32 Within Hansen space, a solute is represented not only by its HSPs with solvents, but also by an interaction radius (R0), thus defining a solubility sphere whose center coordinates are the HSPs (δD, δP and δH). All substances qualified to be good solvents for the solute should stay within this sphere and all considered bad (non-solvents) should lie outside. A useful parameter for comparing two substances is the solubility parameter distance (Ra), based on their respective HSP components (eqn (4)).33 It is obvious that solubility, or high affinity, requires Ra < R0, so a RED (Relative Energy Difference) number is often used to quantify distances Ra relative to the interaction radius R0, as shown in eqn (5).30
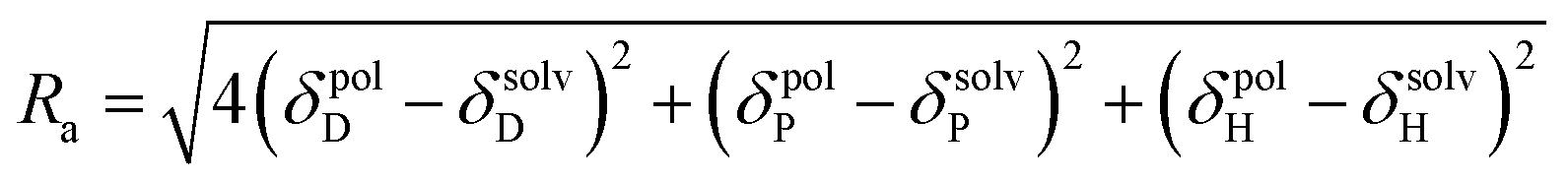 | (4) |
 | (5) |
When RED is smaller than 1, the affinity between the solvent and the polymer is high. If RED is larger than 1, the affinity between the solvent and the polymer is low and, as it grows, the affinity between the two decreases progressively. When RED is equal to 0, there is no difference between the solvent and polymer interaction energies. Therefore, the affinity between the solvent and polymer reaches its maximum. The boundary condition of polymer dissolution occurs when the value is equal or close to 1.
Polar solvents with hydrogen bonding capabilities, such as methanol or ethanol, generally show significantly high polar solubility and hydrogen bonding solubility parameters. These solvents could potentially participate in strong hydrogen bonding interactions in addition to polar–polar interactions with lignin. Lignin has several hydroxyl groups in its structure, which could easily interact with the solvent via interactive forces of hydrogen bonding and dipole moments. Hansen and Björkman34 reported the Hansen solubility parameters (HSPs) for extracted wood lignin with values δD = 21.9 MPa1/2, δP = 14.1 MPa1/2, δH = 16.9 MPa1/2, and R0 = 13.7. Due to the heterogeneity of lignin and the significant variations in final extracted lignin depending on the pretreatment process, these parameters cannot be used universally. Thus, it is important to assume specific values for HSP in the function of the source of the lignin. In our study, we performed solubility experiments with L4 (lignin from wheat straw). Accordingly, we will use a specific set of values of HSP for sugar cane bagasse lignin as determined by Novo et al.33 Lignins from sugar cane (as well as lignins from grasses i.e. wheat straw) are classified as HGS lignin and contain more p-hydroxy phenyl moieties including p-coumarates and ferulates.35 These values are δD = 21.42 MPa1/2, δP = 8.57 MPa1/2, δH = 21.8 MPa1/2, and R0 = 13.56.
Results and discussion
Optimum solvolysis operating window
We optimized the solvolysis of lignin (without catalyst) towards oligomeric fractions by varying the lignin/alcohol ratio, the temperature, and the alcohol solvent. This optimization builds on an earlier techno-economic feasibility study in which we found that a viable catalytic process to convert lignin into solubilized oligomeric lignin fragments requires an ethanol (solvent) conversion below 5%.36 The described catalytic solvolysis process involved two steps. At a lower temperature, in the range of 120–200 °C, lignin is solubilized and some bonds are cleaved leading to lignin fragments with a lower molecular weight (Mw) in the range of 400–900 g mol−1. Cracking of these fragments using a catalyst requires temperatures higher than 300 °C. An inherent problem is the absorption of lignin fragments on the heterogeneous catalyst surface, leading to severe deactivation. Accordingly, we focus here on the first step, avoiding the use of a catalyst and aiming for solubilized lignin fragments.Solvolysis of lignin by ethanol was first conducted at a temperature of 200 °C at a lignin:ethanol ratio of 1:15 w/v and different reaction times (Fig. 1a). The yield of solubilized lignin was approximately 73–75 wt% after 4 h. We observed that the lignin concentration in the resulting crude lignin oil (CLO) increased slightly from 5.5 to 7.5 wt%, when the reaction time was increased from 0.5 h to 4 h. Non-catalytic lignin solvolysis at moderate temperatures in the 200–250 °C range in the absence of stabilizing agents has been studied before.37,38 Reaction time is an important parameter determining the yield and quality of fractionated lignins because of repolymerization reactions of intermediates. Another study emphasized the balance of repolymerization over depolymerization reactions and concluded that depolymerization without a catalyst is only dominant over repolymerization above 300 °C.39Fig. 2a shows the molecular weight distribution of the parent lignin (L4) and the solubilized lignin fractions derive from L4 for different reaction times. Compared to the Protobind lignin, shoulders at the low- and high-MW ends develop in the gel permeation chromatograms. The increased signal in the lower MW range (180–200 g mol−1) with increasing residence time points to the formation of depolymerized lignin fragments. Thermolytic cleavage of weak β-O-4 ether bonds, which according to the literature can already occur at a relatively mild temperature of 200 °C, resulted in low lignin monomer yields. In the absence of reducing and capping agents, repolymerization between reactive fragments will take place, leading to high molecular weight products.40,41 On the other hand, the high-MW shoulder increases during prolonged reaction. In the absence of a capping agent or hydrogenation of reactive double bonds, the rate of repolymerization of intermediate fractions is high, leading to the formation of heavier compounds.
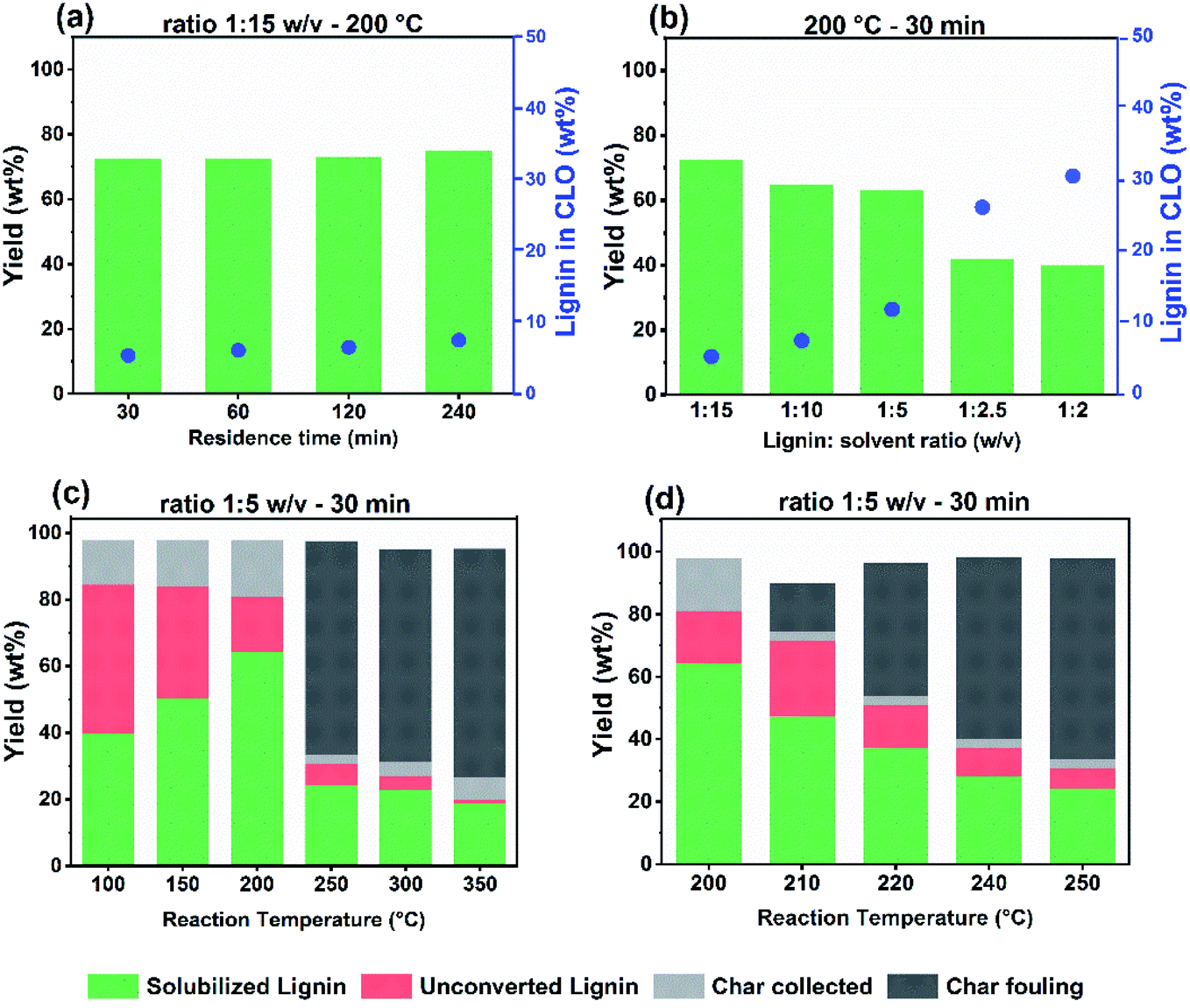 | ||
| Fig. 1 Solubilized lignin yield and lignin composition in the final crude lignin oil (CLO) product after solvolysis of Protobind soda lignin in ethanol at 200 °C and a fixed lignin-to-solvent ratio as a function of reaction time (a), and a fixed temperature of 200 °C and a residence time of 30 min as a function of the lignin-to-ethanol ratio (b). Solubilized lignin yield, unconverted lignin yield, collected residual char yield and residual fouling char are represented as a function of the reaction temperature at fixed lignin-to-ethanol ratio and reaction time (c and d). | ||
 | ||
| Fig. 2 Molecular weight distribution of Protobind soda lignin (THF-soluble fraction) and (a) solubilized lignin fractions obtained after solvolysis of soda lignin in ethanol at 200 °C, lignin:ethanol ratio 1:15 w/v at different reaction times, (b) solubilized lignin fractions obtained after solvolysis of lignin in ethanol at 200 °C, a residence time of 30 min and at various lignin:ethanol ratios. (Depicted chromatograms have been normalized). | ||
We next conducted a series of experiments at a temperature of 200 °C and a reaction time of 0.5 h in which we varied the lignin:ethanol ratio (Fig. 1b). Although the yield of solubilized lignin decreased with increasing lignin content, the concentration of lignin solubilized in the CLO product was higher. The final lignin concentration was 5.5 wt% at a lignin-to-ethanol ratio of 1:15 w/v. This concentration increased to approximately 30 wt% at a ratio of 1:2 w/v. Only a few studies investigated the influence of the lignin-to-alcohol ratio on solvolysis.12,25,42 A direct comparison to our data is however hampered due to differences in the lignin feedstock, the alcohol, and mainly the reaction parameters.
Next, we investigated the effect of solvolysis temperature in the range of 100–350 °C on the product yield at a lignin:ethanol ratio of 1:5 w/v and at a reaction time of 0.5 h. As at high temperature char is expected,25 we analysed the product mixture in more detail by distinguishing solubilized lignin and unconverted lignin residue, residual collected char and residual fouling char. This led to a closure of the mass balance above 90%. Char is a product of repolymerization at high temperature, characterized by a low H/C ratio. The solubilized lignin yield refers to the total amount of lignin in the organic ethanol phase, after separation from remaining solids via filtration. These residual solids were washed with THF to distinguish them in terms of residual collected char and unconverted lignin. The heavy char fractions collected from the reactor walls and stirrer were added to the char fraction, which is named residual fouling char. Reactor fouling is an important consideration with respect to economic viability, as it affects pre-treatment and cleaning requirements, operating conditions, safety, cost and performance.43 An optimum yield of 64 wt% solubilized lignin was obtained at 200 °C (lignin:ethanol ratio of 1:5 w/v). The corresponding yields of unconverted lignin and residual char yields under these conditions are 16 wt% and 17 wt%, respectively (Fig. 1c). At a lower reaction temperature, the concentration of solubilized lignin decreases to values in the range of 40–50 wt%, indicating that the remainder ends up as unconverted material. The amount of char remains at a similarly low level. The decreased concentration of solubilized lignin may indicate a lower solvolytic efficiency of ethanol for lignin at a lower temperature. The influence of temperature on the solubility parameters of solvents is not well understood. Hansen et al.34 reported that an increased temperature leads to higher solubility for entropic reasons. Williams et al.44 reported that increased pressure at a constant temperature will increase the total solubility parameters through an increase in the solvent density. Similarly, an increase in the temperature at constant pressure will decrease the total solubility parameter. The observation that higher lignin loading and temperatures in the range of 250–350 °C led to residual char as the dominant product can be mainly attributed to condensation reactions. This led to lower lignin liquefaction yields of 24 wt% at 250 °C and 18 wt% at 350 °C. The formation of char at higher temperatures was also reported by Nielsen et al.,25 who analyzed, by 13C MAS NMR, residual solids obtained from solvolysis of biorefinery lignin in ethanol (feed concentration 10 g/100 mL ethanol) at different reaction temperatures. Important findings were the removal of oxygen from the biomass above 300 °C and the removal of aliphatic side chains from the aromatic rings above 400 °C. The residual product has a strong char character consisting of polyaromatic hydrocarbons with a low H/C ratio. Similar trends were observed during catalytic ethanolysis in the work of Huang et al.45 under these extreme conditions. Based on the present data, the optimum is most likely in the temperature range of 200–250 °C with a residence time 30 min and a 1:5 lignin-to-ethanol ratio. We observed a rapid increase in the residual fouling with rising temperature in this operation window as shown in Fig. 1d. Fouling started already at 210 °C and gradually increased reaching a yield of 68 wt% at 250 °C. Clearly, the optimum temperature is around 200 °C.
Influence of alcohol
Solvolysis of soda lignin by methanol, ethanol, 1-butanol, 1-propanol, and 1-octanol was conducted at a temperature of 200 °C, a lignin:solvent ratio of 1:5 w/v and a reaction time of 30 min. We correlated lignin solubility with the Hansen solubility parameters using the lignin parameters (δD, δP, δH and Ro) determined by Novo et al.33 The RED and Hansen solubility parameters of the tested solvents are listed in Table 2.
:solvent ratio of 1:5 w/v
| Solvents | Hansen solubility parameters (MPa1/2) | RED (−) | ||
|---|---|---|---|---|
| δ D | δ P | δ H | ||
| Methanol | 15.1 | 12.3 | 22.3 | 0.9781 |
| Ethanol | 15.8 | 8.8 | 19.4 | 0.8477 |
| 1-Propanol | 16 | 6.8 | 17.4 | 0.8725 |
| 1-Butanol | 16 | 5.7 | 15.8 | 0.9378 |
| 1-Octanol | 16 | 5 | 11.2 | 1.1486 |
Fig. 3a shows the correlation between the RED and the yield of solubilized lignin for the investigated solvents. The RED values for methanol, ethanol, 1-propanol and 1-butanol fall in the lignin solubility sphere (smaller than unity), indicating a high solvolytic efficiency. This can explain their use in numerous lignin catalytic depolymerization and biomass fractionation studies.46–51 It is likely that lignin is first solubilized in the solvent before it is adsorbed on the catalyst for further depolymerization. 1-Octanol, however, has a RED value higher than 1, which suggests a lower ability to dissolve lignin. The dissolution rates are strongly dependent on the molar volume of the solvents, which is the lowest for 1-octanol among solvents investigated, because penetration rates increase for smaller solvent size.52Fig. 3b shows a linear correlation between the hydrogen bonding parameter (δH) of the solvents and the yield of solubilized lignin. Methanol has the highest polarity and hydrogen bonding ability and exhibits a RED close to unity, explaining its much better ability to dissolve lignin (61 wt%) than 1-octanol (38 wt%) with the lowest δH. Ethanol, 1-propanol and 1-butanol with δH and δP values between the aforementioned extreme cases of methanol and 1-octanol (Table 2) show a linearly decreasing lignin solubility of 56 wt%, 52 wt% and 51 wt% respectively.
 | ||
| Fig. 3 Lignin solubility yields as a function of the relative energy difference (RED) from the HSP sphere (a), and yields of lignin liquefaction after mild solvolysis treatment of soda lignin in 6 polar organic solvents at 200 °C for 30 min and a lignin:solvent ratio of 1:5 w/v as a function of the hydrogen bonding solubility parameter (b). | ||
31P NMR spectroscopy was employed to quantify aliphatic, aromatic, and carboxylic acid hydroxyl groups in the parent lignin and the soluble lignin fractions obtained by thermal solvolysis in methanol and ethanol under optimized conditions (200 °C, 30 min, lignin:solvent 1:5 w/v). The results collected in Fig. 4 show that the aliphatic and carboxylic hydroxyl contents of lignin solubilized in methanol and ethanol were lower than in the parent lignin. The ethanol-soluble lignin fractions showed the largest reduction of the aliphatic hydroxyl groups with respect to the parent lignin, whereas the aromatic hydroxyl content of the solubilized lignin fractions in methanol and ethanol was higher after thermal solvolysis. Given the complexity and heterogeneity of lignin, it is difficult to explain these differences in detail.53,54 The macromolecules in Protobind lignin have a molecular weight in the range of 500–10000 g mol−1 (see below). Reduction of aliphatic and carboxylic hydroxyl groups in solubilized lignin fractions is not fully understood. Decrease of the carboxylic moieties might occur due to their involvement with methanol in esterification reactions. Previous studies on thermal degradation of lignin (starting in the range of 200–275 °C) revealed decarboxylation and dehydration, releasing CO2 and H2O.55,56 Nielsen et al.25 reported the removal of aliphatic side chains of the aromatic structures in lignin. The observed release of CO2 during lignin ethanolysis at 250 °C also points to decarboxylation. Another hypothesis is that aliphatic groups are involved in condensation reactions between methoxy type of phenolic groups. Tonge et al.57 studied this reaction in detail and reported the formation of ether or methylene bridges between two methoxy groups at a low temperature 60 °C, leading to condensed structures with a high melting point.
 | ||
| Fig. 4 Aliphatic, aromatic and carboxylic hydroxyl content of the original Protobind soda lignin and soluble parts of lignin in methanol and ethanol after thermal solvolysis at 200 °C, a reaction time of 30 min and a lignin:solvent ratio of 1:5 w/v. | ||
The increased content of aromatic hydroxyl groups in methanol and ethanol solubilized lignin fractions may have several causes. The dominant one is the cleavage of phenolic ether linkages. Constant et al.58 reported that the relatively high phenolic to aliphatic hydroxyl ratios in lignin fractions obtained during pretreatment steps such as pulping originate from the cleavage of phenolic ether linkages, which occur together with recondensation reactions, during pretreatment steps such as pulping. Another study on thermal degradation of alkali lignin to phenolic compounds in subcritical and supercritical ethanol and water–ethanol solvent mixtures59 showed an increased phenolic content in the degraded lignin products, which was explained by hydrolysis and hydrogenolytic cleavage of aryl-O-aryl and aryl-O-aliphatic linkages. Xue et al.28 depolymerized lignin in ionic liquids and found that the increased aromatic hydroxyl content was strongly related to a lower molecular weight and lower β-O-4′ content in the depolymerized products. A second contribution for the increased phenolic OH content can be that these groups in the parent lignin are diluted due to the presence of high molecular-weight fragments with relatively few phenolic OH groups. Ragauskas and co-workers described in their investigations of structural alterations of lignin during thermochemical treatment typical interunit linkages and simplified structures for the lignin networks.60,61 Most of the proposed oligomeric structures containing 15–20 monomeric units contain a relatively low amount of phenolic OH groups compared to typically reported phenolic OH group content. Phenolic OH groups are preferably located at the external surface of the lignin networks at places where interunit linkages such as β-O-4′, β-5/α-O-4′, and β-β′ are absent. We speculate that the thermal solvolysis process solubilizes smaller and more polar aromatic chains, whereas heavy, more apolar chains with less phenolic OH groups will be rejected in the insoluble fraction. A similar trend was reported by Argyropoulos et al.,62 for the solubilization of softwood Kraft lignin in acetone at room temperature for 6 h. The lighter acetone-soluble fraction has a 35% higher phenolic OH content than the parent lignin, while the acetone-insoluble fraction with a higher Mw had much less phenolic OH groups.
We characterized the soluble and insoluble parts of the products obtained by thermal solvolysis in methanol, ethanol and 1-butanol in more detail by GPC. The normalized gel permeation chromatograms are shown in Fig. 5. Table 3 shows the average Mn, Mw, and the polydispersity (PD) of these fractions. All the solubilized fractions contained lighter macromolecules than the parent lignin and the insoluble fractions. The lower PD of the soluble lignin fractions points to the formation of lower MW fragments. While the reduction of Mn was minor in methanol, thermal solvolysis in ethanol and 1-butanol led to stronger reduction of the molecular weight. This can be compared with the molecular weight distributions (MWDs) shown in Fig. 5. Compared to the parent lignin, a shoulder at the high-Mw end develops in the gel permeation chromatogram of the methanol-soluble lignin fragments (Fig. 5a). This shoulder, which implies possible condensation of the extracted fractions, is not present after thermal solvolysis in ethanol (Fig. 5b) and 1-butanol (Fig. 5c). In the absence of a capping agent or hydrogenation of reactive double bonds, the rate of repolymerization of intermediate fractions is high, leading to the formation of high Mw compounds.45 Methanol exhibits a higher solvolytic efficacy than ethanol and 1-butanol. Therefore, it is likely that methanol can solubilize a part of the heavier compounds derived from recondensation, while these fragments end up in the insoluble fractions for the less polar solvents. This agrees not only with the observed high Mw of ethanol- and 1-butanol-insoluble lignin fractions (Table 3) but also with the polarity differences between the three solvents. In brief, methanol can solubilize a larger amount of condensed fragments with a high Mw.
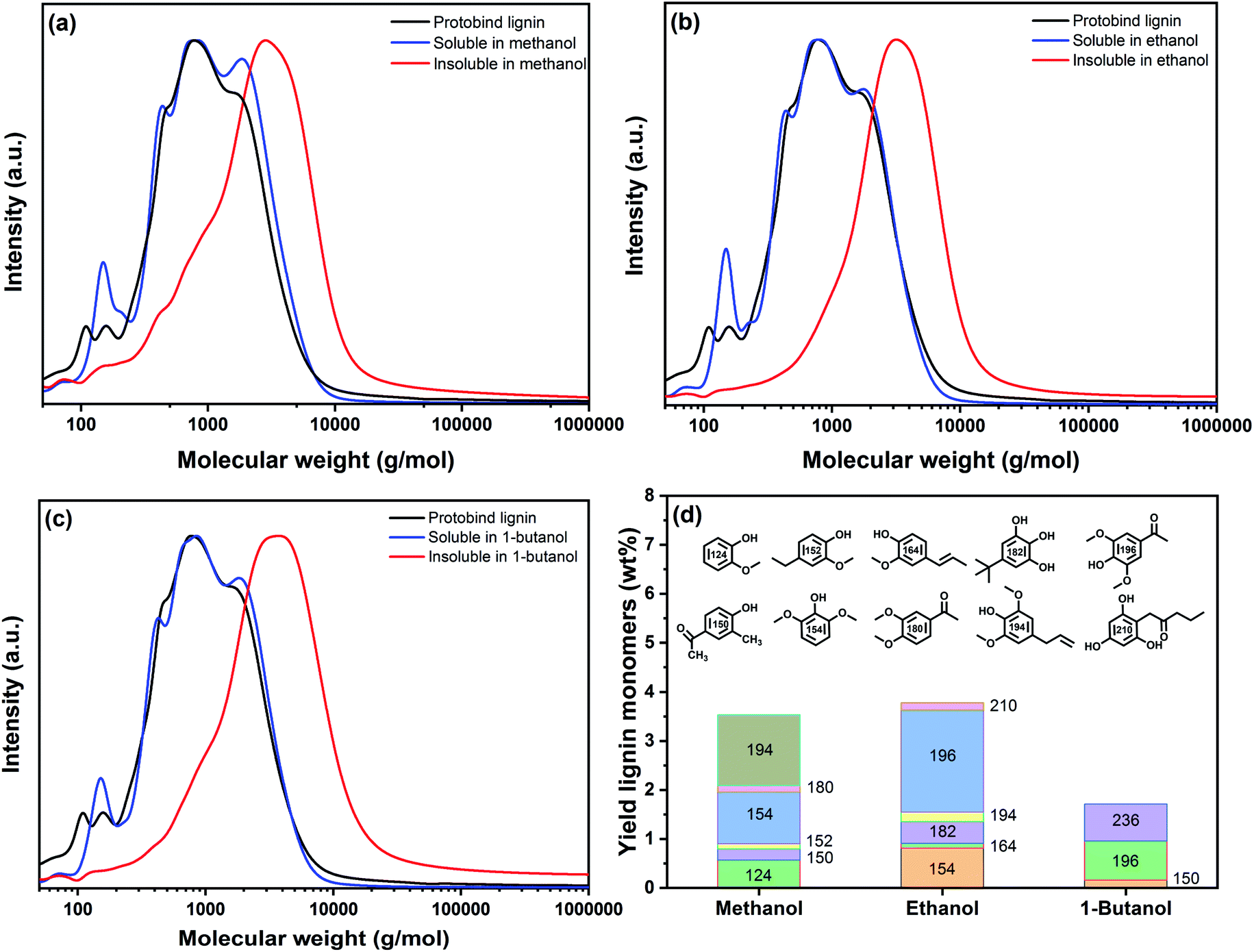 | ||
| Fig. 5 Molecular weight distribution of Protobind lignin together with the soluble and insoluble lignin fractions obtained after thermal solvolysis of soda lignin in (a) methanol, (b) ethanol and (c) 1-butanol at 200 °C, a lignin:solvent ratio 1:5 w/v and a reaction time of 30 min (chromatograms normalized). (d) Yield of lignin monomers in the soluble fraction deriving from thermal solvolysis of Protobind lignin in methanol, ethanol and 1-butanol at the same reaction conditions. | ||
:solvent ratio of 1:5 w/v, as identified by GPC
| Entry | Relative peak area (%) | Average values | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M W (g mol−1) | M n (g mol−1) | M w (g mol−1) | PD (−) | |||||||
| ∼150b | ∼430c | ∼800d | ∼1800e | ∼2842f | ∼3174g | ∼3800h | ||||
|
a The chromatograms were integrated on the basis of distinct contributions based on peak maxima and the peak-start and -end molecular weights are given in the footnote.
b [80–244 g mol−1].
c [244–498 g mol−1].
d [498–1415 g mol−1].
e [1415–18214 g mol−1].
f [96–322 976 g mol−1].
g [99–7761760 g mol−1].
h [97–11608841 g mol−1].
|
||||||||||
| Protobind lignin | 2 | — | 79 | 19 | — | — | — | 773 | 2465 | 3.19 |
| Soluble in methanol | 8 | 15 | 36 | 39 | — | — | — | 662 | 1417 | 2.14 |
| Insoluble in methanol | — | — | — | — | 100 | — | — | 1256 | 3984 | 3.17 |
| Soluble in ethanol | 7 | 17 | 42 | 32 | — | — | — | 554 | 1273 | 2.29 |
| Insoluble in ethanol | — | — | — | — | — | 100 | — | 2025 | 51547 |
25.45 |
| Soluble in 1-butanol | 5 | 16 | 42 | 35 | — | — | — | 623 | 1362 | 2.18 |
| Insoluble in 1-butanol | — | — | — | — | — | — | 100 | 1940 | 78335 |
40.36 |
Fig. 5a–c emphasizes that the MWD of the parent lignin is largely retained in the three solubilized fractions. Integration of the gel permeation chromatograms allowed estimating the contributions of different Mw classes present in the parent lignin and the different fractions after solvolysis (Table 3). The data show that thermal solvolysis results in the dissolution of a significant part of Protobind lignin in alcohol and cleavage of weak ether bonds, resulting in a higher proportion of low-Mw compounds such as monomers, dimers and trimers as compared to the parent lignin. The solubilized fractions contain monomers with a molecular weight below 200 g mol−1 and a significant amount of compounds with a molecular weight around 400 g mol−1. Moreover, a part of the lignin condenses into larger fragments (1700–1800 g mol−1), which is due to reactions between depolymerized compounds. The solubilized fraction is the largest for methanol solvent due to its higher polarity compared to the other two alcohols. The high-Mw fractions from the parent lignin are rejected from the solution. This fraction becomes larger going from methanol to ethanol to 1-butanol with dominant high-Mw compounds around 2800 g mol−1, 3200 g mol−1 and 4000 g mol−1 respectively, as revealed by deconvolution (Table 3). This fraction also includes condensed structures formed between reactive groups formed by cleavage reactions. We investigated in more detail the monomer fraction by GC analysis. Fig. 5d shows typical monomers for the three solvent-soluble fractions. The monomer contents in the solubilized fractions obtained in methanol, ethanol and 1-butanol were 3.6 wt%, 3.8 wt% and 2 wt%, respectively. The main products were syringol-type of monomers alkylated with methyl, ethyl, propyl and/or ketone groups substituents, likely derived from reactions with the solvent.
Table 4 shows the elemental analysis and higher heating value (HHV) of the parent lignin and the solvent soluble and insoluble lignin fractions after solvolysis of Protobind lignin in methanol, ethanol and 1-butanol at 200 °C. The oxygen content of the parent lignin is 34 wt%. Following solvolysis at 200 °C for 30 min and a lignin:solvent ratio of 1:5 w:v, the oxygen content decreased to approximately 29 wt% for methanol, and 28 wt% for ethanol and 1-butanol. The corresponding higher HHV are 29.7, 30.3 and 30.9 GJ per ton, respectively. Moreover, the sulphur content of the soluble lignin fractions is decreased after solvolysis in all three solvents. The reduction was the lowest for methanol (42%), intermediate for ethanol (55%) and the highest for 1-butanol (64%). There is growing interest in using a blend of lignin and alcohols as sustainable shipping fuels.71 In this context, the here proposed thermal solvolysis process can increase the heating value of the lignin part of the marine fuel and also decrease its sulphur content, relevant to more severe legislative restrictions on the sulphur content of marine fuels.75
:solvent ratio of 1:5 w/v
| Sample type | Nitrogen (wt%) | Carbon (wt%) | Hydrogen (wt%) | Sulphur (wt%) | Oxygen (wt%) | HHV (GJ per ton) |
|---|---|---|---|---|---|---|
| Protobind soda lignin | 0 | 59.4 | 5.7 | 0.7 | 34.0 | 27.6 |
|
||||||
| Methanol | ||||||
| Soluble lignin | 0 | 63.9 | 6.2 | 0.4 | 29.4 | 29.7 |
| Insoluble lignin | 0 | 56.5 | 5.1 | 0.3 | 37.1 | 25.9 |
|
||||||
| Ethanol | ||||||
| Soluble lignin | 0 | 64.7 | 6.4 | 0.3 | 28.5 | 30.3 |
| Insoluble lignin | 0 | 64.1 | 6.0 | 0.3 | 29.4 | 29.6 |
|
||||||
| 1-Butanol | ||||||
| Soluble lignin | 0 | 64.8 | 6.9 | 0.2 | 28.0 | 30.9 |
| Insoluble lignin | 0 | 61.9 | 5.6 | 0.5 | 31.7 | 28.3 |
2D HSQC NMR spectroscopy is widely used for the characterization of lignin structures.63,64Fig. 6 shows spectra of the starting and solubilized lignin in methanol, ethanol and 1-butanol under optimized conditions. Peak assignments for the parent lignin are based on the literature.65 Cross-signals from syringyl (S), guaiacyl (G) and hydroxyphenol (H) lignin units can be observed in the aromatic region of the spectrum in Fig. 6a. The measured S/G/H ratio of the original lignin is 50/38/15, which is in good agreement with the reported S/G/H ratio of 48/35/17 for the same Protobind lignin.42 Significant signals of ferulate (FA), p-hydroxybenzoate (PB), cinnamyl (I) and p-coumarate (pCA) units can also be seen. In the side-chain region (Fig. 6a, upper right), three signatures of interunit linkages (β-O-4′, β-5, and β-β′) can be clearly observed. The relative amount of side chains involved in the inter-unit expressed as a number per 100 aromatic units (S + G) is presented in Table 5. The analysis of the soluble fraction in methanol, ethanol and butanol by HSQC NMR shows that most of the β-O-4′ linkages (Aα, Aβ and Aγ) were cleaved. The use of methanol showed a higher propensity to cleave these bonds in comparison to ethanol and 1-butanol. These relatively weak ether linkages will cleave under mild conditions, while other interlinkages are unreactive under our conditions. These findings agree with earlier studies on lignin and lignin model compounds.67 Typically, the β-O-4′ bonds can be broken in the temperature range of 200–300 °C in a variety of solvents without a catalyst.66 Aryl–aryl ether bonds are more stable and can only be cleaved using catalysts at a higher temperature.68 Condensed linkages (β-β′, and β-5) were preserved and increased in all 3 investigated solvents. In particular, resinol (B) and phenylcoumarane (C) units were observed in all soluble lignin fractions, which reveal an increased condensation degree (Cβ & Cγ). Solubilization in the alcohol solvents preserved the G and S units, whereas the signal due to H units was slightly reduced. H units are more prone to condensation reactions because of the reduced steric hindrance compared to S and G units.69 The initially strong signals corresponding to FA, I, PB and pCA units decrease or even disappear due to the solvolysis step.
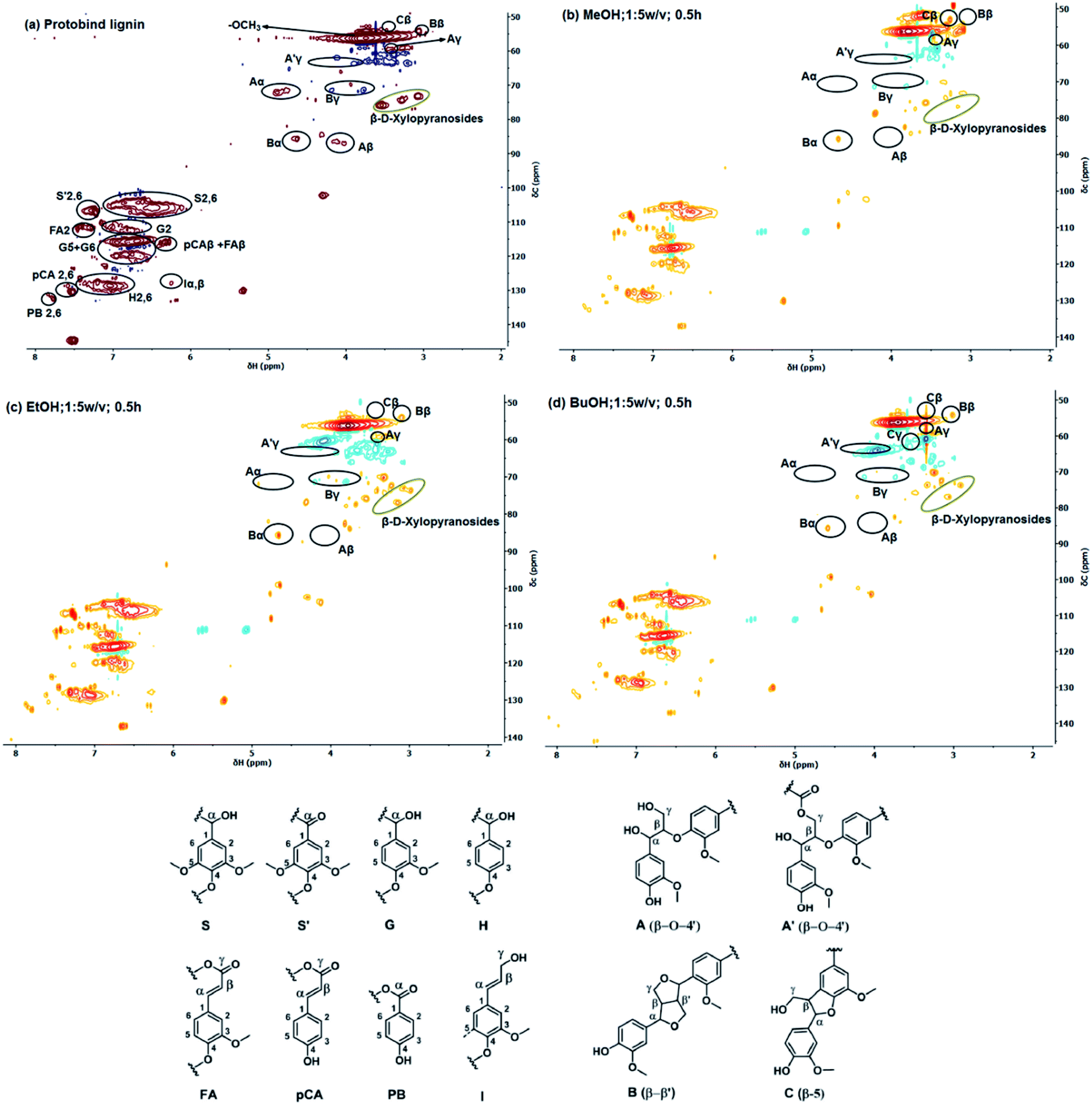 | ||
| Fig. 6 Comparison of the aromatic and side-chain regions of the 1H–13C HSQC NMR spectra of the (a) parent Protobind lignin and the soluble lignin fractions after (b) methanol, (c) ethanol and (d) butanol solvolysis at 200 °C for 30 min and 1:5 w/v lignin:solvent ratio. | ||
:solvent ratio of 1:5 w/v
| Linkages | Amount of linkages per 100 aromatic units | |||
|---|---|---|---|---|
| Protobind lignin | Methanol soluble lignin | Ethanol soluble lignin | 1-Butanol soluble lignin | |
| β-O-4′ (Aα) | 7.6 | 0 | 1.7 | 0 |
| β-O-4′ (Aβ) | 2.2 | 0 | 0 | 0 |
| β-O-4′ (Aγ) | 1.2 | 0.4 | 1.1 | 1.3 |
| β-5 (Cβ) | 0.9 | 1.8 | 0.8 | 1.1 |
| β-5 (Cγ) | 3.8 | 2.9 | 7.0 | 5.9 |
| β-β (Bα) | 0.9 | 3.4 | 2.8 | 2.7 |
| β-β (Bβ) | 2.8 | 2.9 | 3.3 | 4.0 |
| β-β (Bγ) | 3.0 | 5.5 | 3.9 | 2.9 |
Feedstock heterogeneity
In addition to its low reactivity, the heterogeneous nature of lignin makes it a challenging feedstock. This pertains to many aspects such as composition, structure, molecular weight distribution, and hydroxyl group content. Differences are primarily due to the type of biomass from which lignin is derived. Another important factor is the applied pre-treatment method to extract and isolate lignin. Differences in the ratio of methoxylated phenylpropane structures in woody biomass feedstock will influence the quality of the isolated lignins in the following process steps.70 Accordingly, we compared several technical lignins differing in composition (Table 1), MWD and PD (Fig. 7) and the number of functional hydroxyl groups (Fig. 8). We employed the optimum thermal solvolysis conditions using methanol (lignin:methanol ratio 1:5 w/v, 200 °C, 30 min). After reaction, we isolated and analyzed the solubilized lignin fractions by GPC and 31P NMR spectroscopy. Fig. 6 shows the MWD and PD of the parent technical lignins and the soluble fractions after thermal solvolysis in methanol. The parent lignin samples have a Mw in the range of 2121–2650 g mol−1 and a PD in the range of 3.01–3.26. After thermal solvolysis in methanol, the solubilized fractions had a Mw in the range of 1423–1636 g mol−1 and a PD in the range of 1.94–2.24. From the perspective of the molecular weight, the thermal solvolysis step can reduce the heterogeneity of the feedstock.
 | ||
| Fig. 7 Effect of thermal solvolysis of the original lignins (L) in methanol at 200 °C, a residence time of 30 min and a lignin:methanol ratio of 1:5 w/v on Mw and PD of the obtained soluble lignin fractions (SL). | ||
 | ||
| Fig. 8 Aliphatic (a), aromatic (b), carboxylic (c) and total (d) hydroxyl content of the original lignins and their soluble parts in methanol after thermal solvolysis at 200 °C, a residence time of 30 min and a lignin:methanol ratio 1:5 w/v. | ||
Fig. 8 shows the results of hydroxyl group analysis by 31P NMR spectroscopy. The aliphatic hydroxyl content in the soluble part of the lignins was decreased with respect to the original lignins (Fig. 8a). The same trend was observed for the carboxylic acid hydroxyl groups, which in most cases were eliminated completely (Fig. 8c). The phenolic hydroxyl content of methanol-soluble lignin fractions showed an overall increase for all lignin samples (Fig. 8b). For lignin L2, the phenolic OH content of the methanol-soluble fraction was increased to 2.65 mmol g−1, while the Mw was decreased from 2162 g mol−1 in the parent lignin to 1636 g mol−1 in the soluble fraction. Similarly, for the soluble fractions of L3 and L5 the phenolic hydroxyl content was significantly increased to 1.75 mmol g−1 and 3.89 mmol g−1, respectively, and the Mw was decreased to 1423 g mol−1 and 1598 g mol−1, respectively. The methanol-soluble fraction of L1 exhibited a small increase of the phenolic OH content, but similar to the other entries, a reduction of Mw (1544 g mol−1). These findings are in line with the hypothesis postulated for the solubilization of Protobind lignin L4 that methanol can solubilize lower Mw fractions with a higher content of phenolic OH groups. Overall, we observed that the total hydroxyl content of all the soluble lignin fractions, except for L1, was increased. Such reactive lignin fractions can for instance be used as polyols in biobased phenol-formaldehyde resins, polyurethanes, composites or binders.43
Economic feasibility
The proposed thermal solvolysis process to obtain CLO from technical lignin represents the conversion of a renewable feedstock to a platform product for the production of sustainable chemicals and fuels. The CLO can be directly valorized as a marine fuel (base of the value pyramid) or serve as an intermediate product for further processing into products with a higher value. There is significant interest from the shipping industry to explore the environmental and financial viability of advanced biofuels. An example is lignin ethanol oil (LEO), which may contribute to sustainable shipping.71 The CLO process represents a simple method to produce such fuel options like lignin methanol oil (LMO) and LEO. The lignin part of the LMO and LEO fuels can be benchmarked against the only advanced drop-in marine biofuel in the market today, i.e., Used Cooking Oil Methyl Ester (UCOME).76 UCOME is considered as 2G biofuel, because it is produced from non-edible used cooking oils and is currently being used in the shipping industry as a drop-in biofuel with fossil-derived bunker fuels.76 The HHV of UCOME is 40 GJ per ton and the current price on the energy market is at 1045 € per ton.72,73 Thus, UCOME represents an energy cost price of 26 € per GJ. The lignin oligomers dissolved in methanol or ethanol have a HHV of approximately 30 GJ per ton (Table 4), which prices them at about 780 € per ton at a calorie-adjusted price parity with UCOME. On the other hand, industrial lignin-rich feedstocks from 2G biorefineries can have a minimum selling price of € 36–59 per ton (1.3–2.1 € per GJ), as demonstrated in our previous work.74 The conversion of lignin to lignin oligomers thus represents at least an order of magnitude value increase in terms of price and energy, which makes the presented technology attractive for further commercialization.Conclusions
We have demonstrated a process that can fractionate a technical lignin like Protobind soda lignin into lignin oligomers with reduced molecular weight and polydispersity and an increased reactivity in terms of phenolic hydroxyl groups. The process involves a thermal solvolysis step at a moderate temperature (200 °C) in alcohols for short reaction times (∼0.5 h). The process is limited to a lignin:solvent ratio of 1:5 w/v. Under such conditions, high yields of lignin fragments in the solvents can be achieved with limited recondensation, solvent consumption and char formation. Among the linear alcohols investigated, lignin exhibited maximum solubility in methanol (61 wt%), ethanol (56 wt%), 1-propanol (53 wt%) and 1-butanol (51 wt%). 1-Octanol showed a substantially lower solubility towards lignin (38 wt%). Hansen solubility parameters provided a semi-quantitative measure of the solvent efficacy towards the product yield. Methanol is the preferred solvent, which can solubilize a wide range of also heavier lignin molecules, resulting from depolymerization and recondensation reactions. The optimized mild solvolysis conditions were applied to 5 different biorefinery lignins, showing that this approach can be used to fractionate a wide range of technical lignins into a favorable CLO product with reduced Mw and PD. 2D HSQC NMR analysis showed that in addition to fractionation also depolymerization took place to a limited extent, predominantly by cleavage of β-O-4′ linkages during lignin solvolysis in different alcohols. The number of aromatic and total hydroxyl groups in the soluble lignin fractions of the biorefinery lignins was higher than for the parent materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the framework of Chemelot InSciTe and is supported by financial contributions from the European Interreg V Flanders, the European Regional Development Fund (ERDF) within the framework of OP-Zuid, the province of Brabant and Limburg and the Dutch Ministry of Economy. A patent application entitled “A method for obtaining a stable lignin: polar organic solvent composition via mild solvolytic modifications” with international publication number WO2019/053287A1 was filed on September 18, 2018 by the inventors P. D. K., H. O., M. D. B and E. J. M. H.References
- British Petroleum, BP Statistical Review of World Energy, 2015, vol. 48 Search PubMed.
- W. K. Britt, Papermaking, https://www.britannica.com/technology/papermaking, accessed 6 July 2020 Search PubMed.
- H. Ludmila, J. Michal, Š. Andrea and H. Aleš, Wood Res., 2015, 60, 973–986 Search PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS.
- S. Yang, Y. Zhang, T. Q. Yuan and R. C. Sun, J. Appl. Polym. Sci., 2015, 132, 1–8 Search PubMed.
- S. Kalami, M. Arefmanesh, E. Master and M. Nejad, J. Appl. Polym. Sci., 2017, 134, 1–9 CrossRef.
- N. Mahmood, Z. Yuan, J. Schmidt and C. Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef.
- M. P. Pandey and C. S. Kim, Chemical Engineering and Technology, 2011, 34, 29–41 CrossRef CAS.
- J. Y. Kim, S. Oh, H. Hwang, T. su Cho, I. G. Choi and J. W. Choi, Chemosphere, 2013, 93, 1755–1764 CrossRef CAS.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 4941–4950 RSC.
- P. D. Kouris, X. Huang, M. D. Boot and E. J. M. Hensen, Top. Catal., 2018, 61, 1–11 CrossRef.
- A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049 RSC.
- J. Zakzeski and B. M. Weckhuysen, ChemSusChem, 2011, 4, 369–378 CrossRef CAS.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- M. J. Antal, S. G. Allen, D. Schulman, X. Xu and R. J. Divilio, Ind. Eng. Chem. Res., 2000, 39, 4040–4053 CrossRef CAS.
- J. B. Nielsen, A. Jensen, L. R. Madsen, F. H. Larsen, C. Felby and A. D. Jensen, Energy Fuels, 2017, 31, 7223–7233 CrossRef CAS.
- J. B. Nielsen, A. Jensen, C. B. Schandel, C. Felby and A. D. Jensen, Sustainable Energy Fuels, 2017, 1, 2006–2015 RSC.
- a. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Laboratory Analytical Procedure (LAP), 2012, vol. 17 Search PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, J. Agric. Food Chem., 2013, 61, 635–645 CrossRef CAS.
- J. H. Hildebrand, Chem. Rev., 1949, 44, 37–45 CrossRef CAS.
- G. C. Vebber, P. Pranke and C. N. Pereira, J. Appl. Polym. Sci., 2014, 131, 1–12 CrossRef.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- C. M. Hansen and A. Björkman, Holzforschung, 1998, 52, 335–344 CrossRef CAS.
- L. P. Novo and A. A. S. Curvelo, Ind. Eng. Chem. Res., 2019, 58, 14520–14527 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, Second Edition, CRC Press, 2nd edn, 2007 Search PubMed.
- J. C. del Río, A. G. Lino, J. L. Colodette, C. F. Lima, A. Gutiérrez, Á. T. Martínez, F. Lu, J. Ralph and J. Rencoret, Biomass Bioenergy, 2015, 81, 322–338 CrossRef.
- T. Renders, E. Cooreman, S. Van Den Bosch, W. Schutyser, S. F. Koelewijn, T. Vangeel, A. Deneyer, G. Van Den Bossche, C. M. Courtin and B. F. Sels, Green Chem., 2018, 20, 4607–4619 RSC.
- M. Oregui Bengoechea, N. Miletíc, M. H. Vogt, P. L. Arias and T. Barth, Bioresour. Technol., 2017, 234, 86–98 CrossRef CAS.
- E. M. Anderson, M. L. Stone, M. J. Hülsey, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS.
- S. Cheng, I. DCruz, M. Wang, M. Leitch and C. Xu, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- R. Brežný, V. Mihalov and V. Kováčik, Holzforschung, 1983, 37, 199–204 CrossRef.
- S. Mukundan, L. Atanda and J. Beltramini, Sustainable Energy Fuels, 2019, 3, 1317–1328 RSC.
- J. Yip, M. Chen, Y. S. Szeto and S. Yan, Bioresour. Technol., 2009, 100, 6674–6678 CrossRef CAS.
- C. A. Gasser, M. Č. Van, E. M. Ammann, A. Schäffer, P. Shahgaldian and P. F. Corvini, Environ. Biotechnol., 2017, 101, 2575–2588 CrossRef CAS.
- L. L. Williams, J. B. Rubin and H. W. Edwards, Ind. Eng. Chem. Res., 2004, 43, 4967–4972 CrossRef CAS.
- X. Huang, C. Atay, J. Zhu, S. W. L. Palstra, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2017, 5, 10864–10874 CrossRef CAS.
- F. Wang, Y. Z. Yu, Y. Chen, C. Y. Yang and Y. Y. Yang, Biotechnol. Rep., 2019, 24, e00363 CrossRef.
- T. Renders, S. Van Den Bosch, T. Vangeel, T. Ennaert, S. F. Koelewijn, G. Van Den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS.
- X. Ouyang, X. Huang, B. M. S. Hendriks, M. D. Boot and E. J. M. Hensen, Green Chem., 2018, 20, 2308–2319 RSC.
- T. Renders, S. Van Den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- J. S. Papanu, D. W. Hess, D. S. Soane (Soong) and A. T. Bell, J. Appl. Polym. Sci., 1990, 39, 803–823 CrossRef CAS.
- V. Suseela, Ecosystem Consequences of Soil Warming, 2019, ch. 4, pp. 103–124 Search PubMed.
- S. Stiefel, C. Marks, T. Schmidt, S. Hanisch, G. Spalding and M. Wessling, Green Chem., 2016, 18, 531–540 RSC.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44, 353–363 CAS.
- E. Avni, R. W. Coughlin, P. R. Solomon and H. H. King, Fuel, 1985, 64, 1495–1501 CrossRef CAS.
- L. Tonge, Faculty of Engineering and Industrial Sciences, PhD thesis, Swinburne University of Technology, 2007.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. De Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- S. Cheng, C. Wilks, Z. Yuan, M. Leitch and C. Xu, Polym. Degrad. Stab., 2012, 97, 839–848 CrossRef CAS.
- M. Li, Y. Pu and A. J. Ragauskas, Front. Chem., 2016, 4, 1–8 CrossRef.
- Y. Pu, F. Hu, F. Huang and A. J. Ragauskas, BioEnergy Res., 2015, 8, 992–1003 CrossRef CAS.
- D. S. Argyropoulos, H. Sadeghifar, C. Cui and S. Sen, ACS Sustainable Chem. Eng., 2014, 2, 264–271 CrossRef CAS.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- Z. Yuan, S. Cheng, M. Leitch and C. C. Xu, Bioresour. Technol., 2010, 101, 9308–9313 CAS.
- E. Minami, H. Kawamoto and S. Saka, J. Wood Sci., 2003, 49, 158–165 CrossRef CAS.
- A. A. Smirnov, Z. Geng, S. A. Khromova, S. G. Zavarukhin, O. A. Bulavchenko, A. A. Saraev, V. V. Kaichev, D. Y. Ermakov and V. A. Yakovlev, J. Catal., 2017, 354, 61–77 CrossRef CAS.
- Z. Fang, R. L. Smith and X. Qi, Production of Biofuels and Chemicals with Microwave, Springer Netherlands, 2014 Search PubMed.
- W. M. Goldmann, J. M. Anthonykutty, J. Ahola, S. Komulainen, S. Hiltunen, A. M. Kantola, V. V. Telkki and J. Tanskanen, Waste Biomass Valorization, 2020, 11, 3195–3206 CrossRef CAS.
- Maersk, Maersk join forces with industry peers and customers to develop LEO, 2019, https://www.maersk.com/news/articles/2019/10/29/maersk-join-forces-with-industry-peers-and-customers-to-develop-leo, last accessed 14.09.2020 Search PubMed.
- N. Jeyakumar and B. Narayanasamy, J. Therm. Anal. Calorim., 2020, 140, 457–473 CrossRef CAS.
- Greenea, Market and Analysis – Our View on the Waste Based Biodiesel, 2020, https://www.greenea.com/en/market-analysis/, accessed 14.09.2020 Search PubMed.
- S. V. Obydenkova, P. D. Kouris, E. J. M. Hensen, D. M. J. Smeulders, Y. van der Meer and M. D. Boot, Bioresour. Technol., 2019, 291, 12185 CrossRef.
- IMO, The 2020 Global Sulphur Limit, http://www.imo.org/en/MediaCentre/HotTopics/GHG/Documents/2020sulphurlimitFAQ2019.pdf, last accessed 18.09.2020 Search PubMed.
- D. Phillips and J. Tomkinson, NNFCC, 2019, Impilications of Imported Used Cooking Oil (UCO) as a Biodiesel Feedstock. https://www.nnfcc.co.uk/files/mydocs/UCOReport.pdf, last accessed 18.09.2020 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |